

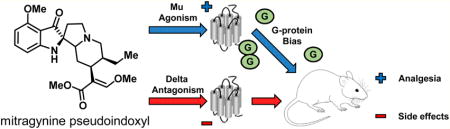
Abstract
Natural products found in Mitragyna speciosa, commonly known as kratom, represent diverse scaffolds (indole, indolenine, and spiro pseudoindoxyl) with opioid activity, providing opportunities to better understand opioid pharmacology. Herein, we report the pharmacology and SAR studies both in vitro and in vivo of mitragynine pseudoindoxyl (3), an oxidative rearrangement product of the corynanthe alkaloid mitragynine. 3 and its corresponding corynantheidine analogs show promise as potent analgesics with a mechanism of action that includes mu opioid receptor agonism/delta opioid receptor antagonism. In vitro, 3 and its analogs were potent agonists in [35S]GTPγS assays at the mu opioid receptor but failed to recruit β-arrestin-2, which is associated with opioid side effects. Additionally, 3 developed analgesic tolerance more slowly than morphine, showed limited physical dependence, respiratory depression, constipation, and displayed no reward or aversion in CPP/CPA assays, suggesting that analogs might represent a promising new generation of novel pain relievers.
Graphical abstract
INTRODUCTION
Opioids, including morphine, are clinically used for the treatment of moderate to severe chronic pain. However, despite their proven efficacy, mu opioid receptor agonists have problematic side effects such as tolerance, physical dependence, and substance abuse.1 Agonists selective for other opioid receptors produce analgesia, but with their own liabilities.2–4 The ultimate goal of opioid-related drug development has been to design and synthesize potent antinociceptive agents that are devoid of adverse side effects. Many approaches have been taken over the years, starting with the development of partial agonists or mixed agonist/antagonists.5–9 A more recent approach takes advantage of biased agonism, in which distinct downstream signaling pathways are activated by different agonists working through the same receptor.10,11 It has been proposed that ligands biased against recruiting β-arrestin-2, or showing preference for activating specific G-protein-mediated signal transduction pathways, will demonstrate diminished side effects.12,13 Oliceridine (TRV130)14,15 is an example of a mu opioid receptor-biased agonist which has recently entered phase-III clinical trials, showing separation between antinociception and some opioid-related side effects. 6′-Guanidinonaltrindole (6′-GNTI),16 22-thiocyanatosalvinorin A (RB-64),17 and two new classes of kappa opioid ligands from the Aube group have also recently been reported in the opioid literature as biased kappa opioid receptor agonists.18,19
Natural products have provided many lead compounds leading to the design of new pharmaceuticals. Natural products and their derivatives account for approximately 50% of approved drugs.20 Morphine, the most commonly employed opioid, and thebaine, the structure on which the vast majority of semisynthetic opiates is based, are natural alkaloids found in the poppy plant, Papaver somniferum. While opioid chemistry has traditionally been dominated by thebaine-derived alkaloids isolated from poppy, there are a growing number of opioid natural products derived from structures other than the traditional morphinan scaffold and thus structurally not closely related to morphine. These include analogs of salvinorin A21–23 such as herkinorin24 and thiocyanatosalvinorin A,17 which have been developed as mu and kappa opioid receptor-biased agonists, respectively, while some peptide analogs such as cyclo[Phe-D-Pro-Phe-Trp] (CJ-15,208)25 are being developed as analgesics and medications against cocaine abuse (Figure 1). Mitragynine (indole core) (1) and its congeners isolated from the Southeast Asian plant Mitragyna speciosa, commonly known as kratom, are monoterpene indole alkaloids structurally not closely related to morphine.26 In addition to its traditional use, kratom has become a quickly emerging substance of abuse. It is currently legal in many parts of the world, and kratom leaves are available for purchase over the Internet. Case studies of fatalities resulting from overdose have been published, although the risk posed by mitragynine remains uncertain, given that simultaneous use or contamination with other substances (including opioids) that may have been involved in the reported deaths.27–30 Both mitragynine (1) and its naturally occurring oxidation product, 7-OH mitragynine (indolenine core) (2), are opioid antinociceptive agents that have been examined both in vitro and in vivo.31–39 Mitragynine pseudoindoxyl (3), a rearrangement product of 2 with a spiro-pseudoindoxyl core, was first isolated in 1974 by Zarembo et al. as a microbial metabolite of 1 by the fungus Helminthosporum sp.40 Yamamoto et al. reported that it acted nonselectively on mu- and delta opioid receptors, while its kappa opioid receptor affinity was negligible.41 In later publications by Takayama et al., the in vivo supraspinal analgesic properties of 3 were briefly discussed.39
Figure 1.
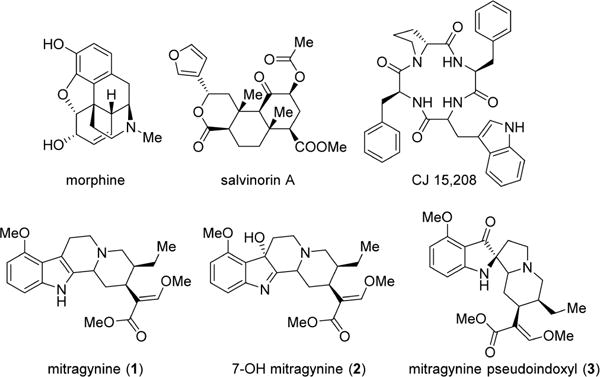
Structure of several important opioid natural product analogs.
In this work, we report the in vitro and in vivo pharmacology and structure–activity relationships (SAR) of mitragynine pseudoindoxyl (3). We demonstrate for the first time that 3 and its C-9 substituted derivatives, corynantheidine pseudoindoxyls, are systemically active mixed mu opioid receptor agonist/delta opioid receptor antagonist compounds in vitro and produce potent antinociception in vivo. Characterization of 3 demonstrated an opioid-mediated analgesia devoid of any place-conditioning effects and a side effect profile far superior to clinically used mu opioid-based antinociceptive agents.
RESULTS
Chemistry
1 was extracted from dry kratom powder using a modified protocol reported by Ponglux et al.42 Compounds 2 and 3 were synthesized from 1 as shown in Scheme 1.39,43 To better understand the pharmacology of this template, SAR studies were carried out by modifying the C-9 and N-1 (indole nitrogen) positions. Six analogs with various substituents in the C-9 positions and 2 analogs at N-1 were synthesized. C-9-substituted corynantheidine pseudoindoxyl derivatives were synthesized starting from 2. To gain access to the C-9 position on the pseudoindoxyl scaffold, 2 was converted to 9-hydroxycorynantheidine pseudoindoxyl (4) using AlCl3 and ethanethiol in DCM. This intermediate was converted to its triflate (5) using triflic anhydride and pyridine, which was subsequently used as the precursor for further reactions.
Scheme 1. Synthesis of Mitragynine Derivatives 2–12a.
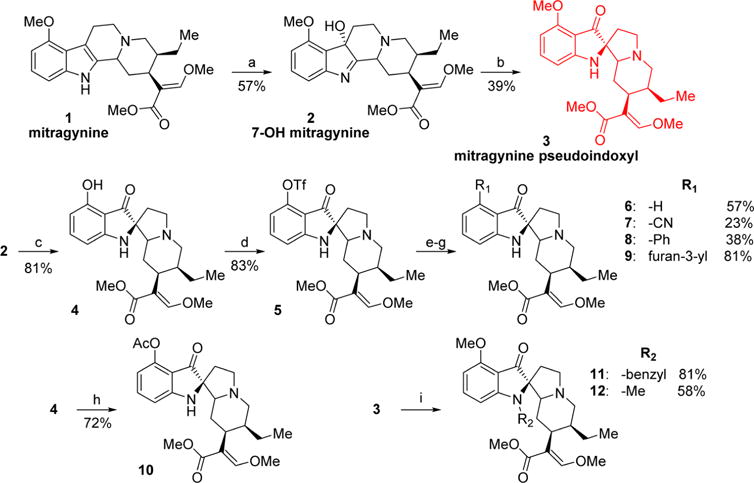
aReagents and conditions: (a) PIFA, H2O, acetonitrile, 0 °C, 1 h; (b) Zn(OTf)2, toluene, 110 °C, 2 h; (c) AlCl3, EtSH, DCM, 0 °C, 5 h; (d) Tf2O, pyridine, DCM, −40 °C, 1 h; (e, yielding 6) Pd(OAc)2, dppp, HCOOH, DMF, 60 °C, 1 h; (f, yielding 7) Zn(CN)2, Pd(PPh3)4, DMF, 80 °C, 2 h; (g, yielding 8 and 9) phenylboronic acid (8) or 3-furanylboronic acid (9), Pd(PPh3)4, K2CO3, MeOH, toluene, 80 °C, 2 h; (h) Ac2O, pyridine, rt, 1 h; (i, yielding 11 and 12) benzyl bromide (11) or iodomethane (12), NaH, acetonitrile, rt, 2 h.
The pseudoindoxyl of corynantheidine (6) was synthesized using palladium-catalyzed removal of the triflate ester by formic acid. The synthesis of the nitrile 7 was accomplished in a palladium-catalyzed reaction of 5 with Zn(CN)2. Compounds 8 and 9 were obtained via Suzuki coupling reactions of 5 and the appropriate boronic acids. 3 was alkylated in the N-1 position with benzyl bromide and iodomethane to synthesize 11 and 12, respectively.
In vitro Pharmacology
Initial investigations used in vitro radioligand binding assays with cell lines stably expressing murine opioid receptors (Figure 2, Table 1). Mitragynine (1) showed poor affinity at all opioid receptors, whereas 7-hydroxymitragynine (2) showed moderate affinity at the mu opioid receptor clone MOR-1, 5-fold higher than 1 and was considerably more potent at the expressed delta opioid receptor clone DOR-1 than 1.44 Mitragynine pseudoindoxyl (3) displayed the highest overall binding affinity for MOR-1 and DOR-1 (Ki 0.8 nM and 3 nM, respectively) while also showing a moderate affinity for KOR-1. These data suggest that conversion of the indole to indolenine ring to the spiro-pseudoindoxyl core dramatically increases affinity for opioid receptors. The binding affinities of 3 at MOR-1 and DOR-1 were comparable to the prototypic mu ligands morphine and DAMGO and delta ligands DPDPE and NTI. Alkyl substitution at the N-1 position of the template (compounds 11, 12) eliminated opioid affinity, suggesting that the unsubstituted indole NH is important for receptor binding. Substitutions at C-9, however, yielded potent derivatives. All six C-9-modified compounds (4, 6–10) maintained the high affinity at both MOR-1and DOR-1 sites as observed with 3 previously. 2 and 3 were also screened across a panel of other nonopioid drug targets using the PDSP screening facility at NIMH.45 2 exhibited no affinity appreciable affinity at these receptors (Ki > 10 μM). 3 had poor affinity at α2A adrenergic receptor, α2C adrenergic receptor, and 5HT7 (Table S1).
Figure 2.
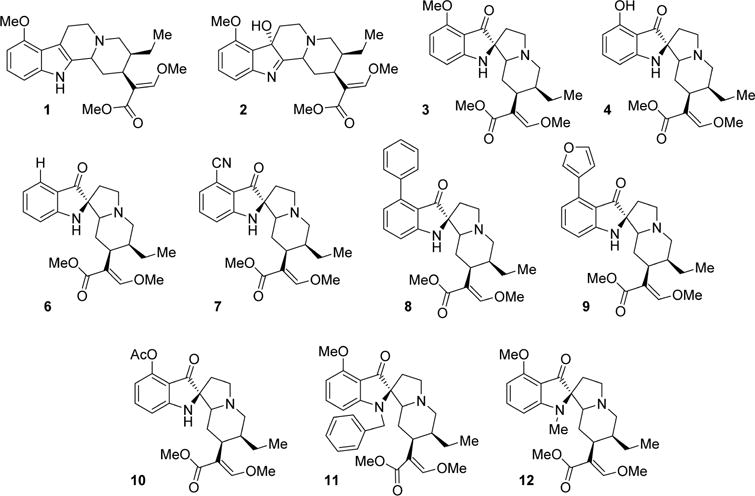
Structure of the studied mitragynine analogs.
Table 1.
Receptor Affinities and Subcutaneous Antinociception in Mice
| compd | affinity (Ki nM)a
|
antinociception sc,b mg/kg (CI) | ||
|---|---|---|---|---|
| MOR-1 | KOR-1 | DOR-1 | ||
| 1 | 230 ± 47 | 231 ± 21 | 1011 ± 49 | 166 (101–283) |
| 2 | 37 ± 4 | 132 ± 7 | 91 ± 8 | 0.46 (0.39–0.71) |
| 3 | 0.8 ± 0.2 | 24 ± 0.9 | 3.0 ± 1.3 | 0.76 (0.56–0.83) |
| 4 | 1.4 ± 0.2 | 170 ± 61 | 6.1 ± 1.1 | 0.18 (0.16–0.20) |
| 6 | 0.46 ± 0.01 | 19 ± 4.7 | 2.9 ± 0.29 | 0.24 (0.20–0.26) |
| 7 | 0.5 ± 0.01 | 47 ± 3.3 | 2.4 ± 0.3 | 0.32 (0.26–0.41) |
| 8 | 0.91 ± 0.06 | 51 ± 9.7 | 0.8 ± 0.13 | 1.0 (0.70–1.35) |
| 9 | 0.94 ± 0.02 | 39 ± 11 | 1.5 ± 0.37 | 1.1 (0.83–1.38) |
| 10 | 2.5 ± 0.6 | 31 ± 14 | 20 ± 1 | 0.38 (0.29–0.49) |
| 11 | 249 ± 41 | 136 ± 8 | 258 ± 34 | |
| 12 | 375 ± 138 | >1000 | >1000 | |
| DAMGO | 3.3 ± 0.43c | – | – | |
| U50,488H | – | 0.73 ± 0.32c | – | |
| DPDPE | – | – | 1.39 ± 0.67c | |
| NTI | – | – | 0.46 ± 0.32c | |
| norBNI | – | 0.23 ± 0.03c | – | |
| morphine | 4.6 ± 1.8c | – | – | 2.5(1.8, 3.4) |
Competition studies were performed with the indicated compounds against 125I-IBNtxA (0.1 nM) in membranes from CHO cells stably expressing the indicated cloned mouse opioid receptors. Results are presented as nM ± SEM from three independent experiments performed in triplicate.
Cumulative dose–response curves were carried out on groups of CD1 mice (n = 10) using radiant heat tail flick assays with indicated compound at the indicated doses (sc), and antinociception was tested 15 min later at peak effect. Results from two independent experiments are shown as mean (95% Cl).
Values from the literature.78 “–” Denotes not determined or not applicable.
In [35S]GTPγS functional assays using opioid transfected cell lines, 1 was a partial agonist with moderate potency at MOR-1 and a weak antagonist at both DOR-1 and KOR-1 (Table 2). 2 was a partial agonist at MOR-1, 4-fold more potent than 1, and a weak KOR-1 and DOR-1 antagonist.44 Analog 3 was a potent full agonist at MOR-1 and an antagonist at both DOR-1 and KOR-1. Most C-9-modified derivatives (4, 6, 7, and 9) were MOR-1 agonists and DOR-1 antagonists in functional assays with the exception of compound 8, which was a dual MOR-1/DOR-1 agonist (Table 2).
Table 2.
[35S]GTPγS Functional Assays in Transfected Cell Lines
| compd | [35S]GTPγS functional assaysa
|
|||||
|---|---|---|---|---|---|---|
| MOR-1
|
KOR-1
|
DOR-1
|
||||
| EC50 (nM) | Emax (%) | EC50 (nM) | IC50 (nM) | EC50 (nM) | IC50 (nM) | |
| 1 | 203 ± 13 | 65 ± 2.8 | – | >4 μM | – | >4 μM |
| 2 | 53 ± 4 | 77 ± 5 | – | 2524 ± 552 | – | 691 ± 434 |
| 3 | 1.7 ± 0.1 | 84 ± 5 | – | 31 ± 3 | – | 61 ± 6 |
| 4 | 2.0 ± 0.1 | 124 ± 2 | – | – | – | 293 ± 129 |
| 6 | 1.4 ± 0.03 | 116 ± 2 | – | 252 ± 48 | – | 193 ± 48 |
| 7 | 0.7 ± 0.2 | 122 ± 2 | – | 721 ± 58 | – | 73 ± 3 |
| 8 | 1.4 ± 0.25 | 123 ± 5 | – | 171 ± 10 | 0.83 ± 0.37 (89 ± 3)b | – |
| 9 | 1.5 ± 0.3 | 100 ± 1 | – | 202 ± 14 | – | 39 ± 5 |
| 10 | 3.9 ± 0.5 | 120 ± 6 | – | – | – | – |
| 11 | – | – | – | – | – | – |
| 12 | – | – | – | – | – | – |
| DAMGO | 19 ± 7 | – | – | – | – | – |
| U50,488H | – | – | 17 ± 6.1 | – | – | – |
| DPDPE | – | – | – | – | 10 ± 2.2 | – |
| NTI | – | – | – | – | – | 0.72 ± 0.11 |
| norBNI | – | – | – | 2.9 ± 0.14 | – | – |
Efficacyata were obtained using agonist induced stimulation of [35S]GTPγS binding assay. Efficacy is represented as EC50 (nM) and percent maximal stimulation (Emax) relative to standard agonist DAMGO (MOR-1), DPDPE (DOR-1), or U50,488H (KOR-1) at 1000 nM. To determine the antagonist properties of a compound, membranes were incubated with 100 nM of the appropriate agonist in the presence of varying concentrations of the compound
Compound 8 is an agonist at DOR-1. Results are presented as nM ± SEM from three independent experiments performed in triplicate. “–” Denotes not determined or not applicable.
Both DAMGO and endomorphin-2 effectively recruited β-arrestin-2 in CHO cells expressing MOR-1, as measured with the DiscoveRx PathHunter assay. In contrast, compounds 1–4 and 6–9 failed to recruit β-arrestin-2 at concentrations as high as 10 μM (Figure 3A). Both 2 and 3 reduced DAMGO-induced β-arrestin-2 stimulation in these cells in a concentration-dependent manner (Figure 3B)44 consistent with their respective binding affinities at the receptor. Thus, 3 and its analogs potently stimulated [35S]GTPγS binding without stimulating β-arrestin-2 recruitment. However, their antagonism of DAMGO stimulation of β-arrestin-2 recruitment revealed that they could still bind to both G-protein and arrestin functional receptor configurations.
Figure 3.
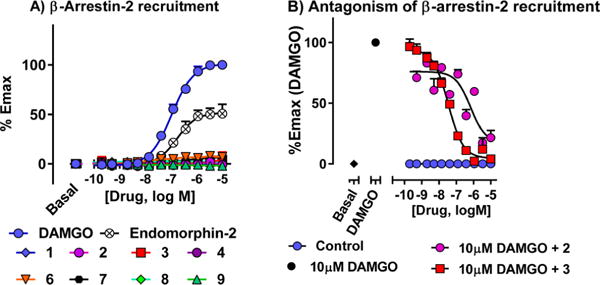
β-arrestin-2 recruitment and antagonism of β-arrestin-2 recruitment. (A) β-arrestin-2 recruitment: β-arrestin-2 recruitment was determined using the DiscoveRx PathHunter enzyme complementation assay using modified MOR-1 in CHO cells. Compounds were found to be completely G-protein biased. (B) Antagonism of β-arrestin-2 recruitment: The same cells were incubated with the antagonist (2, 3) for 30 min at 37 °C prior to the addition of agonist (10 μM DAMGO) at MOR-1. Compounds 2 and 3 were able to antagonize β-arrestin-2 recruitment by DAMGO. IC50 values: 2: 725 ± 292 nM; 3: 34 ± 2 nM.
Antinociception
The antinociceptive effect of mitragynine and its derivatives was evaluated in vivo in mice using the radiant heat tail flick assay (Table 1). After subcutaneous administration, 1 produced antinociception with an ED50 (and 95% CI) value of 166 mg/kg (101, 283), 66-fold less active than morphine. On the other hand, 2 was about 5-fold more potent than morphine and 350-fold more potent than 1 (Figure S1), similar to literature values.32–34 Compound 3 was 1.5-fold more potent than morphine after intracerebroventricular administration (icv, Figures 4A and S2) and 3-fold more potent following subcutaneous (sc, Figures 4B and S4A and S4B) administration. Compound 3 has a shorter duration of antinociceptive effect than morphine with a peak effect at 15 min (Figure S3). Compound 3 proved equally active in CD1, C57BL/6, and 129Sv6 strains of mice subcutaneously (Figure S5). Furthermore, 3 also was active orally, with an ED50 (95% CI) value of 7.5 (4.3–13) mg/kg (Figure 4C). The C-9 derivatives (4 and 6–10) also produced antinociception following systemic administration, with ED50 values comparable to 3 (Table 1). Compound 3 was also active in the hot plate assay of antinociception, with an ED50 (95% CI) value of 0.99 (0.75–1.3) mg/kg (Figures 4D and S4C), comparable to morphine (ED50 = 1.7 (1.3, 2.4) mg/kg) (Figure S4D).
Figure 4.
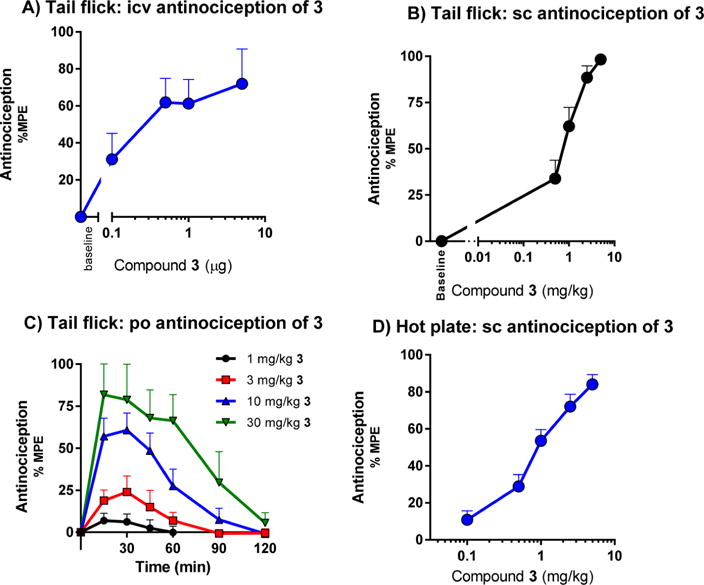
Antinociception of compound 3 upon intracerebroventricular, subcutaneous, and oral administration. (A) Dose–response curves of antinociception of 3 and morphine given supraspinally in CD1 mice. Two independent determinations of the cumulative dose–response curves were performed on groups of mice (n = 5) for antinociception in the tail flick assay with 3 intracerebroventricularly. Animals were tested 15 min later at peak effect to generate the analgesic dose–response curve. Each point represents mean ± SEM for 10 mice. ED50 values (and 95% CI) were 0.38 (0.18- 0.81) μg for 3. (B) Dose–response curves of antinociception of 3 given subcutaneously in CD1 mice. Three independent determinations of the cumulative dose–response curves were performed on groups of mice (n = 10) for antinociception in the tail flick assay, 30 mice in total. ED50 (and 95% CI) = 0.76 (0.56–0.83) mg/kg. (C) Time course of tail flick antinociception of 3 given orally in CD1 male mice. Groups of mice (n = 10) were given different doses of 3 orally by gavage and tested for analgesic response at the indicated time points. ED50 (and 95% CI) = 7.5 (4.3–13) mg/kg. (D) Hot plate. Groups of CD1 mice (n = 10) were assessed for antinociception of 3 at peak effect in two independent experiments (n = 20 total) in a cumulative dose–response paradigm. Analgesia was determined using a 55 °C hot plate, where the latency to respond with a hind paw lick or shake/flutter, whichever came first, was recorded. ED50 (and 95% CI) = 0.99 mg/kg (0.75–1.3).
Opioid Receptor Antagonism
Based on the high affinity and favorable mixed mu agonist/delta antagonist profile in vitro, we examined the activity of compound 3 in greater detail. Naloxone and the mu-selective antagonist β-FNA effectively reversed 3-induced antinociception, whereas the delta-selective antagonist NTI and the kappa antagonist norBNI did not. Yohimbine, an α2 antagonist, had no effect on the antinociception of 3 (Figure 5A).
Figure 5.
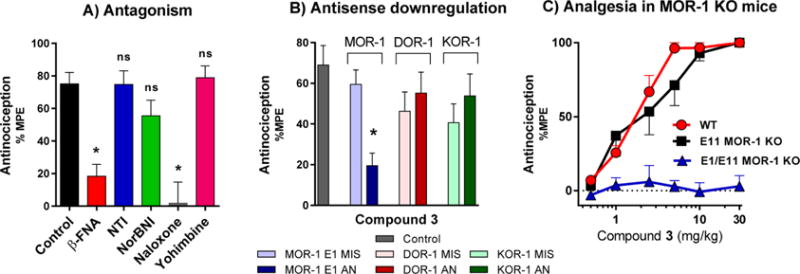
Pharmacological and genetic reversal of antinociception of 3. (A) Reversal of antinociception by selective antagonists. Groups of CD1 mice (n = 10) received 3 (1.5 mg/kg sc) and the indicated antagonist. β-Funaltrexamine (β-FNA; 40 mg/kg sc) and norbinaltorphimine (norBNI; 10 mg/kg sc) were administered 24 h before agonist testing. Naltrindole (NTI; 0.5 mg/kg sc), naloxone (1 mg/kg), and yohimbine (10 mg/kg) were administered 15 min before 3. All antinociception testing was performed 15 min after the administration of 3. Similar results were observed in two independent replications. 3 antinociception is insensitive to NTI, norBNI, and yohimbine, whereas antinociception is antagonized by β-FNA and naloxone (two-way ANOVA followed by Bonferroni post hoc comparisons test, p < 0.05). All values are expressed as the mean ± SEM. (B) Antisense oligodeoxynucleotide injection: Groups of mice (n = 15) received the stated antisense (5–10 μg) or mismatch (5–10 μg) oligodeoxynucleotide icv under light isoflurane anesthesia on days 1, 3, and 5. Tail flick antinociception was tested on day 6. Control groups received no injection prior to testing. On test day, mice received 3 (1.5 mg/kg, sc). All experiments were performed 3 times with similar results observed with each determination. Analgesic response of 3 was only affected in mu receptor downregulated mice (MOR-1 AN) (one-way ANOVA followed by Bonferroni post hoc comparisons test). *Significantly different from control (p < 0.05). Data for agonist controls are shown in the Figure S6 and sequences of AN and MIS oligos are shown in Table 4. (C) Antinociception of 3 in wild-type, exon 11 KO, and exon 1/exon 11 double KO C57 mice. Two independent determinations of the cumulative dose–response curves were performed on groups of mice (n = 5) for antinociception in the tail flick assay with 3 given subcutaneously. Compound 3 displayed similar antinociceptive effects in wild-type (ED50 = 0.83 mg/kg (0.37–1.9)) and exon 11 KO mice (ED50 = 1.4 mg/kg (0.34–5.8)), however, no antinociception was observed in exon 1/exon 11 double KO mice, suggesting that the antinociceptive effect of 3 is mediated by the E1MOR-1 variants.
To examine the selectivity of antinociception further, we used an antisense oligodeoxynucleotide mapping paradigm. The activity of the oligodeoxynucleotide antisense probes for MOR-1, KOR-1, and DOR-1 has been established previously.46–48 Targeting exon 1 of MOR-1, the antisense oligodeoxynucleotide lowered the analgesic actions of morphine, reproducing earlier studies (Figure S6).46 Similarly, the responses of 3 were lowered (Figure 5B). The specificity of the response was established by the inactivity of the control mismatch. Similarly, downregulation of exon 3 of DOR-148 and exon 2 of KOR-149 with antisense oligodeoxynucleotides attenuated antinociception produced by the prototypic delta agonist DPDPE and the prototypic kappa agonist U50,488H, in accordance with previous studies (Figure S6). However, these oligodeoxynucleotides targeting kappa and delta receptors did not alter the antinociception of 3 (Figure 5B).
The mu opioid receptor Oprm1 creates an array of splice variants through alternative splicing with patterns conserved from rodents to humans.50,51 The major sets of variants are full-length 7 transmembrane domain (7TM) variants associated with exon 1 (E1). A second set of variants of truncated six transmembrane domain splice variants (6TM) is generated by an alternative promoter associated with exon 11 (E11) of the Oprm1 gene.52,53 MOR-1 KO mice were used to establish the contributions of 7TM E1-MOR-1 and 6TM E11-MOR-1 variants to 3 antinociception.
Two different types of MOR-1 KO mice were utilized: exon 11 (E11) MOR-1 KO mice, which lack the 6TM E11splice variants of MOR-1 but retain expression of 7TM E1 splice variants of MOR-1, and total mu opioid receptor knockout in which both exons 1 and 11 were disrupted (E1/E11) to eliminate all 7TM and 6TM mu opioid receptor variants of the Oprm1 gene. Morphine antinociception has previously been demonstrated to be independent of the E11-associated 6TM splice variants of MOR-1, maintaining full analgesic activity in the E11 KO,54 but its antinociception was completely eliminated in E1/E11 MOR-1 KO mice,55 suggesting 7TM E1-MOR-1 variant as the primary mechanism of analgesic action. Compound 3 showed similar antinociceptive responses as morphine. Compound 3 antinociception was similar in wild-type (ED50 = 0.83 mg/kg (0.37–1.9)) and exon 11 KO C57/BL6 mice (ED50 = 1.4 mg/kg (0.34–5.8)) in a tail flick assay (Figure 5D). However, antinociception of compound 3 was eliminated in E1/E11 MOR-1 KO mice (ED50 > 30 mg/kg), indicating a mu opioid receptor mechanism.55 Taken together with the antisense results, these in vivo findings indicate that 3 analgesia is mediated by 7TM E1-MOR-1 receptors.
Side Effect Profile
We next evaluated 3 in mouse models of antinociceptive tolerance, dependence, respiratory depression, and inhibition of GI transit (Figure 6). Mice developed antinociceptive tolerance to morphine after twice daily administration for 5 days (5 mg/kg/injection, sc). In contrast, antinociceptive tolerance to 3 developed far more slowly, with twice daily administration of 3 (at an equianalgesic dose as morphine) requiring 29 days instead of 5 days (Figure 6A). After 5 days, the morphine ED50 value was shifted 6-fold to 12.1 (7.6–19.4) mg/kg from 2.0 (1.2–3.3) mg/kg. In contrast, after 5 days, the ED50 value for 3 was shifted <2-fold to 1.1 (0.66–2.0) mg/kg. After 29 days, the ED50 value for 3 was shifted 6-fold (4.5 mg/kg (2.7–7.7) mg/kg) (Figure S7). An independent sample t test was used to test for the group difference between 3 and morphine on day 5. The mean difference of 53.97%MPE between the two groups was found to be statistically significant (p-value <0.0001) at the 5% level of significance. This difference is likely to range between 42.27 and 65.67% MPE as measured by a 95% confidence interval. To highlight 3’s ability to sustain antinociception over repeated dosing, the composite areas under the curve (AUCs) were calculated using the trapezoidal rule on the mean response across 1–29 days for the 3 group and 1–5 days for the morphine group, respectively. The AUC for the morphine group was 205.9, while for the 3 group it was 6-fold larger, 1239.
Figure 6.
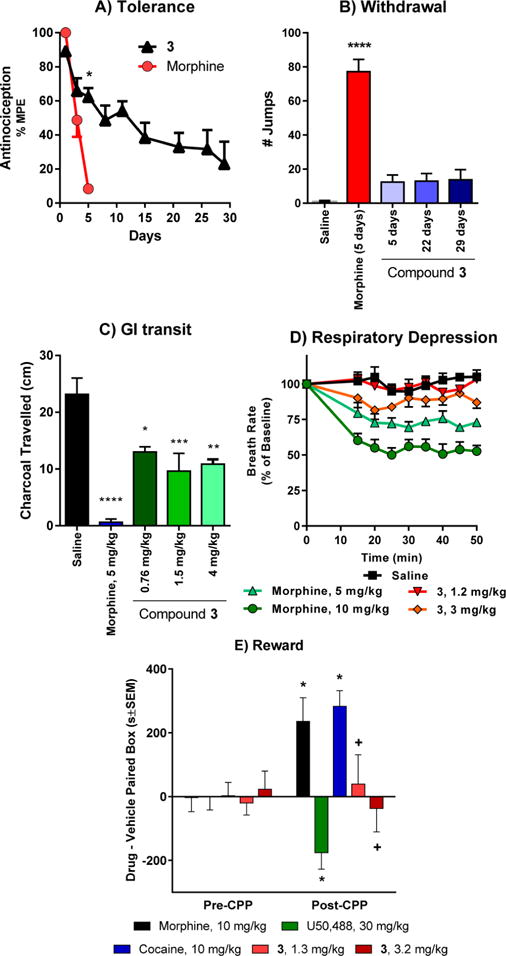
Side effect studies with compound 3. (A) Antinociceptive tolerance: Mice were dosed 2× daily with 2× antinociceptive ED50 with either morphine or 3 until they showed complete analgesic tolerance. 3 showed very slow onset of tolerance compared with morphine. *Significantly different from morphine (p < 0.05). The experiment was replicated at least twice with similar results. (B) Physical dependence: Groups of mice were dosed 2× daily with 2× antinociceptive ED50 with morphine or 3. Separate groups of mice were used for the 5, 22, and 29 day treatment with 3 and the animals within each group were sacrificed following the experiment with naloxone. Animals were challenged with naloxone (1 mg/kg) on day 5 of the morphine group, and days 5, 22, and 29 of the 3 groups. Number of jumps was counted over a 15 min period postinjection. The response of mice treated with 3 on either day was not significantly greater than that of mice treated with saline. *Significantly different from saline. (One-way ANOVA followed by Dunnett’s multiple comparison test, p < 0.05). (C) Gastrointestinal transit. Groups of mice (n = 10) received saline, morphine (5 mg/kg), or 3 (1.5 and 4 mg/kg) before receiving an oral dose of 0.2 mL of charcoal meal by gavage. Animals were sacrificed 30 min later, and the distance traveled by charcoal was measured. 3 lowered transit significantly compared with saline (P < 0.05) but less than morphine at both doses (P < 0.05) as determined by ANOVA followed by Tukey’s multiple-comparison test. The inhibition of gastrointestinal transit seems to plateau even at doses ∼5× higher than the antinociceptive ED50. (D) Respiratory rate. Animals were randomly assigned to receive saline (n = 5), 3 (1.2 and 3 mg/kg, sc, n = 5 at each dose) or morphine (5 and 10 mg/kg, n = 5 at each dose). While morphine caused respiratory depression at both 2× and 5× antinociceptive ED50 dose (5 and 10 mg/kg, respectively), 3 did not depress respiratory rate at ∼2× antinociceptive ED50 dose (1.2 mg/kg) and was not significantly different from saline at any time point, whereas morphine (5 mg/kg) decreased respiratory depression in comparison with 3 (p < 0.05) as determined by repeated-measures ANOVA followed by Tukey’s multiple-comparison test. However, at ∼5× antinociceptive ED50 dose (3 mg/kg), 3 showed signs of respiratory depression albeit significantly less than the equianalgesic dose of morphine (10 mg/kg) at any given time point (repeated measures ANOVA followed by Tukey’s multiple-comparison test). (E) Conditioned place preference and aversion. Compound 3 alone did not produce conditioned place preference or aversion. After determination of initial preconditioning preferences, mice were place conditioned daily for 2 days with morphine (10 mg/kg/d, ip), U50,488 (30 mg/kg/d, ip), cocaine (10 mg/kg/d, ip), or 3 (1.3 mg/kg/d, ip and 3.2 mg/kg/d, ip). Mean difference in time spent on the drug-paired side ± SEM is presented (n = 17–21). * Significantly different from matching preconditioning preference (p < 0.05); + significantly different from cocaine, morphine, and U50,488 preference (two-way repeated measures ANOVA with Sidak’s post hoc test).
Time action studies revealed that 3 has a shorter duration of action than morphine (Figure S3). Since the shorter duration of action of 3 led to a decreased drug exposure, we also examined tolerance in a different dosing paradigm in which 3 was given four times per day to provide similar drug exposures for morphine and 3. In this paradigm, 3 failed to show a significant decreased effect over 5 days, whether examined at a fixed dose over the 5 days (Figure S8A) or dose–response curves at day 5 (Figure S8B). After 5 days, the ED50 value for 3 was shifted from 0.69 (0.46, 1.0) to 1.6 (0.97–2.6) mg/kg (Figure S8C), a shift similar to that observed when 3 was given twice a day (ED50 = 1.1 (0.66–2.0)) on day 5 (Figure S7).
Physical dependence was assessed in mice treated repeatedly with 3 at twice its ED50 dose (1.5 mg/kg) twice daily for 5, 22, or 29 days by administration of the opioid antagonist naloxone. Mice showed only minimal signs of withdrawal following administration of naloxone with 12 ± 4.1, 13 ± 4.4, and 14 ± 6.0 jumps on average, respectively (Figure 6B). These values were not significantly different from saline but differed significantly from mice treated with morphine for 5 days, which showed 77 ± 7.2 jumps (Figure 6B). In summary, compound 3 demonstrated limited antinociceptive tolerance and physical dependence in comparison with morphine following chronic administration.
Differences in effect between 3 and morphine extended to other opioid effects. Mu selective agonists inhibit gastrointestinal transit, a major component of constipation. At a dose twice its analgesic ED50 value (5 mg/kg, sc), morphine almost totally eliminated transit (Figure 6C). An equianalgesic dose of 3 (1.5 mg/kg, sc) also lowered gastrointestinal transit, but not nearly as much as morphine. The effect on GI transit plateaued, with a greater dose (4 mg/kg, sc) also showing no further inhibition (Figure 6C).
Morphine dose-dependently reduced the respiratory rate in mice, with a decrease of approximately 30% by a dose twice its analgesic ED50 value (5 mg/kg, sc) by approximately 50% after a higher dose corresponding to 4-fold its analgesic ED50 value. In contrast, 3 showed no respiratory depression at ∼2× its antinociceptive ED50 dose in C57BL/6 mice (1.2 mg/kg, sc). Although a higher dose (3 mg/kg, sc), transiently lowered the respiratory rate by approximately 15%, this was still significantly less than morphine (Figure 6D).
Compound 3 also failed to show either rewarding or aversive behavior in a conditioned place preference paradigm. In this study, morphine produced significant conditioned place preference (CPP; F(4180) = 5.62, p = 0.003; two-way ANOVA with Tukey HSD post hoc test) and U50,488H produced conditioned place aversion (CPA), but 3 demonstrated neither preference or aversive behavior at doses 2-fold or 5-fold its analgesic ED50 value (n.s.; Tukey HSD post hoc test; Figure 6E). Overall, these results demonstrate that 3 produces potent opioid receptor-mediated antinociception both centrally and systemically, yet shows a separation of antinociception from some classic opioid side effects such as antinociceptive tolerance, dependence, and conditioned place preference. Furthermore, 3 shows a lower propensity to cause respiratory depression and constipation compared with the canonical opioid, morphine.
Modeling
In silico docking studies were carried out to unravel potential differences in receptor interactions of 1–3. The results of in silico prediction of inhibitory constants are listed in Table 3 for all three mitragynine compounds and the opioid receptors. In general, ligands of the lowest energy complexes were located in the binding pocket observed in the crystal structures. Inhibitory constants (Ki) calculated for the lowest energy complexes follow the trend observed in the experiments, but the range of values is much more narrow compared to experimental data, suggesting much lower selectivity of both the ligands and the receptors. The lowest energy complexes, which are considered to reflect specific binding between 1–3, and the receptors are shown in Figures 7 and S14. These in silico Ki values reproduce experimental data with much higher accuracy compared to the ones calculated for the lowest energy complexes in the first pass, where all nonspecific hits were included (for explanation, see the Experimental Section). Receptor side chains in contact with the bound 1–3 are also depicted in Figure 7 and listed in Table 3.
Table 3.
In Vitro and in Silico Binding Affinities and Specific Contacts of Mitragynine and Derivatives
| compd | in vitro Ki (nM) | in silico Ki (nM) nonspecifica | in silico Ki (nM) specificb | receptor side chains in polar contact with the ligand | receptor side chains in nonpolar contact with the ligand | |
|---|---|---|---|---|---|---|
| mu | 1 | 230 ± 47 | 23 | 569 | Gln124, Tyr128, Asp147, Tyr326 | Met151, Trp293, Ile296, Ile322 |
| 2 | 37 ± 4 | 4.0 | 55 | Asp147, Lys233, Trp293 | Tyr148, Met151, Leu219, Leu232, Ile296 | |
| 3 | 0.75 ± 0.18 | 2.5 | 2.5 | Asp147, Tyr148, Lys233 | Ile144, Leu219, Leu232, Trp293, Ile296, His297 | |
| delta | 1 | 1011 ± 49 | 159 | 1565 | Gln105, Lys108, Asp128, Tyr129, Tyr129 | Leu128, Lys214 (aliphatic chain), Val217 |
| 2 | 90 ± 8 | 19 | 19 | Asp128, Tyr129 | Met132, Lys214 (aliphatic chain), Val217, Trp274, Ile277, Ile304 | |
| 3 | 3 ± 1.3 | 17 | 17 | Asp128, Tyr129 | Met132, Lys214 (aliphatic chain), Val217, Trp274, Ile277, Val281 | |
| kappa | 1 | 231 ± 21 | 17 | 69 | Asp138, Tyr139, Ser211 | Trp124, Val134, Leu135 |
| 2 | 131 ± 7 | 8.3 | 53 | Thr111, Asp138, Tyr139, Lys227, Tyr312, Tyr320 | Gln115, Leu135, Ile294 | |
| 3 | 24 ± 9 | 3.2 | 23 | Gln115, Asp138, Tyr139, Ser211, Tyr312 | Phe114, Val118, Ile294, Ile316 |
Inhibitory constants calculated for the lowest energy docked complexes including nonspecifically bound poses.
Inhibitory constants calculated for the lowest energy docked complexes excluding low energy poses which were regarded as false positives.
Figure 7.
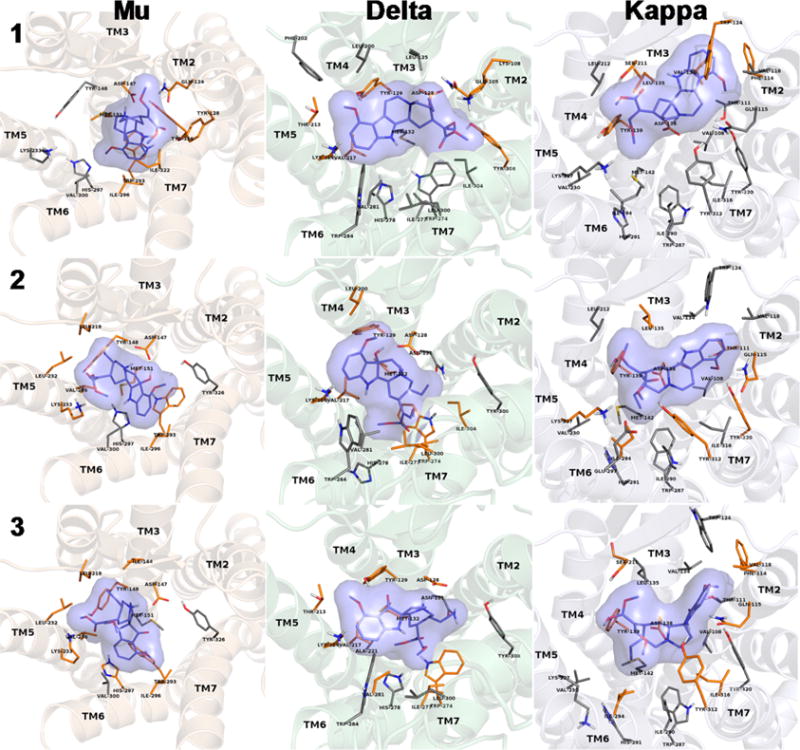
Lowest energy docked complexes of specifically bound mitragynine derivatives (1–3) to opioid receptors. Binding pocket side chains which were identified in the crystal structures to take part in receptor ligand interactions and were kept flexible during docking are shown. Side chains which are in direct contact with the ligands are depicted in orange. Nonpolar hydrogens are omitted for clarity.
DISCUSSION
Although initially described in the scientific literature as early as 1974, very little was known about the pharmacology of 3 prior to this study. Its chemical structure suggested it as an excellent starting point for semisynthetic diversification. Thus, we subjected 3 to detailed pharmacological analysis. In opioid receptor-transfected CHO cell lines, 3 had a high affinity for MOR-1 and DOR-1 sites with a moderate affinity at KOR-1. After subcutaneous administration, 3 was a potent antinociceptive agent in two thermal pain models: radiant heat tail flick and hot plate. It was also active in the mouse tail flick assay when administered supraspinally and orally. We established the agonist selectivity of 3 through pharmacological and genetic approaches, demonstrating that its antinociception was mediated by mu opioid receptors and not kappa, delta, and/or α2 adrenergic receptors. Mu antagonists attenuate the antinociception, but it was insensitive to kappa and delta antagonists. Clonidine, an α2 agonist, is a potent analgesic used for the treatment of various pain conditions.56 Yohimbine, an α2 antagonist, did not affect the antinociception of 3, therefore, its antinociception is likely not related to adrenergic pathways. The nonreversal of analgesia also rules out a role of α2A and α2C adrenergic receptors in mediating analgesia of 3, although the drug had some affinity for these receptors in our initial screening. Antisense downregulation of E1-MOR-1 variants attenuated antinociception, while downregulation of DOR-1 and KOR-1 failed to modify 3 antinociception. In mice lacking E11-MOR-1 variants (E11 MOR-1 KO mice) compound 3, like morphine, still exhibited antinociception comparable to wild-type mice, while in mice lacking all mu opioid receptor splice variants (E1/E11 MOR-1 KO mice), 3 antinociception was completely eliminated. The results from antisense and KO mice experiments implicate traditional 7TM mu opioid receptor aka E1-MOR-1 variants in 3 antinociception.
Of interest, 3 signaling failed to recruit β-arrestin-2 alone and antagonized both DAMGO-induced β-arrestin-2 recruitment and stimulation of [35S]GTPγS binding. To the best of our knowledge, 3 is the first example of a mixed activity mu opioid agonist/delta antagonist ligand which does not recruit β-arrestin-2. Prior evidence in the literature suggests that failure to recruit β-arrestin-2 and delta antagonism may both be successful in separating antinociception from unwanted side effects. Consistent with this, 3 displayed a robust antinociceptive effect in mice and without conditioned place preference or aversion. Two different dosing paradigms (two and four times per day) revealed the slow development of tolerance and a marked decrease in jumping following challenge with naloxone, which is characteristic of physical dependence. Furthermore, 3 showed no respiratory depression at twice its analgesic ED50 dose and far less constipation than morphine.
A number of mixed mu agonist/delta antagonist ligands have been reported in the literature. DIPP-NH2[Ψ],57 5″-(4-chlorophenyl)-6,7-didehydro-4,5α-epoxy-3-hydroxy-17-methylpyrido[2′,3′:6,7]morphinan (SoRi20411),5 and 14-alkoxy pyridomorphinans58 are potent delta antagonists and mu agonists which produce analgesia with reduced tolerance when given supraspinally. It must be noted that a majority of these studies monitored the development of tolerance through the accepted practice of administering the ED80 antinociceptive dose of the test drug twice daily for 5–7 days. The current study tested this approach even more rigorously, yet still found significant reductions in antinociceptive tolerance following administration of 3 for 29 days. DIPP-NH2[Ψ] also shows no physical dependence in treated mice. 4a,9-Dihydroxy-7a-(hydroxymethyl)-3-methyl-2,3,4,4a,5,6-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-7(7aH)-one (UMB425)59 and the cyclic peptide analog of KSK103 (C-terminal Ser(β-Glc)NH2)6 and ([Dmt1]DALDA → CH2CH2NH ← TICP-[Ψ]), which connects Dmt1-DALDA (mu agonist) with the delta antagonist TICP[Ψ] through a spacer, also show reduced acute antinociceptive tolerance compared to morphine when given systemically.60 Administration of delta antagonists or induced downregulation of delta opioid receptors has been reported to prevent morphine tolerance without sacrificing analgesic potency.61,62 Genetic disruption of the opioid system led to similar observations. In DOR-1 antisense knockdown mouse models, tolerance and acute dependence to morphine were eliminated,63 while DOR-1 knockout animals did not develop antinociceptive tolerance to morphine.64 The role of delta opioid receptors in blocking mu opioid-mediated CPP is still unclear.65 CPP is retained unchanged or abolished depending on assay conditions in DOR-1 KO mice, while morphine reportedly was more rewarding in a study with β-arrestin-2 KO mice.66 Additional study of this phenomenon should be possible as additional novel ligands with suitable MOR agonist/DOR antagonist activity profiles such as 3 become available.
Although GPCRs are mostly studied in relation to their ability to activate G-proteins, these transmembrane receptors are capable of recruiting β-arrestins to initiate separate cellular signal transduction pathways.67 β-Arrestin-2 activation has been implicated in the mechanism of receptor desensitization and the occurrence of deleterious side effects.68 Similarly to G-protein signaling, β-arrestin-2 activation is ligand and receptor dependent. Different ligands are able to stabilize GPCRs in a variety of conformations, resulting in the differential activation (bias) of G-protein and β-arrestin-2-mediated signaling pathways. Therefore, ligands interacting with the receptor do not simply have a linear effect on efficacy but also affect the functional quality of the downstream pathways.69 Biased signaling of opioid receptors has been studied in detail by Bohn and co-workers. Compared with wild-type mice, morphine displayed enhanced antinociception and significantly attenuated respiratory depression and inhibition of GI transit in β-arrestin-2 KO mice.70–72 These results suggest that a fully G-protein-biased opioid ligand that does not activate β-arrestin-2 signaling may be able to separate antinociception from some opioid adverse effects. An important example of G-protein-biased opioids is oliceridine, a synthetic mu agonist currently in clinical trials as an alternative to morphine and fentanyl for the treatment of chronic pain. Oliceridine is a potent analgesic that causes less respiratory depression and constipation than morphine at equianalgesic doses in humans.15
Taken together, these previous observations may explain why 3 is able to separate antinociception from typical opioid side effects resulting in low risk of developing tolerance while showing limited physical dependence, respiratory depression, and inhibition of GI transit. However, the relative contributions of its mu agonist/delta antagonist activity and its inability to recruit β-arrestin-2 to this advantageous pharmacological profile are not clear.
In order to investigate the SAR of the mitragynine pseudoindoxyl scaffold, semisynthetic analogs were made starting from 1 (C-9 analogs) and 3 (N-1 analogs). Receptor affinities were not significantly affected by modifications at the C-9 position, although 9-O-acetylation slightly lowered mu and delta affinities. Compounds 4 and 6–9 retained high, subnanomolar affinity at cells expressing MOR-1 with little change in affinity in DOR-1 cells. None of the derivatives stimulated β-arrestin-2 activation. According to previous literature reports, C-9 modifications of mitragynine altered the efficacy at mu receptors. Replacing the C-9 methoxy group with H yields corynantheidine, a mu antagonist, whereas C-9 O-demethylation yields 9-OH corynantheidine, a partial agonist in in vitro assays.39,73 According to our studies, the SAR of the C-9-modified pseudoindoxyl scaffold is quite distinct. Various substituents can be tolerated at this position, maintaining full mu agonism. Neither C-9 O-demethylation (4) nor the removal of the methoxy group (6) affected the efficacies at mu as 4 and 6 were mu full agonists. However, the activity at delta receptors is differentially affected by varying substituents. Compounds 4, 6, 7, 9, 10 retained delta antagonism, while the 9-phenyl analog, 8, was a delta agonist. 8 was a dual mu–delta agonist with similar intrinsic activity and potency at both receptors. Substitution and the introduction of bulky groups at N-1 (the indoxyl nitrogen) were not tolerated. Both the N-benzyl (11) and N-methyl (12) derivatives showed diminished affinities at all three opioid receptors compared to 3. In vivo, the C-9 analogs were active after systemic administration. The 9-OH derivative (4) was more potent than 3. Removal of the methoxy group (6) increased analgesic potency. The analgesic potencies of the corresponding C-9 mitragynine analogs (corynantheidine and 9-OH corynantheidine) are not reported in the literature. These results, in addition to the in vitro data, suggest that the SAR of compounds of the pseudoindoxyl scaffold differs from that of the natural Mitragyna alkaloids. Substitution of the C-9 methoxy group with –CN, phenyl, and furan-3-yl groups afforded products (7–9, respectively) roughly equipotent to 3. Acetylation of 4 had a slightly negative impact on the analgesic potency (10).
In comparison to the mu opioid receptor-bound morphinans,74–76 in silico modeling suggests the mitragynine derivatives (1–3) are likely to have a different binding pose. The salt bridge between Asp147 and the tertiary amine of the ligand and the participation of the phenolic OH in a water molecule-assisted hydrogen-bonded polar network of Tyr148, Lis233, and His297 were described as the main, conserved interactions between morphinan ligands and the binding pocket.77 The presence of a salt bridge between Asp147 and the tertiary amine of 1 and its derivatives was a filtering criterion of docking results, therefore, it is present in the docked complexes of all three mitragynine compounds. The β-methoxy acrylate moiety in the mitragynine compounds docked in the mu receptor pocket occupied the same space as the phenol moiety of the morphinan scaffold in the crystallographic structures.75,77
The main difference between the mu receptor-bound 1, 2, and 3 is that the oxidation products (2 and 3) seem to participate in the polar network formed between Tyr148, Lys233, and His297 by contributing their methoxy (C-9) and methyl ester groups and replacing hydrogen-bond assisting water molecules observed in the crystal structure of the agonist-bound mu receptor (Table 3).76 On the other hand, the beta-methoxy methyl acrylate moiety of 1 forms polar interactions with Gln124 and Tyr128 and does not take part in the aforementioned polar network (Figure S9 and Table 3). In addition to the salt bridge between Asp147 in the mu receptor crystal and the tertiary amine, it seems that there is a hydrogen bond present, formed with the 7-OH group of 2 (Figure S10). There is no significant difference in the relative orientation between the receptor-bound 2 and 3 (Figure S11). Differences in the calculated binding free energies and Ki values are most likely to emerge from the type and number of receptor contacts formed by these two compounds in those particularly similar docked orientations. Such differences in interactions are possibly due to the different heterocyclic scaffolds of 2 and 3. Compared to its derivatives, 1 adopts a different orientation when bound to the delta receptor and forms fewer contacts with the residues constituting the binding pocket, resulting in a loss of affinity for this receptor. Admittedly, the relative orientation and the number and type of contacts formed between 2 and 3 with the delta receptor and the calculated in silico Ki values are highly similar, rendering it difficult to give an accurate explanation for the 1 order of magnitude difference in the experimentally determined binding affinities. A key difference between the two bound geometries is that the beta-methoxy methyl acrylate moiety of 2 is positioned in a hydrophobic environment of Ile277, Leu300, and Ile304, (Figure S12 and Table 3), while in the case of 3, this group is projected into a more hydrophilic part of the binding pocket lined with Asn131, Trp274, and His278 (Figure S13). Furthermore, the 7-OH group of 2 was found to interact with Asp128 (Figure S12) similarly to that observed when docked to the mu (Figure S10). In addition, the 9-methoxy group of 2 formed contact with the phenolic OH of Tyr129. In the delta-bound 3 complex, both the 9-methoxy and the carbonyl groups of the pseudoindoxyl moiety were calculated to be in favorable position for hydrogen-bonding with Tyr129 (Figures 7 and S13).
CONCLUSIONS
In summary, we report for the first time the detailed in vitro and in vivo studies on mitragynine pseudoindoxyl. Mitragynine pseudoindoxyl is a mu agonist/delta antagonist opioid with a signaling bias for G-protein-mediated signaling pathways in vitro and which produced potent antinociception in vivo. Perhaps owing to its mixed mu agonism/delta antagonism activity, mitragynine pseudoindoxyl may avoid some of the major problems of opioid therapy, as we observed no reward or aversion and diminished antinociceptive tolerance, physical dependence, respiratory depression, and GI transit inhibition in mouse models. Upon chemical modification of this scaffold, key SAR features distinct from the mitragynine template were revealed. Among analogs modified at the C-9 position, compounds with differential efficacies within in vitro functional assays and improved in vivo potencies were identified. Docking studies to opioid receptors revealed the characteristic binding modes of mitragynine-type derivatives. It is hoped that these studies will contribute to improved understanding of the mechanism of action of compounds related to mitragynine, while holding the promise to provide novel antinociceptive drug candidates based on the mitragynine/corynantheidine pseudoindoxyl template that separates antinociception from the potential for abuse and other side effects due to their unique pharmacological properties. Observations reported in this paper and past studies on opioid ligands suggest a dual mechanism (mu agonism/delta antagonism coupled with β-arrestin2-nonrecruitment) which may account for the separation of side effects from antinociception seen with this template. The potential contributions from each of these two mechanisms will be explored in future studies on this template.
EXPERIMENTAL SECTION
Drugs and Chemicals
Opiates were provided by the Research Technology Branch of the National Institute on Drug Abuse (Rockville, MD). IBNtxA and [125I]BNtxA were synthesized in our laboratory as previously described.78–80 Na125I and [35S]GTPγS were purchased from PerkinElmer (Waltham, MA). Selective opioid antagonists were purchased from Tocris Bioscience. Miscellaneous chemicals and buffers were purchased from Sigma-Aldrich. Kratom “Red Indonesian Micro Powder” was purchased from Moon Kratom (Austin, TX).
Mice
Male CD1 mice (20–32 g) were obtained from Charles River Laboratories, and C57BL/6J mice (20–32 g each) were obtained from Jackson Laboratories (Bar Harbor, ME). Exon-11 KO79 and Exon-1/Exon-11 KO mice55 were bred in our laboratory. All mice used throughout the manuscript were opioid naïve. All mice were maintained on a 12 h light/dark cycle with Purina rodent chow and water available ad libitum and housed in groups of five until testing. All animal studies were preapproved by the Institutional Animal Care and Use Committees of the Memorial Sloan Kettering Cancer Center or University of Florida, in accordance with the 2002 National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Radioligand Competition Binding Assays
[125I]IBNtxA binding was carried out in membranes prepared from Chinese Hamster Ovary (CHO) cells stably expressing murine clones MOR-1, DOR-1, and KOR-1, as previously described.7–9,80 Binding was performed at 25 °C for 90 min. Binding in MOR-1/CHO was carried out in 50 mM potassium phosphate buffer with 5 mM MgSO4 and 20 μg/mL protein, while binding in KOR-1/CHO and DOR-1/CHO was carried out in 50 mM potassium phosphate pH = 7.0 buffer and 40 μg/mL protein. After the incubation, the reaction was filtered through glass-fiber filters (Whatman Schleicher & Schuell, Keene, NH) and washed three times with 3 mL of ice-cold 50 mM Tris-HCl, pH 7.4, on a semiautomatic cell harvester. Nonspecific binding was defined by the addition of levallorphan (8 μM) to matching samples and was subtracted from total binding to yield specific binding. Ki values were calculated by nonlinear regression analysis (GraphPad Prism, San Diego, CA). Protein concentrations were determined using the Lowry method with BSA as the standard.81
[35S]GTPγS Functional Assay
[35S]GTPγS binding was performed on membranes prepared from transfected cells stably expressing opioid receptors in the presence and absence of the indicated compound for 60 min at 30 °C in the assay buffer (50 mM Tris-HCl, pH 7.4, 3 mM MgCl2, 0.2 mM EGTA, and 10 mM NaCl) containing 0.05 nM [35S]GTPγS; 2 μg/mL each leupeptin, pepstatin, aprotinin, and bestatin; and 30 μM GDP, as previously described.82 After the incubation, the reaction was filtered through glass fiber filters (Whatman Schleicher & Schuell, Keene, NH) and washed three times with 3 mL of ice-cold buffer (50 mM Tris-HCl, pH 7.4) on a semiautomatic cell harvester. Filters were transferred into vials with 3 mL of Liquiscint (National Diagnostics, Atlanta, GA), and the radioactivity in vials was determined by scintillation spectroscopy in a Tri-Carb 2900TR counter (PerkinElmer Life and Analytical Sciences). Basal binding was determined in the presence of GDP and the absence of drug. Data were normalized to 1000 nM DAMGO, DPDPE, and U50,488 for MOR-1, DOR-1, and KOR-1 binding, respectively. EC50, IC50, and %Emax values were calculated by nonlinear regression analysis (GraphPad Prism, San Diego, CA).
β-arrestin-2 Recruitment Assay
β-arrestin-2 recruitment was determined using the PathHunter enzyme complementation assay (DiscoveRx, Fremont, CA) using modified MOR-1 expressed in CHO cells (DiscoveRx). Cells were plated at a density of 2500 cells/well in a 384-well plate as described in the manufacturer’s protocol. The following day, cells were treated with the indicated compound for 90 min at 37 °C, followed by incubation with PathHunter detection reagents for 60 min. Chemiluminescence was measured with an Infinite M1000 Pro plate reader (Tecan, Männedorf, Switzerland). For the antagonist dose–response assay, the cells were incubated with the antagonist for 30 min at 37 °C prior to the addition of agonist. Following antagonist treatment, the cells were treated with 10 μM DAMGO for 90 min at 37 °C, and chemiluminescence was detected using the PathHunter detection reagents.
Antinociception
Tail flick antinociception was determined using the radiant heat tail flick technique using an Ugo Basile model 37360 instrument as previously described.8,9 The intensity was set to achieve a baseline between 2 and 3 s. Baseline latencies were determined before experimental treatments for all mice. Tail flick antinociception was assessed quantally as a doubling or greater of the baseline latency, with a maximal 10 s latency to minimize damage to the tail. Data were analyzed as percent maximal effect, %MPE, and was calculated according to the formula: % MPE [(observed latency − baseline latency)/(maximal latency − baseline latency)] × 100. Compounds were injected subcutaneously (sc) or intracerebroventricularly (icv), and antinociception was assessed 15 min later at the peak effect. Intracerebroventricular dosing (icv) was carried out as previously described.83 Briefly, the mice were anesthetized with isoflurane. A small incision was made, and synthetic opiate analog (2 ul/mouse) was injected using a 10 μL Hamilton syringe fitted to a 27 gauge needle. Injections were made into the right lateral ventricle at the following coordinates: 2 mm caudal to bregma, 2 mm lateral to sagittal suture, and 2 mm in depth. Mice were tested for antinociception 15 min post injection. For oral (po) studies, mice were fasted for 18 h with access to water before administering the drug by oral gavage. For the antagonism studies, β-FNA (40 mg/kg, sc) and norbinaltorphimine (norBNI, 10 mg/kg, sc) were administered 24 h before 3. Naltrindole (NTI, 0.5 mg/kg, sc) was administered 15 min before 3. Antinociception also was assessed using the hot plate test.54 The hot plate (Ugo Basile 35100) consisted of a metal surface (55 °C) with a transparent plexiglass cylinder to contain the mouse. The latency to lick a hind paw or shake/flutter when the mouse was placed on the hot plate was measured, with a maximal latency of 30 s to avoid tissue damage. Baseline latencies were taken for each mouse prior to any drug administration. Mice were tested for analgesia with cumulative subcutaneous doses of the drug until the mouse can withstand the maximal latency. Once the mouse reached the maximal latency, the mouse was no longer given higher doses. In vivo experiments were evaluated using GraphPad Prism, San Diego, CA as described above.
Antisense Assays
Antisense (AN) and mismatch (MIS) oligodeoxynucleotides were designed based on the published sequences of the mouse mu opioid receptor gene (Oprm1), delta opioid receptor gene (Oprd1), and kappa opioid receptor gene (Oprk1) (Table 3). These probes have been previously described and validated.49,84–86 Antisense oligodeoxynucleotide injection: Groups of mice received the stated antisense by icv administration (5–10 μg) or mismatch (5–10 μg) oligodeoxynucleotide icv under light isoflurane anesthesia on days 1, 3 and 5, as previously described.85 Tail flick antinociception was tested on day 6. Control groups received no injection prior to testing. On test day, mice received 3 (1.5 mg/kg, sc), morphine (0.75 μg, icv), DPDPE (10 μg, icv), or U50,488H (5 mg/kg, sc). All experiments were performed 3× with similar results observed with each determination.
Respiratory Depression Assessment
Respiratory rate was assessed in awake, freely moving, adult male C57BL/6 mice with the MouseOx pulse oximeter system (Starr Life Sciences) as described previously.79 Each animal was habituated to the device for 30 min and then tested. A 5 s average breath rate was assessed at 5 min intervals. A baseline for each animal was obtained over a 25 min period before drug injection, and testing began at 15 min postinjection and continued for a period of 35 min. Groups of mice (n = 5) were treated sc with either morphine (5 or 10 mg/kg) or 3 (1.2 or 3 mg/kg). Groups were compared with repeated-measures ANOVA followed by Tukey’s multiple-comparison test.
GI Transit
Gastrointestinal transit was determined as previously described.87 Animals received the indicated drug followed by a charcoal meal (2.5% gum tragacanth in 10% activated charcoal in water) by gavage. Animals were sacrificed 30 min later, and the distance traveled by charcoal was measured. Significance was determined by ANOVA followed by Tukey’s multiple-comparison test.
Conditioned Place Preference/Aversion
Mice were conditioned with a counterbalanced place conditioning paradigm using similar timing as detailed previously.8 The amount of time subjects spent in each of three compartments was measured over a 30 min testing period. Prior to place conditioning, the animals (n = 95) did not demonstrate significant differences in their time spent exploring the left (543 ± 13 s) vs right (571 ± 12 s) compartments (p = 0.15; Student’s t-test), resulting in a combined preconditioning response of −0.1 ± 19 s. During each of the next 2 days, mice were administered vehicle (0.9% saline) and consistently confined in a randomly assigned outer compartment for 40 min, half of each group in the right chamber, half in the left chamber. Four h later, mice were administered morphine (10 mg/kg, ip), U50,488H (30 mg/kg, ip), cocaine (10 mg/kg, ip), or 3 (1.3 or 3.2 mg/kg, ip) and confined to the opposite compartment for 40 min. Conditioned place preference data are presented as the difference in time spent in drug- and vehicle-associated chambers and were analyzed via repeated measures two-way ANOVA with the difference in time spent on the treatment- vs vehicle-associated side as the dependent measure and conditioning status as the between groups factor. Where appropriate, Tukey’s HSD or Sidak’s multiple comparison post hoc tests were used to assess group differences. Effects were considered significant when p < 0.05. All effects are expressed as mean ± SEM.
In Silico Docking
Full sequence target structures of the human mu opioid and delta opioid receptors for docking studies were built and used as described elsewhere,9 using crystal structures of the homologous murine opioid receptors74,75 as templates (PDB codes: 4DKL and 4EJ4, respectively. The X-ray structure of the human kappa receptor (PDB code: 4DJH)88 was used as docking target after missing side chains were added. Dockings were performed with the Autodock 4.2 software. Side chains in contact with the bound ligands observed in the crystal complexes of the mu, delta, and kappa opioid receptors were kept flexible as well as all ligand torsions. Mitragynine and its natural derivatives were docked using the Lamarckian genetic algorithm in an 80 × 80 × 80 Å grid volume with 0.375 Å spacing. This docking volume is large enough to cover the whole receptor region accessible from the extracellular side (Figure S14). In this sense, blind docking studies were performed, and 1000 dockings were done for all compounds and receptor models. The resultant ligand–receptor complexes were clustered and ranked according to the corresponding binding free energies, which were also used to calculate inhibitory constants according to the following equation: ΔG = RTln Ki.
Chemistry
General Methods
All chemicals were purchased from Sigma-Aldrich Chemicals, and were used without further purification. Reactions were carried out in flame-dried reaction flasks under Ar. Reaction mixtures were purified by Silica Flash chromatography on E. Merck 230–400 mesh silica gel 60 using a Teledyne ISCO CombiFlash Rf instrument with UV detection at 280 and 254 nm. RediSep Rf silica gel normal phase columns were used. The yields reported are isolated yields. IR spectra were recorded on a Bruker Optics Tensor 27 FTIR spectrometer with peaks reported in cm−1. NMR spectra were recorded on Bruker Avance III 500, Avance III 600 with DCH CryoProbe instruments. NMR spectra were processed with MestReNova software (ver. 10.0.2.). Chemical shifts are reported in parts per million (ppm) relative to residual solvent peaks rounded to the nearest 0.01 for proton and 0.1 for carbon (CDCl3 1H: 7.26, 13C: 77.3; CD3OD 1H: 3.31, 13C: 49.0; DMSO-d6 13C: 39.5). Peak multiplicity is reported as follows: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet. Coupling constants (J) are expressed in Hz. Mass spectra were obtained at the MSKCC Analytical Core Facility on a Waters Acuity SQD LC-MS by electrospray (ESI) ionization. High-resolution mass spectra were obtained on a Waters Acuity Premiere XE TOF LC-MS by electrospray ionization. Accurate masses are reported for the molecular ion [M + H]+. Purity (≥95%) was confirmed using HPLC: Waters 1525 Binary Pump, Waters 2489 UV– vis detector, Waters XBridge C18 column (5 μm × 150 × 4.6 mm), mobile phase: solvent A: water with 0.1% TFA; solvent B: acetonitrile with 0.1% TFA. Gradient: 5–95% acetonitrile/water. Flow rate: 1 mL/min.
Isolation of Mitragynine (1) from Mitragyna speciosa (Kratom)
Kratom “Red Indonesian Micro Powder” was purchased from Moon Kratom (Austin, TX). Mitragynine (1) was extracted from the powdered leaves by a modified method (added new step: petroleum ether extraction of the acidic aqueous phase) from that reported by Ponglux et al.42 Kratom powder (450 g) was extracted by refluxing with MeOH (5 × 500 mL) for 40 min. The suspension was filtered after each extraction, and the solvent evaporated. The residue was resuspended in 20% acetic acid solution (2 L) and rinsed with petroleum ether (3 × 500 mL). The aqueous layer was then cooled on ice bath and basified (pH ∼ 9) with 50% aqueous NaOH solution. The basified suspension was extracted with DCM (4 × 1 L). The combined organic layers were dried over Na2SO4 and filtered. The solvent was evaporated and the residue purified using flash column chromatography (gradient: 0–50% EtOAc in hexanes). The major constituent was 1 (yield 5.59 ± 0.59 g (1.24%); smaller quantities of speciogynine and paynantheine were also isolated.
(E)-Methyl-2-((2S,3S,12bS)-3-ethyl-8-methoxy-1,2,3,4,6,7,12,12b-octahydroindolo[2,3-a]quinolizin-2-yl)-3-methoxyacrylate (Mitragynine, 1)
IR (NaCl): 3363, 2950, 2796, 1698, 1643, 1570, 1508, 1435, 1310, 1275, 1255, 1148, 1106, 769, 734. 1H NMR (600 MHz, Chloroform-d) δ 7.74 (s, 1H), 7.43 (s, 1H), 6.99 (t, J = 7.9 Hz, 1H), 6.90 (d, J = 8.0 Hz, 1H), 6.45 (d, J = 7.7 Hz, 1H), 3.87 (s, 3H), 3.72 (s, 3H), 3.71 (s, 3H), 3.18–3.08 (m, 2H), 3.06–2.99 (m, 2H), 3.00–2.93 (m, 1H), 2.94–2.90 (m, 1H), 2.57–2.42 (m, 3H), 1.83–1.75 (m, 2H), 1.62 (dt, J = 11.5, 3.2 Hz, 1H), 1.24–1.16 (m, 1H), 0.87 (t, J =7.4 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 169.45, 160.75, 154.69, 137.41, 133.90, 121.98, 117.82, 111.67, 108.03, 104.37, 99.91, 61.74, 61.46, 57.94, 55.52, 53.98, 51.57, 40.87, 40.12, 30.14, 24.14, 19.28, 13.07. HRMS calcd for C23H30N2O4 (MH+), 399.2284; found 399.2285.
(E)-Methyl-2-((2S,3S,7aS)-3-ethyl-7a-hydroxy-8-methoxy-1,2,3,4,6,7,7a,12b-octahydro indolo[2,3-a]quinolizin-2-yl)-3-methoxyacrylate (7-OH Mitragynine, 2)
Mitragynine (1, 2.00 g, 5.02 mmol) was dissolved in acetonitrile (150 mL), then water (50 mL) was added. The resulting suspension was cooled to 0 °C, and the following solution was added slowly over the course of several minutes: PIFA (2.16 g, 1.1 equiv) in 22 mL acetonitrile. The reaction mixture was stirred at 0 °C for 1 h, then saturated aqueous NaHCO3 solution was added, and the mixture extracted with EtOAc. The organic phase was rinsed with brine (60 mL) and dried over anhydrous Na2SO4, and then it was evaporated under reduced pressure. The residue was dissolved in DCM and purified using flash column chromatography (gradient: 0–75% EtOAc in hexanes). The fractions containing the product were evaporated to yield 1075 mg (57%) of 2 as a light brown amorphous powder. IR (NaCl): 3436, 2952, 1702, 1645, 1599, 1487, 1461, 1436, 1270, 1246, 1145, 1078, 795, 738. 1H NMR (600 MHz, Chloroform-d) δ 7.44 (s, 1H), 7.34 (t, J = 8.0 Hz, 1H), 7.24 (d, J = 7.6 Hz, 1H), 6.78 (d, J = 8.3 Hz, 1H), 3.91 (s, 3H), 3.80 (s, 3H), 3.70 (s, 3H), 3.31 (dd, J = 11.1, 2.6 Hz, 1H), 3.03 (ddt, J = 11.5, 5.5, 2.8 Hz, 2H), 2.84–2.75 (m, 3H), 2.67 (ddd, J = 12.3, 4.3, 2.6 Hz, 1H), 2.53–2.46 (m, 1H), 1.98–1.93 (m, 1H), 1.87 (ddd, J = 14.6, 12.2, 4.3 Hz, 1H), 1.70–1.54 (m, 3H), 1.26–1.23 (m, 1H), 0.81 (t, J = 7.3 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 181.25, 169.44, 160.94, 156.02, 154.67, 131.50, 126.24, 114.46, 111.38, 109.65, 69.62, 62.00, 60.73, 58.32, 55.92, 51.54, 50.39, 40.67, 39.32, 36.22, 26.38, 19.10, 13.02. HRMS calcd for C23H30N2O5 (MH+), 415.2233; found 415.2248.
(E)-Methyl-2-((1′S,6′S,7′S)-6′-ethyl-4-methoxy-3-oxo-3′,5′,6′,7′,8′,8a′-hexahydro-2′H-spiro[indoline-2,1′-indolizine]-7′-yl)-3-methoxyacrylate (Mitragynine Pseudoindoxyl, 3)
7-OH-mitragynine (2, 200 mg, 0.48 mmol) was dissolved in dry toluene (6 mL), and Zn(OTf)2 (350 mg, 2 equiv) was added. The reaction was stirred in a sealed tube for 2 h at 110 °C. To the cooled mixture were added 10 mL sat. aqueous NaHCO3 solution and water (20 mL). Extracted with EtOAc (30 mL). The organic layer was rinsed with brine (20 mL) and dried over anhydrous Na2SO4. After evaporation of the solvent under reduced pressure, the residue was redissolved in DCM and purified using flash column chromatography (gradient: 1– 5% MeOH in DCM) to yield: 78 mg (39%) of 3 as a yellow amorphous powder. NMR was identical to that reported in the literature.39 IR (NaCl): 3350, 2947, 2794, 1687, 1615, 1502, 1343, 1269, 1246, 1148, 1079, 757. 1H NMR (600 MHz, Chloroform-d) δ 7.32 (t, J = 8.1 Hz, 1H), 7.27 (s, 1H), 6.40 (d, J = 8.1 Hz, 1H), 6.13 (d, J = 8.1 Hz, 1H), 5.13 (s, 1H), 3.89 (s, 3H), 3.66 (s, 3H), 3.62 (s, 3H), 3.15–3.07 (m, 2H), 2.76 (dt, J = 11.9, 3.5 Hz, 1H), 2.38–2.29 (m, 2H), 2.29–2.18 (m, 1H), 2.14 (dt, J = 10.2, 6.3 Hz, 1H), 1.93–1.84 (m, 1H), 1.63 (dt, J = 11.3, 6.8 Hz, 1H), 1.49 (d, J = 11.3 Hz, 1H), 1.18 (ddd, J = 13.2, 7.8, 2.9 Hz, 1H), 1.11 (dd, J = 11.3, 3.6 Hz, 1H), 0.84 (t, J = 7.4 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 199.73, 169.05, 162.27, 160.40, 158.74, 138.85, 111.85, 109.96, 103.95, 99.21, 75.37, 73.38, 61.61, 55.86, 54.96, 53.35, 51.36, 40.28, 38.57, 35.25, 23.95, 19.47, 13.11. HRMS calcd for C23H30N2O5 (MH+), 415.2233; found 415.2216.
(E)-Methyl-2-((1′S,6′S,7′S)-6′-ethyl-4-hydroxy-3-oxo-3′,5′,6′,7′,8′,8a′-hexahydro-2′H-spiro[indoline-2,1′-indolizine]-7′-yl)-3-methoxyacrylate (9-OH-Corynantheidine Pseudoindoxyl, 4)
7-OH mitragynine (2, 400 mg, 0.97 mmol) was dissolved in dry DCM (20 mL), then AlCl3 (1.29 g, 10 equiv) was added. The mixture was cooled to °0 C, and ethanethiol (1.39 mL, 20 equiv) was added. The mixture was stirred at rt for 5 h. Water (30 mL) was slowly added, then it was separated from the organic layer. The organic layer was rinsed with brine (30 mL), then separated and dried over Na2SO4. Evaporated under reduced pressure. The residue was redissolved in DCM and purified using flash column chromatography (gradient: 1– 3% MeOH in DCM) to yield 342 mg (89%) of 4 as a bright yellow amorphous powder. IR (NaCl): 3211, 2945, 1697, 1628, 1513, 1451, 1348, 1247, 1148, 1120, 1082, 744. 1H NMR (600 MHz, CDCl3) δ 7.29 (m, 2H), 6.30 (d, J = 8.1 Hz, 1H), 6.15 (d, J = 8.0 Hz, 1H), 5.12 (s, 1H), 3.67 (s, 3H), 3.63 (s, 3H), 3.17–3.11 (m, 2H), 2.83–2.75 (m, 1H), 2.36–2.29 (m, 2H), 2.26–2.20 (m, 2H), 2.14 (dd, J = 11.2, 2.8 Hz, 1H), 1.98–1.90 (m, 1H), 1.69–1.57 (m, 2H), 1.51 (d, J = 11.2 Hz, 1H), 1.24–1.16 (m, 1H), 1.14–1.08 (m, 1H), 0.86 (t, J = 7.4 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 203.60, 169.03, 160.52, 160.05, 157.31, 140.24, 111.63, 108.97, 103.68, 102.27, 75.49, 72.99, 61.69, 54.93, 53.32, 51.41, 40.21, 38.63, 34.79, 23.97, 19.43, 13.07. HRMS calcd for C22H28N2O5 (MH+), 401.2076; found 401.2068.
(E)-Methyl-2-((1′S,6′S,7′S)-6′-ethyl-3-oxo-4-(trifluoromethylsulfonyloxy)-3′,5′,6′,7′,8′,8a′-hexahydro-2′H-spiro[indoline-2,1′-indolizine]-7′-yl)-3-methoxyacrylate (9-O-Trifluoro Methane Sulfonyl Corynantheidine Pseudoindoxyl, 5)
4 (200 mg, 0.5 mmol) was dissolved in dry DCM (15 mL), and pyridine (647 uL, 16 equiv) was added. Then the solution was cooled to −40 °C on a dry ice acetone bath, and the following solution was slowly added over 2–3 min: 5 mL DCM and triflic anhydride (340 uL, 4 equiv). The reaction was stirred for 1 h at −40 °C. After warming up to rt, the solution was purified using flash column chromatography immediately (gradient: 20–75% EtOAc in hexanes) to yield 233 mg (83%) of 5 as a brown amorphous solid. 1H NMR (600 MHz, CDCl3) δ 7.41 (t, J = 8.1 Hz, 1H), 7.29 (s, 1H), 6.84 (d, J = 8.3 Hz, 1H), 6.52 (d, J = 7.9 Hz, 1H), 5.58 (s, 1H), 3.68 (s, 3H), 3.63 (s, 3H), 3.15 (d, J = 9.7 Hz, 2H), 2.80 (dd, J = 12.5, 3.5 Hz, 1H), 2.35 (d, J = 7.4 Hz, 2H), 2.27 (d, J = 8.8 Hz, 2H), 2.18–2.13 (m, 1H), 1.66–1.58 (m, 1H), 1.52 (d, J = 10.9 Hz, 1H), 1.22–1.16 (m, 2H), 0.85 (t, J = 7.3 Hz, 4H). 13C NMR (151 MHz, CDCl3) δ 198.15, 171.40, 168.92, 161.46, 160.55, 145.34, 138.13, 111.73, 110.01, 73.58, 61.69, 60.62, 54.84, 53.65, 53.33, 51.38, 40.14, 38.33, 35.12, 23.84, 21.28, 19.41, 14.41, 13.00. HRMS calcd for C23H27F3N2O7S (MH+), 533.1569; found 533.1547.
(E)-Methyl-2-((1′S,6′S,7′S)-6′-ethyl-3-oxo-3′,5′,6′,7′,8′,8a′-hexahydro-2′H-spiro[indoline-2,1′-indolizin]-7′-yl)-3-methoxyacrylate (Corynantheidine Pseudoindoxyl, 6)
5 (10 mg, 0.019 mmol) was dissolved in dry DMF (500 uL) in a sealed tube, and the following reagents were added: Pd(OAc)2 (1.4 mg, 0.3 equiv), dppp (4 mg, 0.5 equiv), triethylamine (52.4 uL, 20 equiv), and formic acid (1 uL, 1.8 equiv). The mixture was stirred at 60 °C for 1h. The reaction mixture was diluted with EtOAc (10 mL) and washed with brine (5 mL) 5 times. The organic layer was dried over Na2SO4 and evaporated under reduced pressure. The residue was redissolved in DCM and purified using preparative TLC (75% EtOAc in hexanes) to yield 4 mg (57%) of 6 as an amorphous solid. IR (NaCl): 3286, 2927, 2360, 1676, 1620, 1437, 1248, 1200, 1138, 755. 1H NMR (600 MHz, CDCl3) δ 7.55 (d, J = 7.7 Hz, 1H), 7.43–7.38 (m, 1H), 7.27 (d, J = 6.7 Hz, 1H), 6.85 (d, J = 8.3 Hz, 1H), 6.77–6.72 (m, 1H), 5.20 (s, 1H), 3.66 (s, 3H), 3.62 (s, 3H), 3.20–3.12 (m, 2H), 2.82–2.76 (m, 1H), 2.38–2.29 (m, 2H), 2.25 (s, 2H), 2.20–2.13 (m, 1H), 1.97–1.89 (m, 1H), 1.69–1.61 (m, 1H), 1.55–1.48 (m, 1H), 1.23–1.17 (m, 1H), 1.05 (s, 1H), 0.86 (t, J = 7.4 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 202.60, 169.02, 160.95, 160.51, 137.30, 124.61, 118.30, 111.85, 111.68, 75.22, 73.52, 61.67, 54.94, 53.42, 51.40, 40.20, 38.59, 35.13, 23.92, 21.29, 19.41, 13.06. HRMS calcd for C22H28N2O4 (MH+), 385.2127; found 385.2120.
(E)-Methyl-2-((1′S,6′S,7′S)-4-cyano-6′-ethyl-3-oxo-3′,5′,6′,7′,8′,8a′-hexahydro-2′H-spiro[indoline-2,1′-indolizine]-7′-yl)-3-methoxyacrylate (9-Cyano Corynantheidine Pseudoindoxyl, 7)
5 (20 mg, 0.038 mmol) was dissolved in dry DMF (500 uL) in a sealed tube, and Pd(PPh3)4 (4.3 mg, 0.1 equiv) and Zn(CN)2 (8.8 mg, 2 equiv) were added. The reaction mixture was stirred at 80 °C for 3 h. After 3 h, the reaction was diluted with EtOAc (20 mL) and washed with brine 5 times. The organic layer was dried over Na2SO4 and evaporated under reduced pressure. The residue was redissolved in DCM and purified using flash column chromatography (gradient: 20– 80% EtOAC in hexanes) to yield 10 mg (65%) of 7 as an amorphous brown solid. IR (NaCl): 3355, 2940, 2233, 1699, 1606, 1501, 1438, 1242, 993, 859, 1082, 776. 1H NMR (600 MHz, CDCl3) δ 7.47–7.43 (m, 1H), 7.28 (s, 1H), 7.07 (d, J = 8.4 Hz, 1H), 7.05 (d, J = 7.2 Hz, 1H), 5.43 (s, 1H), 3.68 (s, 3H), 3.62 (s, 3H), 3.15 (t, J = 10.1 Hz, 2H), 2.79 (dt, J = 12.8, 3.5 Hz, 1H), 2.40–2.33 (m, 2H), 2.31–2.27 (m, 1H), 2.23 (t, J = 12.1 Hz, 1H), 2.19–2.13 (m, 1H), 1.96–1.90 (m, 1H), 1.66–1.59 (m, 1H), 1.51 (d, J = 11.2 Hz, 1H), 1.23–1.16 (m, 1H), 1.07 (d, J = 12.7 Hz, 1H), 0.85 (t, J = 7.4 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 201.47, 169.06, 162.01, 160.51, 142.35, 137.85, 136.72, 129.43, 128.16, 127.88, 120.06, 111.79, 110.73, 75.10, 73.89, 61.70, 55.07, 53.59, 51.40, 40.28, 38.57, 36.87, 36.86, 35.53, 24.92, 24.08, 19.48, 13.08. HRMS calcd for C23H27N3O4 (MH+), 410.2080; found 410.2068.
(E)-Methyl-2-((1′S,6′S,7′S)-6′-ethyl-3-oxo-4-phenyl-3′,5′,6′,7′,8′,8a′-hexahydro-2′H-spiro[indoline-2,1′-indolizine]-7′-yl)-3-methoxyacrylate (9-Phenyl Corynantheidine Pseudoindoxyl, 8)
5 (75 mg, 0.14 mmol) was dissolved in dry toluene (0.5 mL), and the solvent was removed under reduced pressure to ensure azeotropic removal of water residues. Dry methanol (1 mL) and dry toluene (1.5 mL) were added. To the resulting solution were added phenylboronic acid (19 mg, 1.1 equiv), K2CO3 (38.9 mg, 2 equiv), and Pd(PPh3)4 (8.1 mg, 0.05 equiv). The mixture was stirred at 80 °C for 2 h. The solvent was evaporated under reduced pressure, and the residue suspended in DCM, rinsed with water and brine (20 mL), then the organic layer was dried over Na2SO4 and was evaporated. Purified using flash column chromatography (gradient: 20–50% EtOAc in hexanes) to yield: 21 mg (32%) of 8 as a yellow amorphous solid. IR (NaCl): 3364, 2936, 2360, 1698, 1600, 1483, 1436, 1233, 1150, 759. 1H NMR (600 MHz, CDCl3) δ 7.52–7.49 (m, 2H), 7.43–7.34 (m, 4H), 7.30 (s, 1H), 6.82 (d, J = 8.2 Hz, 1H), 6.68 (d, J = 7.3 Hz, 1H), 5.26 (s, 1H), 3.69 (s, 3H), 3.63 (s, 3H), 3.15 (d, J = 10.0 Hz, 2H), 2.79 (dt, J = 13.0, 3.3 Hz, 1H), 2.34–2.25 (m, 3H), 2.21 (s, 1H), 2.17–2.10 (m, 1H), 1.95–1.88 (m, 1H), 1.69–1.63 (m, 1H), 1.53–1.47 (m, 1H), 1.24–1.18 (m, 1H), 1.14–1.09 (m, 1H), 0.86 (t, J = 7.4 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 201.47, 169.06, 162.01, 160.51, 142.35, 137.85, 136.72, 129.43, 128.16, 127.88, 120.06, 111.79, 110.73, 75.10, 73.89, 61.70, 55.07, 53.59, 51.40, 40.28, 38.57, 36.87, 36.86, 35.53, 24.92, 24.08, 19.48, 13.08. HRMS calcd for C28H32N2O4 (MH+), 461.2440; found 461.2422.
(E)-Methyl-2-((1′S,6′S,7′S)-6′-ethyl-4-(furan-3-yl)-3-oxo-3′,5′,6′,7′,8′,8a′-hexahydro-2′H-spiro[indoline-2,1′-indolizine]-7′-yl)-3-methoxyacrylate (9-Furyl Corynantheidine Pseudoindoxyl, 9)
The procedure described for the synthesis of 8 was used. Instead of phenylboronic acid, (furan-3-yl)boronic acid was employed. Yield: 81%. Compound 9 is a bright yellow amorphous powder. IR (NaCl): 3358, 2954, 2795, 2360, 2341, 1691, 1604, 1437, 1316, 1238, 1152, 796, 772. 1H NMR (600 MHz, Chloroform-d) δ 8.49 (d, J = 1.4 Hz, 1H), 7.48 (d, J = 1.7 Hz, 1H), 7.40 (t, J = 7.8 Hz, 1H), 7.31 (s, 1H), 6.87 (d, J = 7.5 Hz, 1H), 6.84 (dd, J = 1.9, 0.9 Hz, 1H), 6.77 (d, J = 8.1 Hz, 1H), 5.22 (s, 1H), 3.70 (s, 3H), 3.65 (s, 3H), 3.22–3.14 (m, 2H), 2.81 (dt, J = 12.6, 3.7 Hz, 1H), 2.40–2.32 (m, 2H), 2.32–2.22 (m, 2H), 2.17 (dd, J = 11.5, 3.2 Hz, 1H), 2.00–1.92 (m, 1H), 1.75–1.65 (m, 1H), 1.57–1.51 (m, 1H), 1.23 (dtd, J = 15.1, 7.4, 2.7 Hz, 1H), 1.15–1.10 (m, 1H), 0.89 (t, J = 7.4 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 201.73, 169.03, 162.40, 160.45, 143.85, 142.48, 137.00, 132.42, 122.74, 118.06, 116.66, 111.77, 110.60, 110.18, 75.11, 74.00, 61.65, 55.07, 53.54, 51.38, 40.28, 38.61, 35.68, 24.01, 19.50, 13.11. HRMS calcd for C26H30N2O5 (MH+), 451.2433; found 451.2215.
(E)-Methyl-2-((1′S,6′S,7′S)-4-acetoxy-6′-ethyl-3-oxo-3′,5′,6′,7′,8′,8a′-hexahydro-2′H-spiro[indoline-2,1′-indolizine]-7′-yl)-3-methoxyacrylate (9-O-Acetyl Corynantheidine Pseudoindoxyl, 10)
4 (20 mg, 0.05 mmol) was dissolved in pyridine (0.5 mL), and acetic anhydride (80 uL) was added. The mixture was stirred at rt for 2 h. The solution was poured into sat. aqueous NaHCO3 solution and extracted with DCM (30 mL). The organic layer was separated, rinsed with brine (10 mL), dried over Na2SO4, and evaporated under reduced pressure. The residue was redissolved in DCM and purified using flash column chromatography (gradient: 0–5% MeOH in DCM) to yield 13 mg (59%) of 10 as a bright yellow amorphous solid. IR (NaCl): 3393, 2956, 2874, 2787, 1761, 1688, 1628, 1503, 1239, 1217, 910, 764. 1H NMR (500 MHz, Chloroform-d) δ 7.38 (t, J = 8.0 Hz, 1H), 7.28 (s, 1H), 6.71 (d, J = 8.2 Hz, 1H), 6.36 (d, J = 7.6 Hz, 1H), 5.73 (s, 1H), 3.68 (s, 3H), 3.62 (s, 3H), 3.20 (dd, J = 8.6, 3.0 Hz, 2H), 2.79 (dd, J = 12.6, 3.9 Hz, 1H), 2.36 (s, 3H), 2.33–2.22 (m, 3H), 2.22–2.14 (m, 1H), 1.95 (dt, J = 15.8, 6.3 Hz, 1H), 1.63 (dt, J = 13.0, 6.1 Hz, 1H), 1.53 (d, J = 11.6 Hz, 1H), 1.30–1.16 (m, 2H), 1.16–1.09 (m, 1H), 0.86 (t, J = 7.3 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 199.57, 169.14, 168.98, 161.57, 160.47, 148.07, 138.28, 113.07, 111.70, 110.70, 109.35, 75.71, 73.70, 61.67, 55.02, 53.49, 51.38, 40.25, 38.51, 35.16, 23.91, 21.01, 19.49, 13.08. HRMS calcd for C24H30N2O6 (MH+), 443.2182; found 443.2174.
(E)-Methyl 2-((1′S,6′S,7′S)-1-benzyl-6′-ethyl-4-methoxy-3-oxo-3′,5′,6′,7′,8′,8a′-hexahydro-2′H-spiro[indoline-2,1′-indolizine]-7′-yl)-3-methoxyacrylate (N-Benzyl Mitragynine Pseudoindoxyl, 11)
Compound 3 was dissolved in dry acetonitrile (0.5 mL), and NaH (6 mg, 10 equiv) was added. The resulting suspension was stirred at rt for 30 min, during which its color turned red. Benzyl bromide (7.2 uL, 2.5 equiv) was added, and the mixture stirred for 2 h at rt. The red color disappeared promptly after the addition of benzyl bromide. After the reaction time, the mixture was carefully poured into cold water (20 mL) and extracted with DCM (30 mL). The organic layer was rinsed with brine (5 mL), separated, dried over Na2SO4, and evaporated under reduced pressure. The residue was redissolved in DCM and purified using flash column chromatography (gradient: 0–5% MeOH in DCM) to yield: 7.8 mg (64%) of 11 as a bright yellow amorphous solid. IR (NaCl): 2940, 2794, 1694, 1610, 1497, 1337, 1265, 1239, 1078, 732. 1H NMR (500 MHz, Chloroform-d) δ 7.42–7.28 (multiple overlapping peaks, 5H), 7.25–7.17 (multiple overlapping peaks, 2H), 6.08 (d, J = 8.1 Hz, 1H), 6.04 (d, J = 8.3 Hz, 1H), 5.35 (d, J = 17.4 Hz, 1H), 4.73 (d, J = 17.3 Hz, 1H), 3.90 (s, 3H), 3.70 (s, 3H), 3.64 (s, 3H), 3.13 (dd, J = 11.1, 2.2 Hz, 1H), 3.02 (t, J = 8.1 Hz, 1H), 2.78–2.72 (m, 1H), 2.33–2.18 (m, 4H), 2.09–2.02 (m, 1H), 1.94 (dt, J =13.7, 8.5 Hz, 1H), 1.66 (dt, J = 19.2, 6.8 Hz, 1H), 1.50 (d, J = 10.9 Hz, 1H),, 1.13 (d, J = 11.8 Hz, 1H), 0.86 (t, J = 7.4 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 199.55, 169.06, 161.60, 160.40, 158.81, 138.88, 138.82, 129.25, 129.02, 128.76, 128.64, 126.95, 126.51, 111.99, 108.49, 101.82, 98.07, 61.63, 55.89, 55.23, 53.92, 51.38, 47.61, 40.46, 38.67, 33.80, 32.06, 24.18, 19.82, 13.13. HRMS calcd for C30H36N2O5 (MH +), 505.2702; found 505.2726.
(E)-Methyl 2-((1′S,6′S,7′S)-6′-ethyl-4-methoxy-1-methyl-3-oxo-3′,5′,6′,7′,8′,8a′-hexahydro-2′H-spiro[indoline-2,1′-indolizine]-7′-yl)-3-methoxyacrylate (N-Methyl Mitragynine Pseudoindoxyl, 12)
The procedure described for the synthesis of 11 was used. Instead of benzyl bromide, iodomethane was employed. Yield: 58%. Compound 12 is a bright yellow amorphous powder. IR (NaCl): 2947, 2778, 2361, 1687, 1611, 1500, 1337, 1273. 1H NMR (600 MHz, Chloroform-d) δ 7.34 (t, J = 8.1 Hz, 1H), 7.28 (s, 1H), 6.25 (d, J =8.2 Hz, 1H), 6.05 (d, J = 8.0 Hz, 1H), 3.88 (s, 3H), 3.69 (s, 3H), 3.62 (s, 3H), 3.17 (s, 3H), 3.16–3.10 (m, 2H), 2.74 (dt, J = 13.0, 3.7 Hz, 1H), 2.34 (q, J = 8.9 Hz, 1H), 2.26 (dd, J = 11.3, 2.6 Hz, 1H), 2.21–2.12 (m, 2H), 2.08–2.03 (m, 1H), 1.92 (dt, J = 13.8, 8.7 Hz, 1H), 1.69–1.62 (m, 1H), 1.48 (dt, J = 11.1, 3.1 Hz, 1H), 1.28–1.23 (m, 1H), 1.20 (ddd, J = 13.3, 7.6, 2.8 Hz, 1H), 1.08 (dt, J = 13.0, 3.1 Hz, 1H), 0.84 (t, J = 7.4 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 199.87, 169.08, 162.01, 160.36, 158.90, 139.01, 112.04, 108.08, 100.37, 97.46, 78.27, 74.62, 61.59, 55.86, 55.14, 53.88, 51.34, 40.54, 38.72, 31.99, 30.02, 24.29, 19.71, 13.17. HRMS calcd for C24H32N2O5 (MH+), 429.2389; found 429.2393.
Supplementary Material
Table 4.
Sequences of Antisense (AN) and Mismatch (MIS) Oligodeoxynucleotides
| target | antisense | mismatch control |
|---|---|---|
| MOR-1 exon 1 | CGCCCCAGCCTCTTCCTCT | CGCCCCGACCTCTTCCCTT |
| DOR-1 exon 3 | AGGGGAAGGTCGGGTAGG | GAGGAGAGGTGCGTGGAG |
| KOR-1 exon 2 | CGCCCCAGCCTCTTCCTCT | CTCCGCGCTCTCACCCTCT |
Acknowledgments
This work was supported by research grants from the National Institute on Drug Abuse (DA034106) to S.M., The Harrington Discovery Institute Scholar Award and a grant from the Peter F. McManus Charitable Trust to G.W.P., DA013997 and DA02944 to Y.X.P., a National Science Foundation Graduate Research Fellowship (DGE-1257284) to G.F.M. The State of Florida, Executive Office of the Governor’s Office of Tourism, Trade, and Economic Development to J.P.M. This research was funded in part through the NIH/NCI Cancer Center Support Grant P30 CA008748 to MSKCC. A.B. was supported by the János Bolyai Research Fellowship. The authors would like to thank Professor Bryan L. Roth and the National Institute of Mental Health’s Psychoactive Drug Screening Program for their assistance in determining binding affinities at nonopioid CNS targets. The authors wish to thank Dr. Antara Majumdar of Nonclinical Biostatistics division at Bristol-Meyers Squibb, NJ for performing the statistical analysis for this manuscript and George Sukenick and Rong Wang of the NMR Analytical Core Facility at MSKCC for their assistance with NMR and MS instruments and experiments.
ABBREVIATIONS USED
- %MPE
percent maximal effect
- 6TM
six transmembrane
- 7TM
seven transmembrane
- CHO
Chinese hamster ovary
- CPA
conditioned place aversion
- CPP
conditioned place preference
- DAMGO
[D-Ala2, N-MePhe4, Gly-ol5]-enkephalin
- DCM
dichloromethane
- DMF
N,N-dimethylformamide
- DOR-1
a delta opioid receptor (Oprd1) clone
- DPDPE
[D-Pen2, D-Pen5]Enkephalin
- dppp
1,3-bis(diphenylphosphino)-propane
- GDP
guanosine diphosphate
- KO
knockout
- KOR-1
a kappa opioid receptor (Oprk1) clone
- MOR-1
a mu opioid receptor (Oprm1) clone
- norBNI
norbinaltorphimine
- NTI
naltrindole
- PIFA
[bis(trifluoroacetoxy)iodo]benzene
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.6b00748.
Experimental details and data (PDF)
Compound data (CSV)
Notes
The authors declare no competing financial interest.
References
- 1.Minami M, Satoh M. Molecular biology of the opioid receptors: structures, functions and distributions. Neurosci Res. 1995;23:121–145. doi: 10.1016/0168-0102(95)00933-k. [DOI] [PubMed] [Google Scholar]
- 2.Eguchi M. Recent advances in selective opioid receptor agonists and antagonists. Med Res Rev. 2004;24:182–212. doi: 10.1002/med.10059. [DOI] [PubMed] [Google Scholar]
- 3.Trescot AM, Datta S, Lee M, Hans H. Opioid pharmacology. Pain Physician. 2008;11:S133–S153. [PubMed] [Google Scholar]
- 4.Pasternak GW. Pharmacological mechanisms of opioid analgesics. Clin Neuropharmacol. 1993;16:1–18. doi: 10.1097/00002826-199302000-00001. [DOI] [PubMed] [Google Scholar]
- 5.Ananthan S. Opioid ligands with mixed mu/delta opioid receptor interactions: an emerging approach to novel analgesics. AAPS J. 2006;8:E118–125. doi: 10.1208/aapsj080114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mosberg HI, Yeomans L, Anand JP, Porter V, Sobczyk-Kojiro K, Traynor JR, Jutkiewicz EM. Development of a bioavailable mu opioid receptor (MOPr) agonist, delta opioid receptor (DOPr) antagonist peptide that evokes antinociception without development of acute tolerance. J Med Chem. 2014;57:3148–3153. doi: 10.1021/jm5002088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Váradi A, Hosztafi S, Le Rouzic V, Tóth G, Urai A, Noszal B, Pasternak GW, Grinnell SG, Majumdar S. Novel 6beta-acylaminomorphinans with analgesic activity. Eur J Med Chem. 2013;69:786–789. doi: 10.1016/j.ejmech.2013.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Váradi A, Marrone GF, Eans SO, Ganno ML, Subrath JJ, Le Rouzic V, Hunkele A, Pasternak GW, McLaughlin JP, Majumdar S. Synthesis characterization of a dual kappa-delta opioid receptor agonist analgesic blocking cocaine reward behavior. ACS Chem Neurosci. 2015;6:1813–1824. doi: 10.1021/acschemneuro.5b00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Váradi A, Palmer TC, Haselton N, Afonin D, Subrath JJ, Le Rouzic V, Hunkele A, Pasternak GW, Marrone GF, Borics A, Majumdar S. Synthesis of carfentanil amide opioids using the ugi multicomponent reaction. ACS Chem Neurosci. 2015;6:1570–1577. doi: 10.1021/acschemneuro.5b00137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Law PY, Reggio PH, Loh HH. Opioid receptors: toward separation of analgesic from undesirable effects. Trends Biochem Sci. 2013;38:275–282. doi: 10.1016/j.tibs.2013.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pradhan AA, Smith ML, Kieffer BL, Evans CJ. Ligand-directed signalling within the opioid receptor family. Br J Pharmacol. 2012;167:960–969. doi: 10.1111/j.1476-5381.2012.02075.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Raehal KM, Bohn LM. The role of beta-arrestin2 in the severity of antinociceptive tolerance physical dependence induced by different opioid pain therapeutics. Neuropharmacology. 2011;60:58–65. doi: 10.1016/j.neuropharm.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bohn LM, Lefkowitz RJ, Caron MG. Differential mechanisms of morphine antinociceptive tolerance revealed in (beta)arrestin-2 knock-out mice. J Neurosci. 2002;22:10494–10500. doi: 10.1523/JNEUROSCI.22-23-10494.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dewire SM, Yamashita DS, Rominger DH, Liu G, Cowan CL, Graczyk TM, Chen XT, Pitis PM, Gotchev D, Yuan C, Koblish M, Lark MW, Violin JD. A G protein-biased ligand at the mu opioid receptor is potently analgesic with reduced gastrointestinal respiratory dysfunction compared with morphine. J Pharmacl Ep Ther. 2013;344:708–717. doi: 10.1124/jpet.112.201616. [DOI] [PubMed] [Google Scholar]
- 15.Soergel DG, Subach RA, Burnham N, Lark MW, James IE, Sadler BM, Skobieranda F, Violin JD, Webster LR. Biased agonism of the mu opioid receptor by TRV130 increases analgesia reduces on-target adverse effects versus morphine: A randomized double-blind placebo-controlled crossover study in healthy volunteers. Pain. 2014;155:1829–1835. doi: 10.1016/j.pain.2014.06.011. [DOI] [PubMed] [Google Scholar]
- 16.Rives ML, Rossillo M, Liu-Chen LY, Javitch JA. 6′-Guanidinonaltrindole (6′-GNTI) is a G protein-biased kappa opioid receptor agonist that inhibits arrestin recruitment. J Bil Chem. 2012;287:27050–27054. doi: 10.1074/jbc.C112.387332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.White KL, Robinson JE, Zhu H, DiBerto JF, Polepally PR, Zjawiony JK, Nichols DE, Malanga CJ, Roth BL. The G protein–biased κ opioid receptor agonist RB-64 is analgesic with a unique spectrum of activities in vivo. J Pharmacl Ep Ther. 2015;352:98–109. doi: 10.1124/jpet.114.216820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lovell KM, Frankowski KJ, Stahl EL, Slauson SR, Yoo E, Prisinzano TE, Aubé J, Bohn LM. structure–activity relationship studies of functionally selective kappa opioid receptor agonists that modulate ERK 1/2 phosphorylation while preserving G protein over βarrestin2 signaling bias. ACS Chem Neurosci. 2015;6:1411–1419. doi: 10.1021/acschemneuro.5b00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou L, Lovell KM, Frankowski KJ, Slauson SR, Phillips AM, Streicher JM, Stahl E, Schmid CL, Hodder P, Madoux F, Cameron MD, Prisinzano TE, Aubé J, Bohn LM. Development of functionally selective small molecule agonists at kappa opioid receptors. J Bil Chem. 2013;288:36703–36716. doi: 10.1074/jbc.M113.504381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li JW, Vederas JC. Drug discovery natural products: end of an era or an endless frontier? Science. 2009;325:161–165. doi: 10.1126/science.1168243. [DOI] [PubMed] [Google Scholar]
- 21.Prisinzano TE. Psychopharmacology of the hallucinogenic sage Salvia divinorum. Life Sci. 2005;78:527–531. doi: 10.1016/j.lfs.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 22.Prisinzano TE. Natural products as tools for neuroscience: discovery and development of novel agents to treat drug abuse. J Nt Prod. 2009;72:581–587. doi: 10.1021/np8005748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tidgewell K, Harding WW, Lozama A, Cobb H, Shah K, Kannan P, Dersch CM, Parrish D, Deschamps JR, Rothman RB, Prisinzano TE. Synthesis of salvinorin A analogues as opioid receptor probes. J Nt Prod. 2006;69:914–918. doi: 10.1021/np060094b. [DOI] [PubMed] [Google Scholar]
- 24.Tidgewell K, Groer CE, Harding WW, Lozama A, Schmidt M, Marquam A, Hiemstra J, Partilla JS, Dersch CM, Rothman RB, Bohn LM, Prisinzano TE. Herkinorin analogues with differential beta-arrestin-2 interactions. J Md Chem. 2008;51:2421–2431. doi: 10.1021/jm701162g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aldrich JV, Kulkarni SS, Senadheera SN, Ross NC, Reilley KJ, Eans SO, Ganno ML, Murray TF, McLaughlin JP. Unexpected opioid activity profiles of analogues of the novel peptide kappa opioid receptor ligand CJ-15 208. ChemMedChem. 2011;6:1739–1745. doi: 10.1002/cmdc.201100113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jansen KL, Prast CJ. Ethnopharmacology of kratom the Mitragyna alkaloids. J Ethnopharmacol. 1988;23:115–119. doi: 10.1016/0378-8741(88)90121-3. [DOI] [PubMed] [Google Scholar]
- 27.Karinen R, Fosen JT, Rogde S, Vindenes V. An accidental poisoning with mitragynine. Forensic Sci Int. 2014;245:e29–e32. doi: 10.1016/j.forsciint.2014.10.025. [DOI] [PubMed] [Google Scholar]
- 28.Kronstrand R, Roman M, Thelander G, Eriksson A. Unintentional fatal intoxications with mitragynine O-desmethyltramadol from the herbal blend krypton. J Anl Toxicol. 2011;35:242–247. doi: 10.1093/anatox/35.4.242. [DOI] [PubMed] [Google Scholar]
- 29.McIntyre IM, Trochta A, Stolberg S, Campman SC. Mitragynine ’kratom’ related fatality: a case report with postmortem concentrations. J Anl Toxicol. 2015;39:152–155. doi: 10.1093/jat/bku137. [DOI] [PubMed] [Google Scholar]
- 30.Neerman MF, Frost RE, Deking J. A drug fatality involving Kratom. J Forensic Sci. 2013;58:S278–S279. doi: 10.1111/1556-4029.12009. [DOI] [PubMed] [Google Scholar]
- 31.Macko E, Weisbach JA, Douglas B. Some observations on the pharmacology of mitragynine. Arch It Pharmacodn Ther. 1972;198:145–161. [PubMed] [Google Scholar]
- 32.Matsumoto K, Hatori Y, Murayama T, Tashima K, Wongseripipatana S, Misawa K, Kitajima M, Takayama H, Horie S. Involvement of mu opioid receptors in antinociception inhibition of gastrointestinal transit induced by 7-hydroxymitragynine isolated from Thai herbal medicine Mitragyna speciosa. Eur J Pharmacol. 2006;549:63–70. doi: 10.1016/j.ejphar.2006.08.013. [DOI] [PubMed] [Google Scholar]
- 33.Matsumoto K, Horie S, Ishikawa H, Takayama H, Aimi N, Ponglux D, Watanabe K. Antinociceptive effect of 7-hydroxymitragynine in mice: Discovery of an orally active opioid analgesic from the Thai medicinal herb Mitragyna speciosa. Life Sci. 2004;74:2143–2155. doi: 10.1016/j.lfs.2003.09.054. [DOI] [PubMed] [Google Scholar]
- 34.Matsumoto K, Horie S, Takayama H, Ishikawa H, Aimi N, Ponglux D, Murayama T, Watanabe K. Antinociception tolerance withdrawal symptoms induced by 7-hydroxymitragynine an alkaloid from the Thai medicinal herb Mitragyna speciosa. Life Sci. 2005;78:2–7. doi: 10.1016/j.lfs.2004.10.086. [DOI] [PubMed] [Google Scholar]
- 35.Matsumoto K, Mizowaki M, Suchitra T, Murakami Y, Takayama H, Sakai S-I, Aimi N, Watanabe H. Central antinociceptive effects of mitragynine in mice: contribution of descending noradrenergic serotonergic systems. Eur J Pharmacol. 1996;317:75–81. doi: 10.1016/s0014-2999(96)00714-5. [DOI] [PubMed] [Google Scholar]
- 36.Matsumoto K, Mizowaki M, Suchitra T, Takayama H, Sakai S, Aimi N, Watanabe H. Antinociceptive action of mitragynine in mice: evidence for the involvement of supraspinal opioid receptors. Life Sci. 1996;59:1149–1155. doi: 10.1016/0024-3205(96)00432-8. [DOI] [PubMed] [Google Scholar]
- 37.Matsumoto K, Yamamoto LT, Watanabe K, Yano S, Shan J, Pang PK, Ponglux D, Takayama H, Horie S. Inhibitory effect of mitragynine an analgesic alkaloid from Thai herbal medicine on neurogenic contraction of the vas deferens. Life Sci. 2005;78:187–194. doi: 10.1016/j.lfs.2005.04.042. [DOI] [PubMed] [Google Scholar]
- 38.Takayama H, Ishikawa H, Kitajima M, Aimi N. Formation of an unusual dimeric compound by lead tetraacetate oxidation of a corynanthe-type indole alkaloid mitragynine. Chem Pham Bull. 2002;50:960–963. doi: 10.1248/cpb.50.960. [DOI] [PubMed] [Google Scholar]
- 39.Takayama H, Ishikawa H, Kurihara M, Kitajima M, Aimi N, Ponglux D, Koyama F, Matsumoto K, Moriyama T, Yamamoto LT, Watanabe K, Murayama T, Horie S. Studies on the synthesis opioid agonistic activities of mitragynine-related indole alkaloids: discovery of opioid agonists structurally different from other opioid ligands. J Md Chem. 2002;45:1949–1956. doi: 10.1021/jm010576e. [DOI] [PubMed] [Google Scholar]
- 40.Zarembo JE, Douglas B, Valenta J, Weisbach JA. Metabolites of mitragynine. J Pham Sci. 1974;63:1407–1415. doi: 10.1002/jps.2600630916. [DOI] [PubMed] [Google Scholar]
- 41.Yamamoto LT, Horie S, Takayama H, Aimi N, Sakai S, Yano S, Shan J, Pang PK, Ponglux D, Watanabe K. Opioid receptor agonistic characteristics of mitragynine pseudoindoxyl in comparison with mitragynine derived from Thai medicinal plant Mitragyna speciosa. Gen Pharmacol. 1999;33:73–81. doi: 10.1016/s0306-3623(98)00265-1. [DOI] [PubMed] [Google Scholar]
- 42.Ponglux D, Wongseripipatana S, Takayama H, Kikuchi M, Kurihara M, Kitajima M, Aimi N, Sakai S. A New Indole Alkaloid 7 alpha-Hydroxy-7H-mitragynine from Mitragyna speciosa in Thailand. Planta Med. 1994;60:580–581. doi: 10.1055/s-2006-959578. [DOI] [PubMed] [Google Scholar]
- 43.Mercado-Marin EV, Garcia-Reynaga P, Romminger S, Pimenta EF, Romney DK, Lodewyk MW, Williams DE, Andersen RJ, Miller SJ, Tantillo DJ, Berlinck RGS, Sarpong R. Total synthesis isolation of citrinalin cyclopiamine congeners. Nature. 2014;509:318–324. doi: 10.1038/nature13273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Váradi A, Marrone GF, Palmer TC, Hunkele A, Le Rouzic V, Pasternak GW, Pan Y-X, McLaughlin JP, Majumdar S. Mitragyna alkaloids revisited: pharmacology of mitragynine and related natural products. Poster Presentation. Proceedings of the International Narcotics Research Conference 2015; Phoenix, AZ. Jun 15–19, 2015. [Google Scholar]
- 45.Besnard J, Ruda GF, Setola V, Abecassis K, Rodriguiz RM, Huang XP, Norval S, Sassano MF, Shin AI, Webster LA, Simeons FR, Stojanovski L, Prat A, Seidah NG, Constam DB, Bickerton GR, Read KD, Wetsel WC, Gilbert IH, Roth BL, Hopkins AL. Automated design of ligands to polypharmacological profiles. Nature. 2012;492:215–220. doi: 10.1038/nature11691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rossi GC, Pan Y-X, Brown GP, Pasternak GW. Antisense mapping the MOR-1 opioid receptor: Evidence for alternative splicing a novel morphine-6b-glucuronide receptor. FEBS Lett. 1995;369:192–196. doi: 10.1016/0014-5793(95)00757-z. [DOI] [PubMed] [Google Scholar]
- 47.Rossi GC, Pan YX, Cheng J, Pasternak GW. Blockade of morphine analgesia by an antisense oligodeoxynucleotide against the mu receptor. Life Sci. 1994;54:PL375–PL379. doi: 10.1016/0024-3205(94)90038-8. [DOI] [PubMed] [Google Scholar]
- 48.Standifer KM, Chien C-C, Wahlestedt C, Brown GP, Pasternak GW. Selective loss of d opioid analgesia binding by antisense oligodeoxynucleotides to a d opioid receptor. Neuron. 1994;12:805–810. doi: 10.1016/0896-6273(94)90333-6. [DOI] [PubMed] [Google Scholar]
- 49.Pasternak KR, Rossi GC, Zuckerman A, Pasternak GW. Antisense mapping KOR-1: evidence for multiple kappa analgesic mechanisms. Brain Res. 1999;826:289–292. doi: 10.1016/s0006-8993(99)01294-9. [DOI] [PubMed] [Google Scholar]
- 50.Pan YX, Pasternak GW. Molecular Biology of Mu Opioid Receptors. In: Pasternak GW, editor. The Opiate Receptors. 2nd. Springer; New York: 2011. pp. 121–160. [Google Scholar]
- 51.Pasternak GW, Pan Y-X. Mu opioids their receptors: Evolution of a concept. Pharmacol Rev. 2013;65:1257–1317. doi: 10.1124/pr.112.007138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pan Y-X, Xu J, Mahurter L, Bolan EA, Xu MM, Pasternak GW. Generation of the mu opioid receptor (MOR-1) protein by three new splice variants of the Oprm gene. Proc Nal Acd Si U S A. 2001;98:14084–14089. doi: 10.1073/pnas.241296098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xu J, Xu M, Hurd YL, Pasternak GW, Pan YX. Isolation and characterization of new exon 11-associated N-terminal splice variants of the human mu opioid receptor gene. J Neurochem. 2009;108:962–972. doi: 10.1111/j.1471-4159.2008.05833.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pan YX, Xu J, Xu M, Rossi GC, Matulonis JE, Pasternak GW. Involvement of exon 11-associated variants of the mu opioid receptor MOR-1 in heroin but not morphine actions. Prc Nal Acd Si U S A. 2009;106:4917–4922. doi: 10.1073/pnas.0811586106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lu Z, Xu J, Rossi GC, Majumdar S, Pasternak GW, Pan Y-X. Mediation of opioid analgesia by a truncated 6-transmembrane GPCR. J Cln Invest. 2015;125:2626–2630. doi: 10.1172/JCI81070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tryba M, Gehling M. Clonidine–a potent analgesic adjuvant. Curr Opn Anaesthesiol. 2002;15:511–517. doi: 10.1097/00001503-200210000-00007. [DOI] [PubMed] [Google Scholar]
- 57.Schiller PW, Fundytus ME, Merovitz L, Weltrowska G, Nguyen TMD, Lemieux C, Chung NN, Coderre TJ. The opioid μ agonist/δ antagonist DIPP-NH2[Ψ] produces a potent analgesic effect no physical dependence less tolerance than morphine in rats. J Md Chem. 1999;42:3520–3526. doi: 10.1021/jm980724+. [DOI] [PubMed] [Google Scholar]
- 58.Ananthan S, Saini SK, Dersch CM, Xu H, McGlinchey N, Giuvelis D, Bilsky EJ, Rothman RB. 14-Alkoxy- 14-acyloxypyridomorphinans: μ agonist/δ antagonist opioid analgesics with diminished tolerance dependence side effects. J Md Chem. 2012;55:8350–8363. doi: 10.1021/jm300686p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Healy JR, Bezawada P, Shim J, Jones JW, Kane MA, MacKerell AD, Coop A, Matsumoto RR. Synthesis modeling pharmacological evaluation of UMB 425 a mixed μ agonist/δ antagonist opioid analgesic with reduced tolerance liabilities. ACS Chem Neurosci. 2013;4:1256–1266. doi: 10.1021/cn4000428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schiller PW, Weltrowska G, Berezowska I, Nguyen TMD, Wilkes BC, Lemieux C, Chung NN. The TIPP opioid peptide family: Development of d antagonists d agonists and mixed m agonist/d antagonists. Biopolymers. 1999;51:411–425. doi: 10.1002/(SICI)1097-0282(1999)51:6<411::AID-BIP4>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 61.Abdelhamid EE, Sultana M, Portoghese PS, Takemori AE. Selective blockage of delta opioid receptors prevents the development of morphine tolerance dependence in mice. J Pharmacl Ep Ther. 1991;258:299–303. [PubMed] [Google Scholar]
- 62.Hepburn MJ, Little PJ, Gingras J, Kuhn CM. Differential effects of naltrindole on morphine-induced tolerance physical dependence in rats. J Pharmacl Ep Ther. 1997;281:1350–1356. [PubMed] [Google Scholar]
- 63.Kest B, Lee CE, McLemore GL, Inturrisi CE. An antisense oligodeoxynucleotide to the delta opioid receptor (DOR-1) inhibits morphine tolerance acute dependence in mice. Brain Res Bull. 1996;39:185–188. doi: 10.1016/0361-9230(95)02092-6. [DOI] [PubMed] [Google Scholar]
- 64.Zhu Y, King MA, Schuller AG, Nitsche JF, Reidl M, Elde RP, Unterwald E, Pasternak GW, Pintar JE. Retention of supraspinal delta-like analgesia loss of morphine tolerance in delta opioid receptor knockout mice. Neuron. 1999;24:243–252. doi: 10.1016/s0896-6273(00)80836-3. [DOI] [PubMed] [Google Scholar]
- 65.Charbogne P, Kieffer BL, Befort K. 15 years of genetic approaches in vivo for addiction research: Opioid receptor peptide gene knockout in mouse models of drug abuse. Neuropharmacology. 2014;76:204–217. doi: 10.1016/j.neuropharm.2013.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bohn LM, Gainetdinov RR, Sotnikova TD, Medvedev IO, Lefkowitz RJ, Dykstra LA, Caron MG. Enhanced rewarding properties of morphine but not cocaine in beta(arrestin)-2 knock-out mice. J Neurosci. 2003;23:10265–10273. doi: 10.1523/JNEUROSCI.23-32-10265.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rankovic Z, Brust TF, Bohn LM. Biased agonism: An emerging paradigm in GPCR drug discovery. Bioorg Md Chm Lett. 2016;26:241–250. doi: 10.1016/j.bmcl.2015.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by beta-arrestins. Science. 2005;308:512–517. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- 69.Correll CC, McKittrick BA. Biased ligand modulation of seven transmembrane receptors (7TMRs): Functional implications for drug discovery. J Md Chem. 2014;57:6887–6896. doi: 10.1021/jm401677g. [DOI] [PubMed] [Google Scholar]
- 70.Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, Lin FT. Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science. 1999;286:2495–2498. doi: 10.1126/science.286.5449.2495. [DOI] [PubMed] [Google Scholar]
- 71.Bohn LM, Gainetdinov RR, Lin FT, Lefkowitz RJ, Caron MG. Mu opioid receptor desensitization by beta-arrestin-2 determines morphine tolerance but not dependence. Nature. 2000;408:720–723. doi: 10.1038/35047086. [DOI] [PubMed] [Google Scholar]
- 72.Raehal KM, Walker JK, Bohn LM. Morphine side effects in beta-arrestin 2 knockout mice. J Pharmacl Ep Ther. 2005;314:1195–1201. doi: 10.1124/jpet.105.087254. [DOI] [PubMed] [Google Scholar]
- 73.Takayama H. Chemistry and pharmacology of analgesic indole alkaloids from the rubiaceous plant, Mitragyna speciosa. Chem Pham Bull. 2004;52:916–928. doi: 10.1248/cpb.52.916. [DOI] [PubMed] [Google Scholar]
- 74.Granier S, Manglik A, Kruse AC, Kobilka TS, Thian FS, Weis WI, Kobilka BK. Structure of the delta opioid receptor bound to naltrindole. Nature. 2012;485:400–404. doi: 10.1038/nature11111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Manglik A, Kruse AC, Kobilka TS, Thian FS, Mathiesen JM, Sunahara RK, Pardo L, Weis WI, Kobilka BK, Granier S. Crystal structure of the mu opioid receptor bound to a morphinan antagonist. Nature. 2012;485:321–326. doi: 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Huang N, Shoichet BK, Irwin JJ. Benchmarking sets for molecular docking. J Md Chem. 2006;49:6789–6801. doi: 10.1021/jm0608356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Huang W, Manglik A, Venkatakrishnan AJ, Laeremans T, Feinberg EN, Sanborn AL, Kato HE, Livingston KE, Thorsen TS, Kling RC, Granier S, Gmeiner P, Husbands SM, Traynor JR, Weis WI, Steyaert J, Dror RO, Kobilka BK. Structural insights into [micro] opioid receptor activation. Nature. 2015;524:315–321. doi: 10.1038/nature14886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Majumdar S, Burgman M, Haselton N, Grinnell S, Ocampo J, Pasternak AR, Pasternak GW. Generation of novel radiolabeled opiates through site-selective iodination. Bioorg Md Chm Lett. 2011;21:4001–4004. doi: 10.1016/j.bmcl.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Majumdar S, Grinnell S, Le Rouzic V, Burgman M, Polikar L, Ansonoff M, Pintar J, Pan YX, Pasternak GW. Truncated G protein-coupled mu opioid receptor MOR-1 splice variants are targets for highly potent opioid analgesics lacking side effects. Proc Nal Acd Si U S A. 2011;108:19778–19783. doi: 10.1073/pnas.1115231108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pickett JE, Váradi A, Palmer TC, Grinnell SG, Schrock JM, Pasternak GW, Karimov RR, Majumdar S. Mild Pd-catalyzed stannylation of radioiodination targets. Bioorg Md Chm Lett. 2015;25:1761–1764. doi: 10.1016/j.bmcl.2015.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Bil Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 82.Bolan EA, Pan YX, Pasternak GW. Functional analysis of MOR-1 splice variants of the mouse mu opioid receptor gene Oprm. Synapse. 2004;51:11–8. doi: 10.1002/syn.10277. [DOI] [PubMed] [Google Scholar]
- 83.Haley TJ, McCormick WG. Pharmacological effects produced by intracerebral injection of drugs in the conscious mouse. Br J Pharmacl Chemother. 1957;12:12–15. doi: 10.1111/j.1476-5381.1957.tb01354.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rossi GC, Brown GP, Leventhal L, Yang K, Pasternak GW. Novel receptor mechanisms for heroin morphine-6b -glucuronide analgesia. Neurosci Lett. 1996;216:1–4. doi: 10.1016/0304-3940(96)12976-1. [DOI] [PubMed] [Google Scholar]
- 85.Rossi GC, Standifer KM, Pasternak GW. Differential blockade of morphine morphine-6b-glucuronide analgesia by antisense oligodeoxynucleotides directed against MOR-1 G-protein a subunits in rats. Neurosci Lett. 1995;198:99–102. doi: 10.1016/0304-3940(95)11977-5. [DOI] [PubMed] [Google Scholar]
- 86.Rossi GC, Su W, Leventhal L, Su H, Pasternak GW. Antisense mapping DOR-1 in mice: further support for delta receptor subtypes. Brain Res. 1997;753:176–179. doi: 10.1016/s0006-8993(97)00081-4. [DOI] [PubMed] [Google Scholar]
- 87.Paul D, Pasternak GW. Differential blockade by naloxonazine of two m opiate actions: analgesia inhibition of gastrointestinal transit. Eur J Pharmacol. 1988;149:403–404. doi: 10.1016/0014-2999(88)90680-2. [DOI] [PubMed] [Google Scholar]
- 88.Wu H, Wacker D, Mileni M, Katritch V, Han GW, Vardy E, Liu W, Thompson AA, Huang XP, Carroll FI, Mascarella SW, Westkaemper RB, Mosier PD, Roth BL, Cherezov V, Stevens RC. Structure of the human kappa opioid receptor in complex with JDTic. Nature. 2012;485:327–332. doi: 10.1038/nature10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.