

Abstract
MLN64 is a late endosomal cholesterol-binding membrane protein that has been implicated in cholesterol transport from endosomal membranes to the plasma membrane and/or mitochondria, in toxin-induced resistance, and in mitochondrial dysfunction. Down-regulation of MLN64 in Niemann-Pick C1 deficient cells decreased mitochondrial cholesterol content, suggesting that MLN64 functions independently of NPC1. However, the role of MLN64 in the maintenance of endosomal cholesterol flow and intracellular cholesterol homeostasis remains unclear. We have previously described that hepatic MLN64 overexpression increases liver cholesterol content and induces liver damage. Here, we studied the function of MLN64 in normal and NPC1-deficient cells and we evaluated whether MLN64 overexpressing cells exhibit alterations in mitochondrial function. We used recombinant-adenovirus-mediated MLN64 gene transfer to overexpress MLN64 in mouse liver and hepatic cells; and RNA interference to down-regulate MLN64 in NPC1-deficient cells. In MLN64-overexpressing cells, we found increased mitochondrial cholesterol content and decreased glutathione (GSH) levels and ATPase activity. Furthermore, we found decreased mitochondrial membrane potential and mitochondrial fragmentation and increased mitochondrial superoxide levels in MLN64-overexpressing cells and in NPC1-deficient cells. Consequently, MLN64 expression was increased in NPC1-deficient cells and reduction of its expression restore mitochondrial membrane potential and mitochondrial superoxide levels. Our findings suggest that MLN64 overexpression induces an increase in mitochondrial cholesterol content and consequently a decrease in mitochondrial GSH content leading to mitochondrial dysfunction. In addition, we demonstrate that MLN64 expression is increased in NPC cells and plays a key role in cholesterol transport into the mitochondria.
Highlights
-
•
MLN64 overexpression induces an increase in mitochondrial cholesterol content.
-
•
MLN64 protein expression is increased in NPC cells.
-
•
Down-regulation of MLN64 restores mitochondrial membrane potential and superoxide levels in NPC cells.
-
•
MLN64 overexpression produces mitochondrial dysfunction.
Graphical abstract
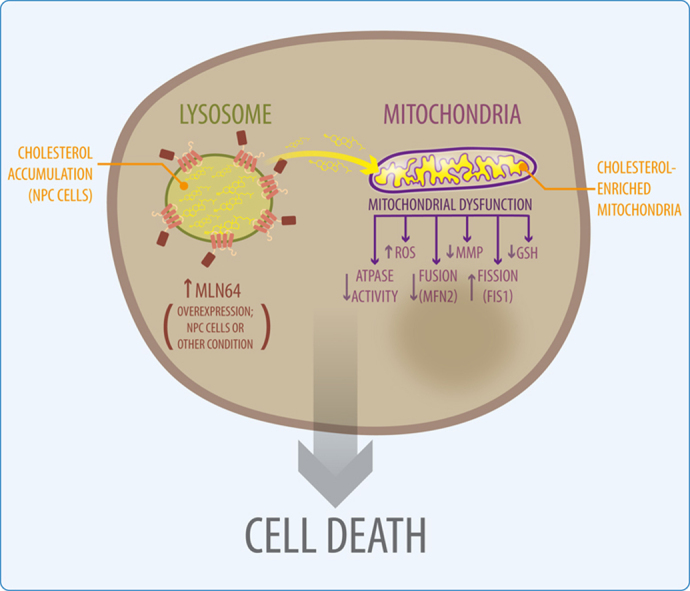
1. Introduction
MLN64 (metastatic lymph node protein 64) is expressed in all tissues [1], [2] and is an integral membrane protein localized primarily in the late endosomes. It has two functional domains, an N-terminal domain consisting of four transmembrane helices and a cytoplasmic C-terminal domain called START (C-terminal steroidogenic acute regulatory protein (StAR)-related lipid transfer) [3], [4]. Its N-terminus named MENTAL (MLN64 N-terminal) domain binds cholesterol [5], [6], and is responsible for the specific localization of the protein in the membrane of late endosomes [3]. Furthermore, crystallographic analysis showed that the START C- terminal domain of MLN64 is a cholesterol-binding domain [3].
Given that the START and MENTAL MLN64 domains are able to mobilize cholesterol [5] a model for endosomal cholesterol egress mediated by MLN64 has been proposed: cholesterol is captured by the MENTAL domain in the late-endosomal membranes, and then transferred by the cytoplasmic START domain to a cytosolic acceptor protein or membrane [5]. However, lack of MLN64 has little effect in mice [7], making it difficult to understand its physiological role.
MLN64 is highly homologous to the steroidogenic acute regulatory (StAR) protein and shares the highly conserved START domain [3]. StAR regulates the rate-limiting step of steroidogenesis, which is the transfer of cholesterol from the outer to the inner mitochondrial membrane, where it is converted into pregnenolone [8]. Overexpression of MLN64 enhances steroidogenesis [2], apparently by stimulating the mobilization of lysosomal cholesterol to the mitochondrial P450 cholesterol side chain cleavage enzyme, whereas mutant MLN64 lacking the START domain was reported to induce cholesterol accumulation into lysosomes [6].
These evidences suggest at least two possibilities: 1) MLN64 transport cholesterol to the mitochondria for steroidogenesis in organs such as brain and placenta that produce steroid hormones but lack the StAR protein [9], and 2) MLN64 complement StAR function in oxysterol synthesis, which also uses cholesterol as a precursor of steroids in the mitochondria.
Interestingly, Charman et al. [10] showed that reduction of MLN64 by RNA interference (siRNA) decreased cholesterol transport to the mitochondrial inner membrane and reduced mitochondrial cholesterol levels in Niemann-Pick C1 (NPC1) deficient cells. The NPC1 protein is involved in cholesterol egress from lysosomes and mutations in the Npc1 gene causes the Niemann-Pick type C (NPC) disease [11], [12] characterized by accumulation of unesterified cholesterol and other lipids within lysosomes.
The results from Charman et al. suggest that MLN64 mediates cholesterol transport to mitochondria, particularly when cholesterol homeostasis is altered such as in NPC disease. The latter suggests that MLN64, even in absence of NPC1, can mobilize endosomal cholesterol into the mitochondria [10]. On the other hand, recently it has been proposed that cholesterol handling in late endosomal compartments has two steps: first cholesterol enters MLN64 positive compartments from where it can be recycled to the plasma membrane or other membranes, and a second step involves cholesterol entering NPC1 endosomes that mediate cholesterol export to the ER [13].
We hypothesize that MLN64 plays a key role in maintaining an adequate cholesterol level in the mitochondrial membranes, which might be relevant for mitochondrial function.
According to this idea it has been recently published that during Anthrax lethal toxin infection of macrophages, MLN64 participates in mitochondrial cholesterol enrichment, mitochondrial hyperpolarization, mitochondrial glutathione (GSH) depletion and ROS generation [14].
Increased mitochondrial cholesterol content can lead to mitochondrial dysfunction, including reduced fluidity of mitochondrial membranes [15], reduced ATP generation [16], [17], and decreased mitochondrial GSH import [18], [19]. Previously, we found that mice with hepatic MLN64 overexpression exhibited significant liver damage and apoptosis [20]. However, whether this outcome relates or not to mitochondrial dysfunction remained unaddressed.
In view of the strong link between MLN64, mitochondrial cholesterol and mitochondrial function we focused on establishing whether MLN64 is capable of enhancing mitochondrial cholesterol transport and if this phenomenon is responsible for mitochondrial dysfunction. In addition, we evaluated the levels of MLN64 in different NPC models and addressed the impact of MLN64 overexpression in hepatic cells as well the effect of reduced MLN64 expression in CHO NPC1-deficient cells by RNA interference.
Our results show increased cholesterol levels in the mitochondrial fraction, resulting in decreased GSH levels and alterations in mitochondrial dynamics in MLN64 overexpressing cells. Furthermore, we found decreased mitochondrial membrane potential (MMP), increased mitochondrial superoxide production and mitochondrial fragmentation in MLN64 overexpressing cells. MLN64 reduction in NPC1-deficient cells restored MMP and oxidative stress. Moreover, our data presented here shows that mitochondrial cholesterol content is increased in MLN64 overexpressing cells and this leads to mitochondrial dysfunction in both in vivo and in vitro models suggesting that MLN64 plays a key role in cholesterol transport into the mitochondria.
2. Materials and methods
2.1. Cell culture
HepG2 cells were purchased from American Type Culture Collection (ATCC) (Vancouver, Canada) and were maintained in Advanced MEM medium (ADMEM) supplemented with 10% fetal bovine serum (FBS).
CHO-K1 cells were purchased from ATCC. CHO-K1 NPC1 deficient cells were generously provided by Laura Liscum from Tufts University School of Medicine (Boston, Massachusetts, USA). Both cell lines were maintained in F12 medium supplemented with 7.5% FBS.
2.2. Animals and diets
C57BL/6 J mice originally purchased from Jackson Laboratory (Bar Harbor, ME) were bred to generate our own colony. All mice had free access to water and a chow diet (<0.02% cholesterol; Prolab RMH 3000, PMI Feeds, St. Louis, MO). Protocols were performed according to accepted criteria for the humane care of experimental animals, and were approved by the review board for animal studies of our institution. (Bioethics Committee of the School of Medicine, Pontificia Universidad Católica de Chile; protocol number CEBA 10–017).
2.3. Preparation and administration of recombinant adenoviruses
The recombinant adenovirus encoding the full-length murine MLN64 cDNA (Ad. MLN64), under control of the cytomegalovirus (CMV) promoter, was generated by recombination using the AdEAsy system in bacterial cells (generously provided by Dr. Bert Vogelstein, The Johns Hopkins University, Baltimore, MD) [21]. The control adenovirus Ad. E1Δ, containing no transgene, was kindly donated by Dr. Karen Kozarsky (SmithKline Beecham Pharmaceutics, King of Prussia, PA). Large-scale production of recombinant adenoviruses was done in infected HEK 293 cells as described previously [22].
For viral administration, 8-weeks-old mice were anesthetized by isoflurane inhalation and 1x1011 viral particles (in 0.1 ml of isotonic saline buffer) of control or recombinant adenoviruses were administered by tail vein injection. An additional control group received 0.1 ml of saline buffer only. Animals were analyzed 48 h after adenoviral infection.
HepG2 cells were infected with 2.8x109 viral particles per 2.5x106 cells in ADMEM for 48 h.
2.4. Liver sampling
Mice were anesthetized by intraperitoneal injection of ketamine (80–100 mg/kg) and xylazine (5–10 mg/kg). The liver was used immediately for mitochondrial isolation or frozen at −80 °C for western blot analyses.
2.5. Mitochondria isolation from mice livers
Highly purified mitochondria from mice livers were prepared by rapid centrifugation on Percoll density gradients, as previously described [15].
2.6. Western blot analyses
Livers were disrupted in 500 µl of homogenization buffer (2 mM MgCl2, 0.25 M sucrose in 20 mM Tris buffer, pH 7.5) plus protease inhibitors (10 μg/ml leupeptin, 2 μg/ml pepstatin and 50 μg/ml PMSF). The homogenates were centrifuged at 3.000g for 10 min and the supernatants were used for protein quantification.
Cells were lysed with ice-cold lysis buffer (PBS, 1% NP40, 0.5% sodium deoxycholate, 0.1% SDS) plus proteases inhibitors. After centrifugation at 14.000g for 10 min, total protein in the supernatants was quantified using the bicinchoninic acid method (BCA kit, Thermo Scientific).
Western Blot analyses were performed using 30–50 μg of protein/lane in 12% denaturing polyacrylamide gel and electrophoresed on a PVDF membrane for detection using primary and secondary antibodies. Membranes were incubated with rabbit polyclonal antibody to MLN64 (1:1000; ab3478, Abcam), rabbit polyclonal antibody to OPA-1 (1:500; ab42364, Abcam) rabbit polyclonal antibody to TCC11, (1:250; ab71498, Abcam), rabbit polyclonal antibody to MFN-2 (1:500; ab50838, Abcam), rabbit polyclonal anti-β-actin (1:5000, Santa Cruz Biotechnology), rabbit polyclonal anti-ε-COP (1:5000, obtained from Dr Monty Krieger; Massachusetts Institute of Technology, Cambridge, MA, USA). After incubation with the primary antibody, the membrane was washed and incubated with peroxidase-conjugated secondary antibodies (1:5000 dilution; Amersham-Pharmacia), and the bound antibody was visualized with ECL detection on Kodak X-OMAT film (Eastman Kodak).
2.6.1. Quantitative real-time reverse transcriptase-polymerase chain reaction assay
Total RNA was extracted from liver, pretreated with DNase I (Invitrogen, Carlsbad, CA, USA), and then reverse-transcribed to cDNA using random primers (Invitrogen). The real-time polymerase chain reaction (PCR) was performed by SYBR Green I chemistry (SYBR Green PCR Master Mix, Applied Biosystems, Naerum, Denmark) with 25 ng of template cDNA and MLN64 gene-specific primers ( GACCAACTCAGAAAGGATGAC Reverse; TCGACATCTTTGTTCTGGCT Forward) present at 5 μM using the ABI 7500 sequence detection system (Applied Biosystems). Thermal cycling involved 35 cycles at 94 °C for 30 s, 55 °C for 30 s, and 72 °C for 30 s. The melting curves were performed according to the manufacturer’s procedure. The mRNA levels were quantified using the mathematical model described by Pfaffl et al. [23] and normalized to those of 18S rRNA.
2.7. Laser confocal imaging
For immunofluorescence, cultured HepG2 cells were fixed in 4% paraformaldehyde in PBS for 10 min. Cells were incubated for 2 h with 25 μg/ml filipin, rinsed with PBS, followed by incubation with primary antibodies, mouse anti-cytochrome C antibody (1:200, ab13575, Abcam) and rabbit anti-MLN64 polyclonal antibody (1:500), rinsed with PBS, and incubated for 2 h with secondary antibodies (1:1000). All steps after the addition of filipin were performed in the dark. After the final washes in PBS, cells were mounted and confocal images acquired using a Leica SP2 laser scanning confocal microscope.
For mitochondrial function analyses, cells were exposed to Mitosox (5 μM), MitoTracker Green (100 nM) and MitoTracker Red CMXRos (25 nM) in DMEM Medium for 30 min, washed with PBS, and imaged live.
For determination of mitochondrial shape, MEF cells that stably overexpress the Cerulean fluorescent protein targeted to the outer mitochondrial membrane (fused to the C-terminal 20 amino acids of human Bcl-xL, and kindly donated by Cliff Guy, St Jude Children's Research Hospital) were imaged in live using confocal microscopy. The mitochondrial sphericity index was measured from tridimensional confocal reconstructions using the IMARIS software.
2.8. Cholesterol and GSH levels quantifications
The amount of cholesterol in mitochondria was measured using high performance liquid chromatography (HPLC) with a Waters μBondapak C18 10-μm reversed-phase column (30 cm×4 mm inner diameter) as described previously [24].
GSH levels in either cytosol or mitochondria were determined as reported previously [25].
2.8.1. ATPase activity quantification
ATPase activity was measured as the hydrolysis rate of ATP determined by the production of inorganic Phosphorus (Pi) over time. This method is based on colorimetric determination of Pi following the Sumner method [26]. In brief, a 2 ml reaction buffer (KCL 100 mM, Tris/HCL 10 mM, pH 7,3) was combined with 1 mg of mitochondrial proteins. The sample was incubated 5 min at 37 °C. The reaction was initiated by adding ATP (at a final concentration of 4 mM), incubated for 15 min at 37 °C and then stopped by adding trichloroacetic acid at a final concentration of 6%. Then, the sample was centrifuged 10 min at 2500×g and the Pi produced was determined in the supernatant using a luminescence spectrometer at 660 nm.
2.9. RNA interference
Transfection with siRNA was performed according to the manufacturer’s protocol using DharmaFECT 4 (Dharmacon). siRNA (Dharmacon) was added to the cells at a final concentration of 50 nM for siNT (non-targeting) or siMLN64 for 96 h using F12 medium.
2.10. Statistical analysis
Mean and standard error of the mean values with the corresponding number of experiments are indicated in the figure legends. Probability values of the data for Student t-tests and ANOVA tests with Bonferroni’s post-test were calculated using the GraphPad Prism 5 software (Graph Pad Software, Inc., San Diego, USA).
3. Results
3.1. MLN64 overexpression increases mitochondrial cholesterol content in mice liver and cultured hepatocytes
We previously showed the overexpression of MLN64 in mouse liver following an adenovirus-mediated overexpression strategy [20]. The increase in hepatic MLN64 protein levels induced by the intravenous infusion of Ad. MLN64 was time-dependent reaching its peak between 24 h and 48 h after infection and decreasing to undetectable levels after 96 h [20]. Thus, all the subsequent experiments were performed after 48 h of Ad. MLN64 infection. To determine the effects of hepatic MLN64 overexpression, C57BL/6 mice were infected with Ad. MLN64 and the control Ad. E1Δ adenovirus. Western blot analysis of total liver homogenates demonstrated that MLN64 protein increased several times in the livers of Ad. MLN64-treated C57BL/6 mice 48 h after infection (Fig. 1A).
Fig. 1.

MLN64 overexpression increases mitochondrial cholesterol content in mice liver (A) MLN64 immunoblot. Total liver extracts (50 μg protein/lane) from Ad.E1Δ and Ad.MLN64 mouse livers 48 h after infection, run on 12% SDS-PAGE, transferred to PVDF membranes. (B) Mitochondria were isolated from Ad.E1Δ and Ad.MLN64 mouse livers 48 h after infection by Percoll density ultracentrifugation. The purity of mitochondria was confirmed by determining the distribution of specific markers such as CoxIV (mitochondria), Lamp1 and cathepsin B (lysosome) using immunoblot analysis. (C) Mitochondrial cholesterol content was quantified in the pure mitochondria fractions using HPLC. * Indicates statistically significant differences (p<0.05). (n Ad.E1Δ=5; n Ad.MLN64=5).
Then, we evaluated total and unesterified hepatic cholesterol levels in mice with adenovirus-mediated hepatic MLN64 overexpression and found a slight increase in hepatic unesterified cholesterol content relative to control animals (data not shown). Next, using HPLC we analyzed mitochondrial cholesterol content from mitochondria purified using a percoll gradient. The purity of our mitochondrial fraction was tested by western blot. Results showed that the isolated mitochondrial fractions were almost completely negative for the endolysosomal compartment antigens Cathepsin-B or Lamp1, while they were strongly positive for the mitochondrial protein CoxIV (Fig. 1B). Interestingly, we found a significant increase in mitochondrial cholesterol content in livers of MLN64-overexpressing mice compared to those infected with Ad. E1Δ (Fig. 1C).
To determine whether MLN64 overexpression yields the same results in cultured cells, we measured cholesterol content in MLN64-overexpressing HepG2 cells. Filipin staining show that these cells exhibited increased mitochondrial cholesterol accumulation that colocalized with a mitochondrial marker (Fig. 2A, and B). Therefore, both in vitro and in vivo results suggest that MLN64 is involved in cholesterol transport into mitochondria.
Fig. 2.

MLN64 overexpression increases mitochondrial cholesterol and decreases mitochondrial glutathione and ATPase activity in cultured hepatocytes (A) Immunofluorescence and filipin staining analysis of Ad.E1Δ and Ad.MLN64 HepG2 cells stained with anti-MLN64 (MLN64, red), anti-cytochrome c (Cyt-C, green) and filipin (cyan) 48 h after infection. Colocalization of Cyt-C and filipin indicating of mitochondrial cholesterol is depicted (white). Merge and zoom of the merge. Scale bar: 20 µm. (B) The graph shows the Pearson’s coefficient that measures the percentage of colocalization of filipin and Cyt-C. The photographs shown are representative confocal images, (n=3). (C) Mitochondrial glutathione content. Mitochondria were isolated by Percoll density ultracentrifugation from Ad.E1Δ and Ad.MLN64 HepG2 cells 48 h after infection. Glutathione in mitochondria was determined as described in the Materials and Methods section, (n=4). (D) ATPase activity in mitochondrial fraction from HepG2 cells (n=4) was quantified by measuring the hydrolysis rate of ATP. *Indicates statistically significant differences (p<0.05). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Rise in mitochondrial membrane cholesterol leads to decreased mitochondrial glutathione (mGSH) levels [18] and decreased ATPase activity [17]. Therefore, we evaluated mGSH levels and ATPase activity in Ad. MLN64 infected cells. The mGSH levels (Fig. 2C) and the ATPase activity (Fig. 2D) were significantly decreased in cells infected with Ad. MLN64 compared with those infected with the control Ad. E1Δ virus. Together, these results suggest that MLN64 mediates cholesterol transport into mitochondria leading to alterations in mitochondrial antioxidant defense and ATP synthesis.
3.2. MLN64 overexpression produces mitochondrial dysfunction in hepatocytes
So far, we found decreased mGSH levels in MLN64-overexpressing cells. Since mGSH is the principal mechanism for eliminating reactive oxygen species (ROS) in mitochondria [27], we used the fluorescent dye MitoSOX to explore mitochondrial superoxide production. Interestingly, MLN64-overexpressing cells showed increased MitoSOX fluorescence indicating higher levels of mitochondrial superoxide production compared with Ad. E1Δ virus infected cells (Fig. 3A, and B).
Fig. 3.

MLN64 overexpression produces mitochondrial dysfunction in hepatocytes (A) 48 h after Ad.E1Δ and Ad.MLN64 infection HepG2 cells were stained with MitoSOX Red to measure superoxide production and with Mitotracker Green to stain mitochondria respectively. Scale bar: 20 µm. (B) The graph shows the fluorescence intensity of MitoSOX as indicative of superoxide production. * p<0.05 (n=5). (C) MLN64 (green) and Mitotracker Red (red) staining of HepG2 cells 48 h after adenoviral infection. MMP levels were determined with Mitotracker red (MTred) staining and confocal imaging. MLN64 immunofluorescence was performed after cells were stained with MTred. The graph shows the quantification of MTred fluorescence intensity. * p<0.05 (n=5). Scale bar: 20 µm. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Then, we evaluated the effect of MLN64 overexpression over the MMP in living HepG2 cells using MitoTracker Red. This red-fluorescent dye stains mitochondria in live cells and its accumulation is dependent on MMP. We found that MLN64 overexpressing HepG2 cells show decreased MMP (Fig. 3C, and D). Interestingly, detailed observation of mitochondria reveal that besides presenting functional alterations, MLN64 overexpressing HepG2 cells showed morphological alterations, and presented mainly smaller and rounded mitochondria.
In order to further establish the impact of MLN64 overexpression on mitochondrial morphology, we analyzed mitochondrial sphericity in MEF cells expressing the cerulean fluorescent protein coupled to mitochondria. While Ad. E1Δ infected cells exhibited tubular mitochondria, we observed a dramatic shortening of mitochondria induced by MLN64 overexpression (Fig. 4A), associated to a significant increase in the mitochondrial sphericity, as calculated using the IMARIS software (Fig. 4B). Thus, we conclude that MLN64 overexpression induced mitochondrial fragmentation.
Fig. 4.

MLN64 overexpression triggers mitochondrial fragmentation in MEF and HepG2 cells. (A) MEF cells were infected with Ad.E1Δ and Ad.MLN64. Cerulean fluorescent protein in the outer mitochondrial membrane was analyzed 48 h after infection. Scale bar: 10 µm, zoom 5 µm. (B) The sphericity index was calculated from analysis of mitochondrial tridimensional morphology using the IMARIS software, (n=3). (C) Western blot analysis of mitochondrial dynamics proteins of HepG2 cells 48 h after adenoviral infection. OPA-1 and MFN-2 are mitochondrial fusion related proteins. FIS1 is a mitochondrial fission related protein. These proteins were analyzed by western blot and the bands intensity was normalized using the structural protein actin. * Indicates statistically significant differences (p<0.05) (n=3).
To further clarify the mechanism by which MLN64 induced mitochondrial fragmentation we analyzed the expression of several proteins involved in mitochondrial fusion and fission processes in HepG2 cells. As indicated in Fig. 4C the fusion protein MFN-2 was decreased in MLN64-overexpressing cells, without changes in the expression of other proteins involved in mitochondrial dynamics, such as FIS1 or OPA-1. These results are consistent with our previous observation described above of increased number of smaller and rounder mitochondria in MLN64-overexpressing cells compared with those infected with the control Ad. E1Δ virus.
Together, these results suggest that MLN64 overexpression produces alterations in mitochondrial function and shape.
3.3. NPC cells show increased MLN64 expression and increased mitochondrial fragmentation
Recent findings suggest that MLN64 is responsible for the increase in mitochondrial cholesterol in NPC cells [10], [28]. Thus, we studied the expression of MLN64 in livers from Npc1-/- (NPC) mice and in CHO NPC1-deficient (NPC) cells. MLN64 protein expression was increased in livers from NPC mice (Fig. 5A, and B) and CHO NPC cells (Fig. 5D-G). MLN64 mRNA levels showed a tendency to be increased in livers from NPC compared to WT mice, however the difference was not significant (Fig. 5C). As expected, CHO NPC cells showed an increased filipin staining in a punctuate perinuclear pattern consistent with cholesterol accumulation in lysosomes (Fig. 5F).
Fig. 5.

Npc1-/- mice liver and NPC cells show increased MLN64 expression. (A) To detect protein expression 30 μg of protein from liver homogenates were subjected to SDS-PAGE and western blotting for MLN64. ε-COP was used as a loading control. The Figure shows a western blot representative image, n=5. (B) Western blots bands intensity quantification. (C) Real time PCR analysis of MLN64 mRNA from mouse liver, n=4. (D) 30 μg of protein of CHO-WT and CHO-NPC cell extracts were analyzed by western blot against MLN64 and ε-COP. A representative image is shown, n=3. (E) Western blots bands intensity quantification. (F) MLN64 immunofluorescence analysis (Top). CHO-WT and CHO-NPC cells were immunostained with an anti-MLN64 antibody. Filipin staining (Bottom). CHO-WT and CHO-NPC cells were fixed, and cholesterol accumulation was detected by filipin staining. Scale bar: 20 µm (G) Graph shows quantifications of fluorescence. * Indicates statistically significant differences (p<0.05).
Given that MLN64 overexpression induces alterations in mitochondrial shape and that NPC cells show increased MLN64 expression we analyzed whether mitochondrial dynamics were altered in CHO-NPC cells and in MLN64-siRNA treated cells. We found that the mitochondrial fusion proteins MFN-2 and OPA-1 have the same levels of expression in the four groups tested (Fig. 6A). Interestingly, the mitochondrial fission protein FIS1 was increased in CHO-NPC cells compared to WT cells (Fig. 6A, and B). We evaluated if these alterations in mitochondrial dynamics have an impact on mitochondrial shape using mitotracker and confocal microscopy. Fig. 6C shows that NPC cells have smaller and rounded mitochondria compared to WT cells. Interestingly, MLN64 down-regulation had no effect on mitochondrial shape in NPC CHO cells (Fig. 6C). These findings suggest that the changes in mitochondrial dynamics are independent of MLN64 in NPC cells.
Fig. 6.

NPC cells show mitochondrial morphological alterations and MLN64 down-regulation improves mitochondrial function in NPC cells. CHO Wild-type (WT) and Npc1-/- (NPC) cells were transfected with nontargeting siRNA (siNT) and a siRNA directed against MLN64 (siMLN64) for 96 h. (A) 30 μg of proteins from cell homogenates were subjected to SDS-PAGE and analyzed by western blot using antibodies against the fission (FIS1) and fusion (MFN-2, OPA-1) related proteins . (B) The FIS1 bands intensities were measured using ImageJ and normalized with the ε-Cop protein. *p<0.05 (n=3) (C) Cells were immunostained with anti-MLN64 antibody (red), mitotracker green and nuclei were stained with Hoechst. (D) Cells were stained using Mitotracker red to measure the MMP or Mitosox (E) to quantify mitochondrial superoxide production 96 h post transfection. aStatistically different from WT. bStatistically different from NPC. cStatistically different from NPC+siNT. (n=3). Scale bar: 10 µm, zoom 5 µm.
3.4. MLN64 down-regulation in NPC cells improves mitochondrial function
Since MLN64 overexpression decreases MMP and increases mitochondrial superoxide production, we evaluated the effect of MLN64 down-regulation on MMP and mitochondrial superoxide levels in CHO-NPC cells.
Fig. 6A and C shows a reduction of MLN64 levels in cells treated with the MLN64-siRNA compared with the control-siRNA treated cells. Interestingly, MLN64-siRNA treated cells shows MMP and mitochondrial superoxide levels comparable to those of WT cells (Fig. 6D, and E). These results suggest that MLN64 contributes to mitochondrial dysfunction in NPC cells.
Taken together, our results suggest that MLN64 mediates cholesterol transport to the mitochondria producing mitochondrial dysfunction in hepatic cells and in NPC cells being in this last dissociated from mitochondrial dynamics.
4. Discussion
This work demonstrates the role of the endosomal protein MLN64 in mitochondrial function. Moreover, we show for the first time that MLN64 expression is increased in NPC cells and our results suggest that this protein could mediate cholesterol transport to the mitochondria leading to mitochondrial dysfunction. Therefore, aberrant mitochondrial function, mediated at least in part by MLN64, may contribute to cell damage in NPC disease.
It is well known that lysosomal cholesterol accumulation is detrimental in NPC cells [29], [30], [31]. However, the consequences of imbalances in mitochondrial cholesterol homeostasis and the mechanisms leading to an increase in mitochondrial cholesterol levels in NPC cells are not well characterized. This work shows the existence of mitochondrial alterations in NPC models and strongly suggests that the mechanism by which these changes occur involves MLN64-mediated increased mitochondrial cholesterol content. Indeed, mitochondrial cholesterol content was increased in MLN64 overexpressing cells, including mouse livers and hepatocytes (Figs. 1 and 2A). These results are in accordance with previous data that postulate a role for MLN64 in cholesterol transport to the inner mitochondrial membrane increasing steroidogenesis [2]. Because mitochondria also synthesize oxysterols from cholesterol, increasing cholesterol in mitochondria could induce an increase in circulating oxysterols. According to this idea, NPC cells show an increase in mitochondrial cholesterol [16], [32] and also of circulating oxysterols [33], which are differentially affected by recovery of mitochondrial GSH levels [32]. Therefore, MLN64 could promote this increase in oxysterol levels in NPC cells.
MLN64 overexpression and mitochondrial cholesterol overloading negatively impacted mGSH stores (Fig. 2C) and ATPase activity (Fig. 2D). These results were found in other models of mitochondrial cholesterol overloading as cancer [34] and in isolated mitochondria incubated with cholesterol [17], respectively. Interestingly, recent findings have reported that mGSH replenishment restored mitochondrial function in cerebellum of Npc1-/- mice and protected Purkinje cells against oxidative stress [32].
The source of the cholesterol that MLN64 transports into the mitochondria is controversial. One possibility is that the cholesterol increased in mitochondria comes from the plasma membrane, and another possibility is that comes from lysosomes. The first option is supported by the fact that the major source of cholesterol that reaches mitochondria comes from the plasma membrane [35], [36], mainly from HDL. Furthermore, an active transport of cholesterol from the plasma membrane into the mitochondria has been described [37]. Another study shows that MLN64 cycles between late endosomes and the plasma membrane [38], [39].
The second possibility is that augmented levels of cholesterol in mitochondria come from lysosomes. Recently Charman et al. [10] proposed that cholesterol transported by MLN64 into the mitochondria in NPC cells would originate from cholesterol accumulated in lysosomes, because the decrease of MLN64 levels does not alter mitochondrial cholesterol levels in WT cells.
On the other hand we have demonstrated that overexpression of MLN64 in WT mice livers and in hepatocytes increases mitochondrial cholesterol content. When MLN64 is overexpressed in WT cells the cholesterol that reaches the mitochondria could also come from lysosomes, since overexpression of an MLN64 domain causes a NPC like phenotype [4].
We believe that under physiological conditions MLN64 has no essential participation in cholesterol homeostasis and may cycle between the plasma membrane, lysosomes and mitochondria as a cholesterol “sensor”. However, under pathological conditions, such as NPC disease, in which MLN64 is overexpressed and trapped in lysosomes [40], MLN64 may facilitate the clearance of cholesterol from lysosomes to other organelles.
The mechanism by which MLN64 carries cholesterol into the mitochondria is uncertain. It was reported [14] that mitochondrial translocation of MLN64 results in mitochondrial cholesterol enrichment. Also, the START domain of MLN64 could be proteolyzed and reach the mitochondrial membrane [2], [6], [41]. Another possible mechanism is that the MLN64 MENTAL domain might serve to maintain cholesterol in the lysosomal membrane prior to its shuttling through the START domain to cytoplasmic acceptor(s), as it occurs with the family of oxysterol binding proteins (OSBP) and STAR family proteins.
Previously, we reported that MLN64 hepatic mRNA and protein levels are increased in mice with liver damage and that MLN64 overexpression triggered apoptosis and increased hepatic cholesterol content in mice liver [20]. However, we did not investigate the role of MLN64 in mitochondrial function. Here, we demonstrate that MLN64 overexpression leads to mitochondrial alterations such as decreased MMP and increased superoxide production (Fig. 3). Together our previous results and the results showed in this work suggest that MLN64 overexpression promotes cell death by carrying cholesterol into mitochondria that leads to mitochondrial dysfunction and subsequent apoptosis. In this regard, Ha et al [14] reported that the Anthrax lethal toxin induces rapid cell death by mitochondrial translocation of MLN64, resulting in cholesterol enrichment, membrane hyperpolarization, reactive oxygen species (ROS) generation, and depletion of mGSH.
The overexpression of MLN64 also leads to morphological alterations in mitochondria (Fig. 4). These morphological alterations could be explained by the fact that functional mitochondrial alterations could also have an impact on the morphology and dynamics of this organelle [42]. Therefore, although MLN64 is not a mitochondrial residing protein its overexpression impacts negatively both the function and the morphology of the mitochondria.
The increased mitochondrial fragmentation found in MLN64 overexpresing cells could be due to the decreased expression of Mitofusin-2 (MFN-2) (Fig. 4). MFN-2 helps to regulate the morphology of mitochondria by controlling the fusion process and also participates in the regulation of MPP, cellular metabolism and apoptosis [43], [44]. It is noteworthy that the overexpression of MLN64 was maintained only 48 h. The observation that the expression of the MFN-2 protein was decreased under these conditions suggests that MLN64 overexpression had a profound impact on mitochondrial dynamics. The mechanism by which MLN64 regulates MFN-2 expression is unclear. Research in this field suggests that MLN64 regulates late endosomal trafficking by altering the protein composition and function of ER-late endosome membrane contact sites (MCSs) [45]. Interestingly, other evidence suggests that MFN-2 localizes to both ER and mitochondria and participates in ER–mitochondria MCSs [46]. Consequently, the interaction between MLN64 and MFN-2 may take place at this MCSs and MLN64 could also alter MFN-2 expression.
The increased MLN64 expression found in NPC mouse liver and CHO cells (Fig. 5) could be the responsible factor for the mitochondrial alterations reported previously in NPC cells [10], [47]. It is noteworthy that we found higher levels of the MLN64 protein that were not associated with a significant increase in MLN64 mRNA levels. This could be due to alterations in posttranscriptional regulation of MLN64 expression, including defects on the degradation of MLN64 and/or protein accumulation in the endo-lysosome system due to lysosomal dysfunction in NPC disease [48].
To evaluate whether MLN64 triggers mitochondrial dysfunction in NPC cells we down-regulated MLN64 expression using siRNA in CHO-NPC cells. We found that by down-regulating MLN64 expression we restored MMP and mitochondrial superoxide production to near wild type levels (Fig. 6). Our results are in agreement with a recent publication in which decreased MLN64 levels restored mitochondrial cholesterol levels and mitochondrial function in NPC cells [28].
When we evaluated if there were changes in mitochondrial dynamics and morphology we found that decreased MLN64 expression in NPC cells produced no changes in the expression of proteins involved in mitochondrial dynamics or in establishing mitochondrial shape, indicating that mitochondrial dysfunction in NPC disease seems to be independent of mitochondrial dynamics or probably there are other factors that negatively impact on mitochondrial shape besides MLN64 and an increase in cholesterol content.
Finally, this work gives new insights into the function of MLN64 in cholesterol homeostasis and mitochondrial function. Our results suggest that MLN64, which is overexpressed in NPC cells, could mediate cholesterol transport to mitochondria leading to mitochondrial dysfunction. Therefore, MLN64 mediated cholesterol transport represents a possible target for the treatment of diseases where mitochondrial dysfunction is caused by increased mitochondrial cholesterol.
Acknowledgments
We gratefully acknowledge FONDECYT (grant # 1161374 to G.I.C, grants #1110310 and 1150816 to S.Z and grants #1120512 and #1161065 to A.R.A Also Basal Funding (CONICYT-PFB-12/2007 to A.R.A.). E.B. acknowledges support from CONICYT fellowship 24121660 and Vicerrectoría de Investigación (VRI) from Pontificia Universidad Católica de Chile. The authors declare no competing financial interests. Part of the work was supported by grant SAF2014-57674R and SAF2015-69944-R from Plan Nacional de I+D, Spain; and the center grant P50-AA-11999 (Research Center for Liver and Pancreatic Diseases, NIAAA/NIH).
Contributor Information
Elisa Balboa, Email: ebalboa@gmail.com.
Silvana Zanlungo, Email: silvana@med.puc.cl.
References
- 1.Soccio R.E., Breslow J.L. StAR-related lipid transfer (START) proteins: mediators of intracellular lipid metabolism. J. Biol. Chem. 2003;278:22183–22186. doi: 10.1074/jbc.R300003200. [DOI] [PubMed] [Google Scholar]
- 2.Watari H., Arakane F., Moog-Lutz C., Kallen C.B., Tomasetto C., Gerton G.L., Rio M.C., Baker M.E., Strauss J.F., 3rd MLN64 contains a domain with homology to the steroidogenic acute regulatory protein (StAR) that stimulates steroidogenesis. Proc. Natl. Acad. Sci. USA. 1997;94:8462–8467. doi: 10.1073/pnas.94.16.8462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alpy F., Stoeckel M.E., Dierich A., Escola J.M., Wendling C., Chenard M.P., Vanier M.T., Gruenberg J., Tomasetto C., Rio M.C. The steroidogenic acute regulatory protein homolog MLN64, a late endosomal cholesterol-binding protein. J. Biol. Chem. 2001;276:4261–4269. doi: 10.1074/jbc.M006279200. [DOI] [PubMed] [Google Scholar]
- 4.Alpy F., Wendling C., Rio M.C., Tomasetto C. MENTHO, a MLN64 homologue devoid of the START domain. J. Biol. Chem. 2002;277:50780–50787. doi: 10.1074/jbc.M208290200. [DOI] [PubMed] [Google Scholar]
- 5.Alpy F., Tomasetto C. MLN64 and MENTHO, two mediators of endosomal cholesterol transport. Biochem Soc. Trans. 2006;34:343–345. doi: 10.1042/BST0340343. [DOI] [PubMed] [Google Scholar]
- 6.Zhang M., Liu P., Dwyer N.K., Christenson L.K., Fujimoto T., Martinez F., Comly M., Hanover J.A., Blanchette-Mackie E.J., Strauss J.F., 3rd MLN64 mediates mobilization of lysosomal cholesterol to steroidogenic mitochondria. J. Biol. Chem. 2002;277:33300–33310. doi: 10.1074/jbc.M200003200. [DOI] [PubMed] [Google Scholar]
- 7.Kishida T., Kostetskii I., Zhang Z., Martinez F., Liu P., Walkley S.U., Dwyer N.K., Blanchette-Mackie E.J., Radice G.L., Strauss J.F., 3rd Targeted mutation of the MLN64 START domain causes only modest alterations in cellular sterol metabolism. J. Biol. Chem. 2004;279:19276–19285. doi: 10.1074/jbc.M400717200. [DOI] [PubMed] [Google Scholar]
- 8.Stocco D.M. StAR protein and the regulation of steroid hormone biosynthesis. Annu Rev. Physiol. 2001;63:193–213. doi: 10.1146/annurev.physiol.63.1.193. [DOI] [PubMed] [Google Scholar]
- 9.Sugawara T., Holt J.A., Driscoll D., Strauss J.F., 3rd, Lin D., Miller W.L., Patterson D., Clancy K.P., Hart I.M., Clark B.J. Human steroidogenic acute regulatory protein: functional activity in COS-1 cells, tissue-specific expression, and mapping of the structural gene to 8p11.2 and a pseudogene to chromosome 13. Proc. Natl. Acad. Sci. USA. 1995;92:4778–4782. doi: 10.1073/pnas.92.11.4778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Charman M., Kennedy B.E., Osborne N., Karten B. MLN64 mediates egress of cholesterol from endosomes to mitochondria in the absence of functional Niemann-Pick Type C1 protein. J. Lipid Res. 2010;51:1023–1034. doi: 10.1194/jlr.M002345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patterson M.C., Hendriksz C.J., Walterfang M., Sedel F., Vanier M.T., Wijburg F. Recommendations for the diagnosis and management of Niemann-Pick disease type C: an update. Mol. Genet Metab. 2012;106:330–344. doi: 10.1016/j.ymgme.2012.03.012. [DOI] [PubMed] [Google Scholar]
- 12.Carstea E.D., Morris J.A., Coleman K.G., Loftus S.K., Zhang D., Cummings C., Gu J., Rosenfeld M.A., Pavan W.J., Krizman D.B., Nagle J., Polymeropoulos M.H., Sturley S.L., Ioannou Y.A., Higgins M.E., Comly M., Cooney A., Brown A., Kaneski C.R., Blanchette-Mackie E.J., Dwyer N.K., Neufeld E.B., Chang T.Y., Liscum L., Strauss J.F., 3rd, Ohno K., Zeigler M., Carmi R., Sokol J., Markie D., O'Neill R.R., van Diggelen O.P., Elleder M., Patterson M.C., Brady R.O., Vanier M.T., Pentchev P.G., Tagle D.A. Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science. 1997;277:228–231. doi: 10.1126/science.277.5323.228. [DOI] [PubMed] [Google Scholar]
- 13.van der Kant R., Zondervan I., Janssen L., Neefjes J. Cholesterol binding molecules MLN64 and ORP1L mark distinct late endosomes with transporters ABCA3 and NPC1. J. Lipid Res. 2013 doi: 10.1194/jlr.M037325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ha S.D., Park S., Han C.Y., Nguyen M.L., Kim S.O. Cellular adaptation to anthrax lethal toxin-induced mitochondrial cholesterol enrichment, hyperpolarization, and reactive oxygen species generation through downregulating MLN64 in macrophages. Mol. Cell Biol. 2012;32:4846–4860. doi: 10.1128/MCB.00494-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Colell A., Garcia-Ruiz C., Lluis J.M., Coll O., Mari M., Fernandez-Checa J.C. Cholesterol impairs the adenine nucleotide translocator-mediated mitochondrial permeability transition through altered membrane fluidity. J. Biol. Chem. 2003;278:33928–33935. doi: 10.1074/jbc.M210943200. [DOI] [PubMed] [Google Scholar]
- 16.Yu W., Gong J.S., Ko M., Garver W.S., Yanagisawa K., Michikawa M. Altered cholesterol metabolism in Niemann-Pick type C1 mouse brains affects mitochondrial function. J. Biol. Chem. 2005;280:11731–11739. doi: 10.1074/jbc.M412898200. [DOI] [PubMed] [Google Scholar]
- 17.Echegoyen S., Oliva E.B., Sepulveda J., Diaz-Zagoya J.C., Espinosa-Garcia M.T., Pardo J.P., Martinez F. Cholesterol increase in mitochondria: its effect on inner-membrane functions, submitochondrial localization and ultrastructural morphology. Biochem. J. 1993;289(Pt 3):703–708. doi: 10.1042/bj2890703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mari M., Caballero F., Colell A., Morales A., Caballeria J., Fernandez A., Enrich C., Fernandez-Checa J.C., Garcia-Ruiz C. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab. 2006;4:185–198. doi: 10.1016/j.cmet.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 19.Garcia-Ruiz C., Mari M., Colell A., Morales A., Caballero F., Montero J., Terrones O., Basanez G., Fernandez-Checa J.C. Mitochondrial cholesterol in health and disease. Histol. Histopathol. 2009;24:117–132. doi: 10.14670/HH-24.117. [DOI] [PubMed] [Google Scholar]
- 20.Tichauer J.E., Morales M.G., Amigo L., Galdames L., Klein A., Quinones V., Ferrada C., Alvarez A.R., Rio M.C., Miquel J.F., Rigotti A., Zanlungo S. Overexpression of the cholesterol-binding protein MLN64 induces liver damage in the mouse. World J. Gastroenterol. 2007;13:3071–3079. doi: 10.3748/wjg.v13.i22.3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.He T.C., Zhou S., da Costa L.T., Yu J., Kinzler K.W., Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc. Natl. Acad. Sci. USA. 1998;95:2509–2514. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kozarsky K.F., Jooss K., Donahee M., Strauss J.F., 3rd, Wilson J.M. Effective treatment of familial hypercholesterolaemia in the mouse model using adenovirus-mediated transfer of the VLDL receptor gene. Nat. Genet. 1996;13:54–62. doi: 10.1038/ng0596-54. [DOI] [PubMed] [Google Scholar]
- 23.Pfaffl M.W., Horgan G.W., Dempfle L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucl. Acids Res. 2002;30:e36. doi: 10.1093/nar/30.9.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Duncan I.W., Culbreth P.H., Burtis C.A. Determination of free, total, and esterified cholesterol by high-performance liquid chromatography. J. Chromatogr. 1979;162:281–292. doi: 10.1016/s0378-4347(00)81515-7. [DOI] [PubMed] [Google Scholar]
- 25.Lluis J.M., Colell A., Garcia-Ruiz C., Kaplowitz N., Fernandez-Checa J.C. Acetaldehyde impairs mitochondrial glutathione transport in HepG2 cells through endoplasmic reticulum stress. Gastroenterology. 2003;124:708–724. doi: 10.1053/gast.2003.50089. [DOI] [PubMed] [Google Scholar]
- 26.Sumner J.B. A method for the colorimetric determination of phosphorus. Science. 1944;100:413–414. doi: 10.1126/science.100.2601.413. [DOI] [PubMed] [Google Scholar]
- 27.Fernandez-Checa J.C. Redox regulation and signaling lipids in mitochondrial apoptosis. Biochem. Biophys. Res. Commun. 2003;304:471–479. doi: 10.1016/s0006-291x(03)00619-3. [DOI] [PubMed] [Google Scholar]
- 28.Kennedy B.E., Madreiter C.T., Vishnu N., Malli R., Graier W.F., Karten B. Adaptations of energy metabolism associated with increased levels of mitochondrial cholesterol in Niemann-Pick type C1-deficient cells. J. Biol. Chem. 2014;289:16278–16289. doi: 10.1074/jbc.M114.559914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liao G., Yao Y., Liu J., Yu Z., Cheung S., Xie A., Liang X., Bi X. Cholesterol accumulation is associated with lysosomal dysfunction and autophagic stress in Npc1 -/- mouse brain. Am. J. Pathol. 2007;171:962–975. doi: 10.2353/ajpath.2007.070052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ballabio A., Gieselmann V. Lysosomal disorders: from storage to cellular damage. Biochim Biophys. Acta. 2009;1793:684–696. doi: 10.1016/j.bbamcr.2008.12.001. [DOI] [PubMed] [Google Scholar]
- 31.Ballabio A. Disease pathogenesis explained by basic science: lysosomal storage diseases as autophagocytic disorders. Int J. Clin. Pharm. Ther. 2009;47(Suppl 1):S34–S38. doi: 10.5414/cpp47034. [DOI] [PubMed] [Google Scholar]
- 32.Torres S., Matias N., Baulies A., Nunez S., Alarcon-Vila C., Martinez L., Nuno N., Fernandez A., Caballeria J., Levade T., Gonzalez-Franquesa A., Garcia-Roves P., Balboa E., Zanlungo S., Fabrias G., Casas J., Enrich C., Garcia-Ruiz C., Fernandez-Checa J.C. Mitochondrial GSH replenishment as a potential therapeutic approach for Niemann Pick type C disease. Redox Biol. 2016;11:60–72. doi: 10.1016/j.redox.2016.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reunert J., Fobker M., Kannenberg F., Du Chesne I., Plate M., Wellhausen J., Rust S., Marquardt T. Rapid diagnosis of 83 patients with Niemann Pick type C disease and related cholesterol transport disorders by Cholestantriol screening. EBioMedicine. 2016;4:170–175. doi: 10.1016/j.ebiom.2015.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ribas V., Garcia-Ruiz C., Fernandez-Checa J.C. Mitochondria, cholesterol and cancer cell metabolism. Clin. Transl. Med. 2016;5:22. doi: 10.1186/s40169-016-0106-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gocze P.M., Freeman D.A. Plasma membrane cholesterol is utilized as steroidogenic substrate in Y-1 mouse adrenal tumor cells and normal sheep adrenal cells. Exp. Cell Res. 1993;209:21–25. doi: 10.1006/excr.1993.1279. [DOI] [PubMed] [Google Scholar]
- 36.Freeman D.A., Romero A., Choi Y.S. Plasma membrane steroidogenic cholesterol: the relative importance of membrane internalization rate and cholesterol extraction rate of internalized membrane. Endocr. Res. 1998;24:619–622. doi: 10.3109/07435809809032657. [DOI] [PubMed] [Google Scholar]
- 37.Lange Y., Steck T.L., Ye J., Lanier M.H., Molugu V., Ory D. Regulation of fibroblast mitochondrial 27-hydroxycholesterol production by active plasma membrane cholesterol. J. Lipid Res. 2009;50:1881–1888. doi: 10.1194/jlr.M900116-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van der Kant R., Zondervan I., Janssen L., Neefjes J. Cholesterol-binding molecules MLN64 and ORP1L mark distinct late endosomes with transporters ABCA3 and NPC1. J. Lipid Res. 2013;54:2153–2165. doi: 10.1194/jlr.M037325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ren S., Hylemon P., Marques D., Hall E., Redford K., Gil G., Pandak W.M. Effect of increasing the expression of cholesterol transporters (StAR, MLN64, and SCP-2) on bile acid synthesis. J. Lipid Res. 2004;45:2123–2131. doi: 10.1194/jlr.M400233-JLR200. [DOI] [PubMed] [Google Scholar]
- 40.Strauss J.F., 3rd, Kishida T., Christenson L.K., Fujimoto T., Hiroi H. START domain proteins and the intracellular trafficking of cholesterol in steroidogenic cells. Mol. Cell Endocrinol. 2003;202:59–65. doi: 10.1016/s0303-7207(03)00063-7. [DOI] [PubMed] [Google Scholar]
- 41.Bose H.S., Baldwin M.A., Miller W.L. Evidence that StAR and MLN64 act on the outer mitochondrial membrane as molten globules. Endocr. Res. 2000;26:629–637. doi: 10.3109/07435800009048583. [DOI] [PubMed] [Google Scholar]
- 42.Ahmad T., Aggarwal K., Pattnaik B., Mukherjee S., Sethi T., Tiwari B.K., Kumar M., Micheal A., Mabalirajan U., Ghosh B., Sinha Roy S., Agrawal A. Computational classification of mitochondrial shapes reflects stress and redox state. Cell Death Dis. 2013;4:e461. doi: 10.1038/cddis.2012.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pich S., Bach D., Briones P., Liesa M., Camps M., Testar X., Palacin M., Zorzano A. The Charcot-Marie-tooth type 2A gene product, Mfn2, up-regulates fuel oxidation through expression of OXPHOS system. Hum. Mol. Genet. 2005;14:1405–1415. doi: 10.1093/hmg/ddi149. [DOI] [PubMed] [Google Scholar]
- 44.Pang W., Zhang Y., Zhao N., Darwiche S.S., Fu X., Xiang W. Low expression of Mfn2 is associated with mitochondrial damage and apoptosis in the placental villi of early unexplained miscarriage. Placenta. 2013;34:613–618. doi: 10.1016/j.placenta.2013.03.013. [DOI] [PubMed] [Google Scholar]
- 45.Phillips M.J., Voeltz G.K. Structure and function of ER membrane contact sites with other organelles. Nat. Rev. Mol. Cell Biol. 2016;17:69–82. doi: 10.1038/nrm.2015.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.de Brito O.M., Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456:605–610. doi: 10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- 47.Wos M., Szczepanowska J., Pikula S., Tylki-Szymanska A., Zablocki K., Bandorowicz-Pikula J. Mitochondrial dysfunction in fibroblasts derived from patients with Niemann-Pick type C disease. Arch. Biochem. Biophys. 2016;593:50–59. doi: 10.1016/j.abb.2016.02.012. [DOI] [PubMed] [Google Scholar]
- 48.Bi X., Liao G. Autophagic-lysosomal dysfunction and neurodegeneration in Niemann-Pick Type C mice: lipid starvation or indigestion? Autophagy. 2007;3:646–648. doi: 10.4161/auto.5074. [DOI] [PubMed] [Google Scholar]