

Abstract
Chronic thromboembolic pulmonary hypertension (CTEPH) occurs when thromboemboli travel to the pulmonary vasculature, fail to resolve, and cause elevated pulmonary arterial pressure. Untreated, this disease leads to progressive right heart failure and death. It develops in approximately 1% to 5% of patients who suffer an acute pulmonary embolism (PE) and has an overall incidence of 3 to 30 per million in the general population. While it is not entirely evident why most but not all people are able to clear this clot burden, there are known risk factors for the development of CTEPH. These include signs of right heart strain at the time of incident PE, inherited coagulopathies, inflammatory conditions, hypothyroidism, and a history of splenectomy. Since CTEPH can be treated both surgically and medically, it is critical to understand the pathophysiology of the disease so affected patients can be identified and diagnosed appropriately.
Keywords: pulmonary hypertension, chronic thromboembolic pulmonary hypertension, pulmonary embolism, pulmonary arterial hypertension
Introduction
Chronic thromboembolic pulmonary hypertension (CTEPH) is characterized by thromboembolic obstruction of the pulmonary arteries and vascular arteriopathy leading to vascular remodeling and pulmonary hypertension (PH). Untreated, this results in progressive right ventricular failure and death.1 According to the updated clinical classification of PH, CTEPH continues to be designated as Group 4 PH due to its unique pathophysiologic mechanisms.2 Although the exact incidence of the disease is unknown, registry data suggest that it affects between 3 to 30 people per million.3 CTEPH causes a significant burden on society due to excessive health care utilization, high-cost therapy, and increased mortality.4 Importantly, many patients with the disease can be surgically cured with pulmonary endarterectomy, which is the treatment of choice. Patients who undergo this operation have improved survival compared to patients treated with medical therapy alone.5 This review gives an overview of disease prevalence, pathophysiology, and clinical risk factors to enhance physician understanding and help them accurately identify and diagnose patients with this disease.
Epidemiology
Registry data suggest that the prevalence of CTEPH is approximately equal in men and women, with an average age of onset in the sixth decade of life.6 Although CTEPH is thought to be a sequela of chronic thromboembolic disease, a subset of patients ultimately diagnosed with the condition do not have a known prior history of venous thromboembolic disease or pulmonary embolism (PE). Some initial studies reported that up to 63% of patients seen at a center specializing in the treatment of CTEPH did not report a history of venous thromboembolism.7 However, a more recent large registry from Europe showed that 75% of patients did report a history of PE.6 Given that patients diagnosed with CTEPH may not have a prior history of thromboembolic disease, all patients with PH should undergo a work-up to exclude CTEPH.
The incidence of CTEPH after acute PE remains unclear. Historically, between 0.1% and 0.5% of patients with PE were thought to develop CTEPH.1 This range came into question when a study looking at serial echocardiographic data obtained at the time of PE and at 1 year of follow-up found that RV dysfunction was present in up to 8% of patients with acute PE.8 A landmark study published in 2004 enrolled 223 serial patients with acute PE who did not have a history of heart disease, lung disease, or other conditions that could have predisposed them to the development of PH. On follow-up, patients with unexplained shortness of breath were evaluated with a right heart catheterization (RHC). A total of 3.8% of patients developed CTEPH after 2 years of follow-up.9 Another study published 2 years later showed a rate of 1% after acute PE, but this study excluded patients with known permanent risk factors for thromboembolic disease such as cancer.10 Subsequent studies have reported a prevalence ranging between 0.4% and 4.8%.11–17 Differences in rates are likely due to differences in populations studied, screening methods for CTEPH, and length of follow-up.
Most studies suggest that CTEPH develops within 2 years of the acute PE.9,10,16,17 At least one study has evaluated the utility of a formal screening program for CTEPH after an incidental PE, but the program was ineffective in diagnosing additional patients with CTEPH as compared to those receiving usual care.12 Current guidelines state that there is insufficient evidence to recommend screening patients with acute PE for CTEPH.18 A claim database analysis suggests that less than 50% of patients with PE are evaluated with a follow-up echocardiogram.19 Whether or not this is sufficient remains to be determined.
Pathophysiology
Although various pathophysiologic mechanisms have been proposed to explain the development of CTEPH, questions remain as to why this condition only occurs in a minority of patients following an acute PE. Historically, two competing hypotheses were proposed. One early hypothesis was that local in situ thrombosis of smaller arteries resulted in a distal arteriopathy and subsequent PH.20 An important critique of this proposed mechanism is that it does not explain the proximal vascular occlusion commonly seen in CTEPH. The alternate hypothesis, therefore, is the thromboembolic theory: in this model, emboli from the systemic circulation become lodged within the pulmonary vasculature and fail to resolve. Various factors cause these emboli to progress and stabilize. A distal microvascular arteriopathy can develop subsequently due to elevated pulmonary pressures or from other effects triggered by the presence of these stabilized thrombotic lesions.3
A significant amount of knowledge regarding the disease process has been gained by evaluating pathologic specimens of lungs obtained from CTEPH patients. Such studies have shown a number of different types of vascular lesions in these patients. One characteristic finding is organized thrombus; these lesions are described as fibrotic obstructing lesions within the vessel lumen, which may or may not have channels of recanalization. Other vascular lesions mimic those seen in pulmonary arterial hypertension (PAH), including medial hypertrophy of the muscularis layer of pulmonary arterioles, concentric laminal internal fibroelastosis, and plexiform lesions. These lesions were even seen in vessels located in regions of the lung where there were no proximal thrombotic lesions causing decreased perfusion. These results generated additional hypotheses. First, they highlight the possibility that there are some mechanistic similarities between PAH and CTEPH. Second, the presence of vascular lesion in regions of the lung not affected by thrombotic disease suggests that systemic vascular mediators could be triggering the development of these lesions.21 In general, these pathologic findings support the thromboembolic hypothesis.3
One of the first important steps towards the pathogenesis of CTEPH is the non-resolution of thromboemboli that enter the pulmonary vasculature. In normal physiology, the first step in clot resolution is fibrinolysis. Next, an inflammatory response recruits neutrophils that continue the process of breaking down the clot. Endothelial progenitor cells and monocytes are also recruited to the region to promote angiogenesis and reorganization of the clot, respectively.22 In CTEPH, a number of the steps involved in thrombus breakdown could be abnormal. An early hypothesis suggested that the process of fibrinolysis itself was deranged; however, studies evaluating this process showed it was relatively normal in CTEPH patients.23 The structure of fibrin appears to be abnormal in certain patients, suggesting that dysfibrinogenemia could contribute to the pathogenesis of the disease.24,25 Other studies suggest that inflammation itself could affect the normal process of clot breakdown given that levels of C-reactive protein and certain cytokines have been shown to be elevated in CTEPH patients.26,27 The process of angiogenesis, which is thought to be an important step in the recanalization of the clot,27 may also be deranged in CTEPH.28 Some data suggest that a superimposed infection of the thromboembolus could play a role in its lack of resolution.29
The pathologic lesions in CTEPH show that the thromboemboli in the pulmonary vasculature not only fail to resolve but also proliferate and stabilize over time. The clots become organized and fibrous, replacing the normal intima of the arterial wall.22 One study showed that multipotent mesenchymal progenitor cells have been found within endarterectomized tissue; these could contribute to the fibrosis and stabilization of the clot.30
Vascular changes were noted not only in large vessels directly affected by clots but also in small distal arteries.21 This aspect of the disease is of crucial importance, as patients with primarily distal disease are considered poor surgical candidates. Additionally, small-vessel occlusion is thought to contribute to residual post-endarterectomy PH.3 Since there are small-vessel lesions found in vascular beds not affected by thromboembolism, the pathogenesis of this process could be secondary to the release of abnormal mediators from platelets or endothelial cells or due to dysregulated pulmonary blood flow.31 Precapillary and postcapillary vasculature has been implicated.32 A recent study highlights the importance of this small-vessel involvement by demonstrating how hemodynamic data obtained before and after endarterectomy correlated with the degree of pulmonary arteriopathy.33
The timeline for developing PH in patients with CTEPH is unclear. As noted above, studies monitoring the development of CTEPH after initial PE suggest that most cases are identified within 2 years. One of these studies, however, suggested that the process of thromboembolic disease may have been ongoing prior to the initial diagnosis of PE. Computed tomography scans were used to look for signs of CTEPH such as organized mural thrombi, arterial webs or bands, dilated bronchial arteries, and mosaicism of the pulmonary parenchymal perfusion. Interestingly, patients diagnosed with CTEPH had at least two of these signs at the time of index PE.16
Risk Factors
Many risk factors for the development of CTEPH have been identified (Table 1). These clinically derived predisposing conditions provide support for the pathogenic model of the disease described above. Given the now-accepted thromboembolic hypothesis of the disease, it is perhaps unsurprising that having a prior thromboembolic event is a risk factor for the development of this condition.34,35 Although a subset of patients eventually diagnosed with CTEPH have no history of thromboembolic disease, older data suggests that these patients comprise a relative minority.6
Table 1.
Risk factors for chronic thromboembolic pulmonary hypertension.
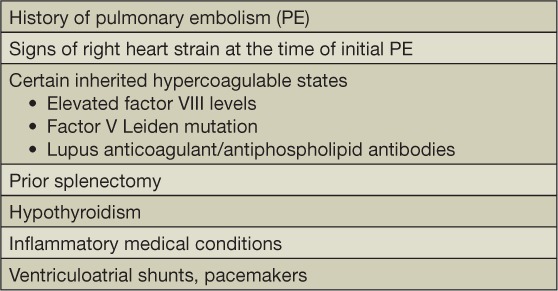
Certain hypercoagulable states are shown to confer a higher risk of CTEPH development, including elevated factor VIII levels,36,37 lupus anticoagulant/antiphospholipid antibodies,38 and factor V Leiden mutations.37 There is no evidence supporting the idea that deficiencies of antithrombin, protein C, or protein S are linked to CTEPH; in addition, prothrombin gene mutations are not shown to occur with higher frequency in those who develop this disease.3 Splenectomy, which in itself is thought to create a prothrombotic state, has been identified in a disproportionate number of CTEPH patients in multiple studies.34,35,39 History of malignancy confers an increased risk of developing this condition,34 as does being on thyroid hormone replacement, although it is unclear if this is due to the possible prothrombotic characteristics associated with the hypothyroid state or a direct effect of the thyroid hormone replacement.34
While traditionally there were no known familial or genetic links in CTEPH, some recent studies have suggested that a genetic predisposition could be present. One case of familial CTEPH has been reported,40 and BMPR2 mutations have been found in patients with CTEPH.41 A recent Chinese study found that patients with CTEPH had a higher frequency of mutations in the known PAH-related genes.42 Although these results are preliminary, they offer insight into the pathophysiology of this disease.
Certain medical conditions characterized by high levels of inflammation have been linked to CTEPH. Specifically, inflammatory bowel disease and osteomyelitis have been found to increase the odds of developing this condition.43 The link between inflammatory conditions and non-resolution of thrombotic disease is supported by data suggesting that inflammatory markers are high in patients with CTEPH compared to controls.27 Having foreign structures implanted in the heart also appears to increase the risk of CTEPH. In a number of studies, the presence of ventriculoatrial shunts for the treatment of hydrocephalus has been shown to increase the chance of developing this condition.34,35,43 The presence of pacemaker wires also increases risk.34,35 It is hypothesized that superimposed infection of these exogenous structures could contribute to the delayed resolution of thromboemboli within the pulmonary vascular bed.29
A number of studies have evaluated the risk of developing CTEPH based on characteristics noted at the time of PE. Elevated pulmonary artery systolic pressure on echocardiogram and larger perfusion defects at the time of initial PE were shown to be predictive for the development of CTEPH.8,14,16,17 Additionally, increased right ventricular/left ventricular ratio and high B-type natriuretic peptide levels were also predictive of CTEPH development.16,44 Older patients were more likely to develop CTEPH,8,16 as were those who had an idiopathic unprovoked PE.9,12,14,44 One group of researchers developed a clinical risk predictive score to identify patients with PE who are at high risk for developing CTEPH. While such tools are promising, further validation will be required before they can be adopted in clinical practice.44
Conclusion
Although it is a rare disease seen in 3 to 30 people per million, CTEPH uses extensive health care resources and is associated with high mortality. Approximately 0.4% to 4.8% of patients diagnosed with acute PE will go on to develop CTEPH. The pathophysiology of the disease is characterized by thrombi within the pulmonary vasculature that fail to resolve, persist, and stabilize with time. Although this results in large-vessel pathology, small-vessel disease plays an important role in the pathophysiology of this disease, particularly in patients who have persistent PH after endarterectomy and in those with primarily distal disease who may not be deemed surgical candidates. Known risk factors for CTEPH include prior thromboembolic disease, certain hypercoagulable states, chronic inflammation, splenectomy, and history of malignancy. Pulmonary artery endarterectomy is the recommended treatment of choice for patients deemed to be surgical candidates. With an improved understanding of the prevalence, pathogenesis, and risk factors for CTEPH, we hope that physicians will be better able to promptly identify and treat affected patients.
Key Points
Chronic thromboembolic pulmonary hypertension (CTEPH) is a rare disease seen in 3 to 30 per million and is associated with a high mortality.
Approximately 0.4% to 4.8% of patients diagnosed with acute pulmonary embolism will go on to develop CTEPH.
Known risk factors for CTEPH include prior thromboembolic disease, certain hypercoagulable states, chronic inflammation, splenectomy, and history of malignancy.
Early recognition of this disease and appropriate treatment is the key to improve outcome. Pulmonary artery endarterectomy is the recommended treatment of choice for patients deemed to be surgical candidates.
Conflict of Interest Disclosure
Dr. Safdar is a consultant, on the speakers' bureau, and an advisory board member for Actelion Pharmaceuticals, Gilead Sciences, Bayer Pharmaceuticals, United Therapeutics, Genetech, and Boehringer Ingelheim.
References
- 1. Fedullo PF, Auger WR, Kerr KM, Rubin LJ.. Chronic thromboembolic pulmonary hypertension. N Engl J Med. 2001. November 15; 345( 20): 1465– 72. [DOI] [PubMed] [Google Scholar]
- 2. Simonneau G, Gatzoulis MA, Adatia I, . et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2013. December 24; 62( 25 Suppl): D34– 41. [DOI] [PubMed] [Google Scholar]
- 3. Lang IM, Pesavento R, Bonderman D, Yuan JX.. Risk factors and basic mechanisms of chronic thromboembolic pulmonary hypertension: a current understanding. Eur Respir J. 2013. February; 41( 2): 462– 8. [DOI] [PubMed] [Google Scholar]
- 4. Schweikert B, Pittrow D, Vizza CD, . et al. Demographics, clinical characteristics, health resource utilization and cost of chronic thromboembolic pulmonary hypertension patients: retrospective results from six European countries. BMC Health Serv Res. 2014. June 9; 14: 246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Delcroix M, Lang I, Pepke-Zaba J, . et al. Long-Term Outcome of Patients With Chronic Thromboembolic Pulmonary Hypertension: Results From an International Prospective Registry. Circulation. 2016. March 1; 133( 9): 859– 71. [DOI] [PubMed] [Google Scholar]
- 6. Pepke-Zaba J, Delcroix M, Lang I, . et al. Chronic thromboembolic pulmonary hypertension (CTEPH): results from an international prospective registry. Circulation. 2011. November 1; 124( 18): 1973– 81. [DOI] [PubMed] [Google Scholar]
- 7. Lang IM. Chronic thromboembolic pulmonary hypertension--not so rare after all. N Engl J Med. 2004. May 27; 350( 22): 2236– 8. [DOI] [PubMed] [Google Scholar]
- 8. Ribeiro A, Lindmarker P, Johnsson H, Juhlin-Dannfelt A, Jorfeldt L.. Pulmonary embolism: one-year follow-up with echocardiography doppler and five-year survival analysis. Circulation. 1999. March 16; 99( 10): 1325– 30. [DOI] [PubMed] [Google Scholar]
- 9. Pengo V, Lensing AW, Prins MH, . et al. Incidence of chronic thromboembolic pulmonary hypertension after pulmonary embolism. N Engl J Med. 2004. May 27; 350( 22): 2257– 64. [DOI] [PubMed] [Google Scholar]
- 10. Becattini C, Agnelli G, Pesavento R, . et al. Incidence of chronic thromboembolic pulmonary hypertension after a first episode of pulmonary embolism. Chest. 2006. July; 130( 1): 172– 5. [DOI] [PubMed] [Google Scholar]
- 11. Miniati M, Monti S, Bottai M, . et al. Survival and restoration of pulmonary perfusion in a long-term follow-up of patients after acute pulmonary embolism. Medicine (Baltimore). 2006. September; 85( 5): 253– 62. [DOI] [PubMed] [Google Scholar]
- 12. Klok FA, van Kralingen KW, van Dijk AP, Heyning FH, Vliegen HW, Huisman MV.. Prospective cardiopulmonary screening program to detect chronic thromboembolic pulmonary hypertension in patients after acute pulmonary embolism. Haematologica. 2010. June; 95( 6): 970– 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Poli D, Grifoni E, Antonucci E, . et al. Incidence of recurrent venous thromboembolism and of chronic thromboembolic pulmonary hypertension in patients after a first episode of pulmonary embolism. J Thromb Thrombolysis. 2010. October; 30( 3): 294– 9. [DOI] [PubMed] [Google Scholar]
- 14. Korkmaz A, Ozlu T, Ozsu S, Kazaz Z, Bulbul Y.. Long-term outcomes in acute pulmonary thromboembolism: the incidence of chronic thromboembolic pulmonary hypertension and associated risk factors. Clin Appl Thromb Hemost. 2012. June; 18( 3): 281– 8. [DOI] [PubMed] [Google Scholar]
- 15. Giuliani L, Piccinino C, D'Armini MA, . et al. Prevalence of undiagnosed chronic thromboembolic pulmonary hypertension after pulmonary embolism. Blood Coagul Fibrinolysis. 2014; 25( 7): 649– 53. [DOI] [PubMed] [Google Scholar]
- 16. Guerin L, Couturaud F, Parent F, . et al. Prevalence of chronic thromboembolic pulmonary hypertension after acute pulmonary embolism. Prevalence of CTEPH after pulmonary embolism. Thromb Haemost. 2014. September 2; 112( 3): 598– 605. [DOI] [PubMed] [Google Scholar]
- 17. Yang S, Yang Y, Zhai Z, . et al. Incidence and risk factors of chronic thromboembolic pulmonary hypertension in patients after acute pulmonary embolism. J Thorac Dis. 2015. November; 7( 11): 1927– 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Galiè N, Humbert M, Vachiery JL, . et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Respir J. 2015. October; 46( 4): 903– 75. [DOI] [PubMed] [Google Scholar]
- 19. Tapson VF, Platt DM, Xia F, . et al. Monitoring for Pulmonary Hypertension Following Pulmonary Embolism: The INFORM Study. Am J Med. 2016. September; 129( 9): 978– 85. [DOI] [PubMed] [Google Scholar]
- 20. Peacock A, Simonneau G, Rubin L.. Controversies, uncertainties and future research on the treatment of chronic thromboembolic pulmonary hypertension. Proc Am Thorac Soc. 2006. September; 3( 7): 608– 14. [DOI] [PubMed] [Google Scholar]
- 21. Moser KM, Bloor CM.. Pulmonary vascular lesions occurring in patients with chronic major vessel thromboembolic pulmonary hypertension. Chest. 1993. March; 103( 3): 685– 92. [DOI] [PubMed] [Google Scholar]
- 22. Lang I. Chronic thromboembolic pulmonary hypertension: a distinct disease entity. Eur Respir Rev. 2015. June; 24( 136): 246– 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Olman MA, Marsh JJ, Lang IM, Moser KM, Binder BR, Schleef RR.. Endogenous fibrinolytic system in chronic large-vessel thromboembolic pulmonary hypertension. Circulation. 1992. October; 86( 4): 1241– 8. [DOI] [PubMed] [Google Scholar]
- 24. Morris TA, Marsh JJ, Chiles PG, Auger WR, Fedullo PF, Woods VL Jr.. Fibrin derived from patients with chronic thromboembolic pulmonary hypertension is resistant to lysis. Am J Respir Crit Care Med. 2006. June 1; 173( 11): 1270– 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li JF, Lin Y, Yang YH, . et al. Fibrinogen Aα Thr312Ala polymorphism specifically contributes to chronic thromboembolic pulmonary hypertension by increasing fibrin resistance. PLoS One. 2013. July 22; 8( 7): e69635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zabini D, Heinemann A, Foris V, . et al. Comprehensive analysis of inflammatory markers in chronic thromboembolic pulmonary hypertension patients. Eur Respir J. 2014. October; 44( 4): 951– 62. [DOI] [PubMed] [Google Scholar]
- 27. Quarck R, Wynants M, Verbeken E, Meyns B, Delcroix M.. Contribution of inflammation and impaired angiogenesis to the pathobiology of chronic thromboembolic pulmonary hypertension. Eur Respir J. 2015. August; 46( 2): 431– 43. [DOI] [PubMed] [Google Scholar]
- 28. Waltham M, Burnand KG, Collins M, McGuinness CL, Singh I, Smith A.. Vascular endothelial growth factor enhances venous thrombus recanalisation and organisation. Thromb Haemost. 2003. January; 89( 1): 169– 76. [PubMed] [Google Scholar]
- 29. Bonderman D, Jakowitsch J, Redwan B, . et al. Role for staphylococci in misguided thrombus resolution of chronic thromboembolic pulmonary hypertension. Arterioscler Thromb Vasc Biol. 2008. April; 28( 4): 678– 84. [DOI] [PubMed] [Google Scholar]
- 30. Firth AL, Yao W, Ogawa A, Madani MM, Lin GY, Yuan JX.. Multipotent mesenchymal progenitor cells are present in endarterectomized tissues from patients with chronic thromboembolic pulmonary hypertension. Am J Physiol Cell Physiol. 2010. May; 298( 5): C1217– 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Galiè N, Kim NH.. Pulmonary microvascular disease in chronic thromboembolic pulmonary hypertension. Proc Am Thorac Soc. 2006. September; 3( 7): 571– 6. [DOI] [PubMed] [Google Scholar]
- 32. Dorfmuller P, Gunther S, Ghigna MR, . et al. Microvascular disease in chronic thromboembolic pulmonary hypertension: a role for pulmonary veins and systemic vasculature. Eur Respir J. 2014. November; 44( 5): 1275– 88. [DOI] [PubMed] [Google Scholar]
- 33. Jujo T, Sakao S, Ishibashi-Ueda H, . et al. Evaluation of the Microcirculation in Chronic Thromboembolic Pulmonary Hypertension Patients: The Impact of Pulmonary Arterial Remodeling on Postoperative and Follow-Up Pulmonary Arterial Pressure and Vascular Resistance. PLoS One. 2015. August 7; 10( 8): e0133167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bonderman D, Wilkens H, Wakounig S, . et al. Risk factors for chronic thromboembolic pulmonary hypertension. Eur Respir J. 2009. February; 33( 2): 325– 31. [DOI] [PubMed] [Google Scholar]
- 35. Condliffe R, Kiely DG, Gibbs JS, . et al. Prognostic and aetiological factors in chronic thromboembolic pulmonary hypertension. Eur Respir J. 2009. February; 33( 2): 332– 8. [DOI] [PubMed] [Google Scholar]
- 36. Bonderman D, Turecek PL, Jakowitsch J, . et al. High prevalence of elevated clotting factor VIII in chronic thromboembolic pulmonary hypertension. Thromb Haemost. 2003. September; 90( 3): 372– 6. [DOI] [PubMed] [Google Scholar]
- 37. Wong CL, Szydlo R, Gibbs S, Laffan M.. Hereditary and acquired thrombotic risk factors for chronic thromboembolic pulmonary hypertension. Blood Coagul Fibrinolysis. 2010. April; 21( 3): 201– 6. [DOI] [PubMed] [Google Scholar]
- 38. Wolf M, Boyer-Neumann C, Parent F, . et al. Thrombotic risk factors in pulmonary hypertension. Eur Respir J. 2000. February; 15( 2): 395– 9. [DOI] [PubMed] [Google Scholar]
- 39. Kimmig LM, Palevsky HI.. Review of the Association between Splenectomy and Chronic Thromboembolic Pulmonary Hypertension. Ann Am Thorac Soc. 2016. June; 13( 6): 945– 54. [DOI] [PubMed] [Google Scholar]
- 40. Desmarais J, Elliott CG.. Familial Chronic Thromboembolic Pulmonary Hypertension. Chest. 2016. April; 149( 4): e99– e101. [DOI] [PubMed] [Google Scholar]
- 41. Feng YX, Liu D, Sun ML, . et al. BMPR2 germline mutation in chronic thromboembolic pulmonary hypertension. Lung. 2014. August; 192( 4): 625– 7. [DOI] [PubMed] [Google Scholar]
- 42. Xi Q, Liu Z, Zhao Z, Luo Q, Huang Z.. High Frequency of Pulmonary Hypertension-Causing Gene Mutation in Chinese Patients with Chronic Thromboembolic Pulmonary Hypertension. PLoS One. 2016. January 28; 11( 1): e0147396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bonderman D, Jakowitsch J, Adlbrecht C, . et al. Medical conditions increasing the risk of chronic thromboembolic pulmonary hypertension. Thromb Haemost. 2005. March; 93( 3): 512– 6. [DOI] [PubMed] [Google Scholar]
- 44. Klok FA, Dzikowska-Diduch O, Kostrubiec M, . et al. Derivation of a clinical prediction score for chronic thromboembolic pulmonary hypertension after acute pulmonary embolism. J Thromb Haemost. 2016. January; 14( 1): 121– 8. [DOI] [PubMed] [Google Scholar]