

Summary
The individual molecular pathways downstream of Cdc42, Rac, and Rho GTPases are well documented, but we know surprisingly little about how these pathways are coordinated when cells move in a complex environment in vivo. In the developing embryo, melanoblasts originating from the neural crest must traverse the dermis to reach the epidermis of the skin and hair follicles. We previously established that Rac1 signals via Scar/WAVE and Arp2/3 to effect pseudopod extension and migration of melanoblasts in skin. Here we show that RhoA is redundant in the melanocyte lineage but that Cdc42 coordinates multiple motility systems independent of Rac1. Similar to Rac1 knockouts, Cdc42 null mice displayed a severe loss of pigmentation, and melanoblasts showed cell-cycle progression, migration, and cytokinesis defects. However, unlike Rac1 knockouts, Cdc42 null melanoblasts were elongated and displayed large, bulky pseudopods with dynamic actin bursts. Despite assuming an elongated shape usually associated with fast mesenchymal motility, Cdc42 knockout melanoblasts migrated slowly and inefficiently in the epidermis, with nearly static pseudopods. Although much of the basic actin machinery was intact, Cdc42 null cells lacked the ability to polarize their Golgi and coordinate motility systems for efficient movement. Loss of Cdc42 de-coupled three main systems: actin assembly via the formin FMNL2 and Arp2/3, active myosin-II localization, and integrin-based adhesion dynamics.
Keywords: Rho GTPases, migration, actin cytoskeleton, cell motility, melanocyte, myosin, actin, adhesion, integrin, Cdc42
Graphical Abstract
Highlights
-
•
Loss of Cdc42 in the melanocyte lineage causes pigmentation defects in mice
-
•
Cdc42 null melanoblasts are elongated, with severe motility and proliferation defects
-
•
Cdc42 and Rac1 have distinct roles for in vivo migration
-
•
Cdc42 coordinates actin dynamics, active myosin-II, and integrin-based adhesion
Woodham and Paul et al. describe an important role for the small GTPase Cdc42 in cell proliferation and migration in mouse embryo melanoblasts. Loss of Cdc42 leads to severe pigmentation defects, and reveals Cdc42 as a coordinator of multiple cell-migration systems in the melanocyte lineage, including actin, contractility, and adhesion.
Introduction
During development of the mouse embryo, melanoblasts derive from neural crest cells following closure of the neural tube. They migrate through the dermis and cross the basement membrane into the epidermis at around embryonic day 12.5 (E12.5) [1] before homing to hair follicles [2, 3]. Melanoblasts use Rac1 to drive actin assembly and extension of pseudopods, which are stabilized by the actin-bundling protein fascin [4, 5]. Migration in the epidermis is individual and seemingly random [4, 6]. Epidermal melanoblasts move between keratinocytes along the epidermal basement membrane [7]. Here we explored the role of Cdc42 in melanoblast migration and population of mouse embryo skin.
Our insight into Cdc42 function in multicellular organisms is still limited, but generally supports a role in epithelial polarity and actin dynamics. Cdc42 is important for epithelial morphogenesis, early cell polarity, and spindle orientation during the development of Drosophila melanogaster and Caenorhabditis elegans [8, 9]. Global Cdc42 knockout in mice caused embryonic lethality before E5.5 [10]. Cdc42 null embryonic stem cells proliferated normally but had cytoskeletal defects [10]. Knockout of Cdc42 in the murine neural crest, using Wnt-1 Cre, allowed survival until E13.5 with severe craniofacial and cardiac abnormalities [11]. These defects were attributed at least in part to aberrant actin dynamics, altered cell migration, and bone morphogenetic protein 2 signaling [12]. However, loss of Cdc42 or Rac1 did not prevent neural crest cells from reaching their targets by E10.5 or growing out of the neural tube in culture [11]. Thus, Cdc42 is implicated in development, but its role in migration in vivo is not clear.
Here we describe a distinct role for Cdc42 in the regulation of pseudopod dynamics and adhesion during melanoblast migration. Cdc42 null melanoblasts extended long blebbing pseudopods, which were not very dynamic. Despite their static nature, Cdc42 null pseudopods showed regular bursts of actin assembly and elevated levels of Rac signal activation but ineffective protrusion. Loss of Cdc42 also caused a severe defect in focal adhesion assembly and dynamics and a de-localization of active myosin. Thus, we propose that in addition to a strong role in cell proliferation, Cdc42 has a coordinating role in melanocytic cell migration, impacting on multiple systems that need to work together for effective cell translocation.
Results
Loss of Cdc42 in the Melanocyte Lineage Leads to Coat Color Defects
Cdc42 was removed from the melanocyte lineage by crossing mice in a C57BL/6 background carrying two floxed alleles of Cdc42 (Cdc42 f/f) [13] with C57BL/6 mice expressing Cre recombinase under the control of the tyrosinase promoter (Tyr::CreB+) [14] (Figure 1A). Cdc42 f/f; Tyr::CreB+ mice were born at the expected Mendelian ratio. They were culled between postpartum day 14 (P14) and P30, as they were slightly shaky and runted, similar to Rac1 f/f Tyr::Cre mice [4].
Figure 1.

Loss of Cdc42 in the Melanoblast Lineage Leads to Coat Color Defects in Adult Mice
(A) Schematic of the gene-targeting strategy for Cdc42 f/f; Tyr::CreB+ mice.
(B) Coat color of control Cdc42 wt/wt (Ctr) mice and Cdc42 f/f; Tyr::CreB+ mice at P22. Yellow arrows indicate dorsal white patches.
(C) Sections from dorsal and ventral Ctr and Cdc42 f/f; Tyr::CreB+ pup skin stained with anti-DCT (melanocyte, red) and hematoxylin counterstain (blue). Scale bars, 100 μm. The far-right panels show hair follicles. N = 20 mice.
See also Figure S1.
All Cdc42 f/f; Tyr::CreB+ mice (N = 20) displayed a white patch running down the ventral midline, covering half to most of the underside (Figure 1B). Small white patches often appeared along the dorsal midline (Figure 1B, yellow arrows). Pigmented areas contained frequent white hairs, and the paws and tails were hypopigmented (Figure 1B). Skin cross-sections from P13 mice revealed a lack of melanocytes in hair follicles of white patches but not pigmented areas in both control (Ctr) and Cdc42 f/f; Tyr::CreB+ mice (Figure 1C, pink stain). Thus, Cdc42 nulls had fewer melanocytes in hair follicles in areas distal to the neural tube but fairly normal melanocyte distribution on the back, similar to Rac1 deletion [4].
In contrast to the dramatic phenotype of Cdc42 f/f; Tyr::CreB+ mice, deletion of RhoA using Tyr::CreB+ had no overt effect on coat color in mice up to P14 (Figure S1A). RhoA f/f; Tyr::CreB+ mice were born at the expected Mendelian ratio and showed no coat color defects, shakiness, or runty phenotype, suggesting that RhoA is dispensable for melanocyte development.
Cdc42 Null Melanoblasts Fail to Fully Populate the Developing Mouse Embryo
To investigate the cellular mechanisms driving pigmentation defects, Cdc42 f/f; Tyr::CreB+ melanoblasts were visualized using the reporter DCT::LacZ [2]. Tyrosinase expression begins around E10.5 [14, 15], when melanoblasts appear around the neural tube in the migration staging area. Control and Cdc42 f/f; Tyr::CreB+; DCT::LacZ (Cdc42 f/f) embryos had similar numbers of melanoblasts at E11.5 (Figures 2A and 2B). At E13.5, the leading wave of melanoblasts in controls and Cdc42 flox heterozygotes Cdc42 f/+; Tyr::CreB+ had reached midway around the embryo and part of the way down the developing limbs, whereas Cdc42 f/f; Tyr::CreB+ embryos consistently showed a deficit of melanoblasts in all areas (boxes, Figures 2C–2F and Figure S1B).
Figure 2.

Loss of Cdc42 in the Melanoblast Lineage Delays Dorsal-to-Ventral Population of the Skin after E11.5
Photos show X-gal-stained Ctr and Cdc42 f/f embryos at various stages as indicated. In all panels, Ctr indicates Cdc42 wt/wt; DCT::LacZ controls, whereas Cdc42 f/f indicates Cdc42 f/f; Tyr::CreB+; DCT::LacZ.
(A) E11.5 embryos; dashed lines denote the quantification area. Scale bar, 1 mm.
(B) Number of melanoblasts in E11.5 embryos.
(C) E13.5 embryos; dashed lines denote melanoblast progress. Scale bar, 1 mm.
(D) Quantification area example.
(E) E13.5 embryos with quantification area. Scale bar, 1 mm.
(F) Melanoblasts from box 1 (most dorsal) to box 6 (ventral) at E13.5.
(G) E15.5. Scale bar, 1 mm. The dashed line indicates the border where the furthest migrating melanoblasts from the neural tube have appeared.
(H) Melanoblasts counted in boxes 1–6 as shown in (D) at E15.5.
(I) Zoom-in of E15.5 embryos. Scale bar, 200 μm.
Mean ± SEM. N ≥ 4 embryos from three litters. Kruskal-Wallis one-way ANOVA test, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; n.s., not significant. See also Figure S2.
By E15.5, melanoblasts in control embryos had spread around the entire embryo and down the developing limbs, but in Cdc42 f/f; Tyr::CreB+ embryos, melanoblasts had only populated partway around the belly and halfway down the developing limb (Figures 2G and 2I; Figure S1C). Cdc42 null cells accumulated around/in hair follicles in places but less efficiently than controls (Figure 2I). Thus, Cdc42 is a major controller of melanoblast proliferation and spread throughout the skin in vivo.
Although loss of Cdc42 caused severe defects in melanoblast population of the skin, there is also striking resiliency within the system, as some of the knockout cells could clearly populate hair follicles and give rise to pigmentation of areas of the mouse skin. Knockout cells also traversed the basement membrane from the dermis to the epidermis normally, with around 20% of cells still in the dermis at E13.5 and >90% of cells in the epidermis by E15.5 (Figure S2).
Cdc42 Controls Melanoblast Cell-Cycle Progression and Cytokinesis
Because Cdc42 mutant embryos showed a paucity of melanoblasts, we investigated possible defects in cell-cycle progression and cytokinesis. Staining for Ki67 revealed normal proportions of Cdc42 f/f cells in cycle (Figure 3A; yellow arrows, +Ki67; white arrow, −Ki67), but BrdU staining at 2 and 24 hr revealed slower cell-cycle progression in Cdc42 f/f (Figures 3B and 3C; yellow arrows, +BrdU; white arrows, −BrdU). We saw no cleaved caspase-3-positive (apoptotic) cells in control or Cdc42 null skins. Thus, loss of Cdc42 delays the cell cycle of melanoblasts.
Figure 3.

Cdc42 Null Melanoblasts and Melanocytes Show Cell-Cycle Progression Defects
(A–C) Representative images of transverse sections of control (Ctr, Cdc42 wt/wt; Tyr::CreB; Z/EG; top) and Cdc42 f/f (Cdc42 f/f; TyrCreB; Z/EG; bottom) E15.5 embryo skin stained with DAPI (blue), DCT (green), and either Ki67 (red) or BrdU (red) at 2 or 24 hr as indicated. Yellow arrows indicate proliferating melanoblasts; white arrows indicate non-proliferating melanoblasts. N ≥ 3 separate embryos. t test with Welch’s correction. Scale bar, 10 μm.
(A) Ki67-positive cells as indicated; n.s., p = 0.38.
(B) 2 hr BrdU-positive cells as indicated; n.s., p = 0.06.
(C) 24 hr BrdU-positive cells as indicated; ∗p = 0.014.
(D) Western blot showing OHT-induced Cdc42 deletion (GAPDH loading control) in EW1 and EW7 Cdc42 f/f; ROSA26::Cre-ERT2; CDKN2−/−cells.
(E) Melanocyte proliferation assay of control (DMSO-treated) and Cdc42 f/f (OHT-treated) cell lines (EW1 and EW7). Error bars show SEM from N = 3 experiments in triplicate.
(F) Representative plot from flow-cytometry analysis of EW7 following mock treatment (DMSO) or deletion (OHT) of Cdc42.
(G) Quantification of cells in cycle. N = 3. Error bars show SEM. ∗∗p = 0.0039 (G1 phase), ∗∗p = 0.0017 (S phase).
See also Figure S3.
We developed immortalized melanocyte cell lines cultured from P1 pups with Cdc42 f/f; Rosa26::Cre-ERT2; CDKN2−/− to allow conditional deletion of Cdc42 with OHT (4-hydroxytamoxifen) in culture [4]. Hereafter, we refer to these lines as EW1, EW2.1, EW2.2, and EW7. These lines behaved similarly, and many experiments were repeated with at least two lines. Addition of OHT induced a loss of Cdc42 protein over 5 days (Figure 3D). Cdc42 null melanocytes had an increased doubling time in vitro (Figure 3E) and accumulated in G1 (Figures 3F and 3G). Thus, cell-cycle progression defects likely contribute to the paucity of melanoblasts in Cdc42 f/f; Tyr::CreB+ embryos.
In addition, time-lapse imaging of live cells in embryo skin explants from Cdc42 f/f; Tyr::CreB+; Z/EG (driving melanoblast-specific expression of GFP) [16] revealed that Cdc42 null melanoblasts have an approximately 3-fold increased division time (Figures S3A and S3B; Movie S1) from cleavage furrow formation (Figure S3A, green boxes) to separation of daughter cells (Figure S3A, red boxes; Figure S3C). Both EW1 and EW7 also spent 2.5 times longer from rounding up to cytokinesis (Figures S3D and S3E; Movie S2). Thus, Cdc42 null melanoblasts struggle to physically separate from each other after division. These problems likely compound the reduction in numbers of melanoblasts in embryos and contribute to the coat color defects of adults.
Loss of Cdc42 Uncouples Actin Dynamics and Pseudopod Extension from Migration
To investigate whether Cdc42 null melanoblasts had migration defects, we imaged melanoblasts migrating in ex vivo skin sections from Cdc42 f/f; Tyr::CreB+; Z/EG. Control melanoblasts in E15.5 embryo skin displayed multiple, highly dynamic pseudopods that extended between surrounding keratinocytes (Figure 4A, yellow arrows). Cdc42 null melanoblasts, in contrast, displayed long wavy non-dynamic pseudopods (Figure 4A, red arrow; Movie S3). Cdc42 null melanoblasts showed an increase of around 40% spread area (Figure 4B). Additionally, Cdc42 null cells frequently had only one or two pseudopods, with 56% appearing bipolar (Figure 4A, orange arrow; Figure 4C). The length-to-width ratio of Cdc42 nulls was increased (Figure 4D), as was pseudopod length (Figure 4E). We were unable to reliably measure persistence, as the density of melanoblasts in wild-type skin was too high. Cdc42 null cells showed very long lived pseudopods, with 10% lasting the entirety of the 4-hr time-lapse movie, compared with 5–80 min for controls (Figure 4F). Normally, elongation of cells is a hallmark of rapid migration, but Cdc42 null cells displayed a 2-fold decrease in speed from around 0.4 to 0.2 μm/min (Figure 4G). Thus, Cdc42 null cells are elongated, but move only slowly in epidermis.
Figure 4.

Cdc42 Null Melanoblasts Have Migration Defects and Altered Pseudopod Dynamics
(A) Still images of Ctr (Cdc42 wt/wt; Tyr::CreB+; Z/EG) and Cdc42 f/f (Cdc42 f/f; Tyr::CreB+; Z/EG) skin explants (see Movie S3). Yellow arrows indicate pseudopods; the red arrow indicates a blebby pseudopod; and the orange arrow denotes the “bipolar” phenotype. Scale bars, 10 μm.
(B–F) N = 125 cells per genotype, from 301 control pseudopods and 239 Cdc42 f/f pseudopods.
(B) Cell area.
(C) Number of pseudopods.
(D) Length-to-width ratio.
(E) Pseudopod length in microns.
(F) Pseudopod lifetime in minutes.
(G) Migration speed. Dashed vertical lines indicate average speed. N = 115 cells per genotype.
(H) Representative images of melanoblast F-actin in control Lifeact-GFP lox-stop-lox; Tyr::CreB+ (Ctr) skin explants and Lifeact-GFP lox-stop-lox; Cdc42 f/f; Tyr::CreB+ (Cdc42 f/f) skin explants (see Movie S4). Scale bar, 10 μm.
(I) Number of minutes between actin bursts in Ctr (9.0 ± 0.8 min SEM) and Cdc42 f/f (11.6 ± 0.7 min SEM) pseudopods. N = 100 pseudopods per genotype.
(J) Action of pseudopods within three frames (3 min) following an actin burst. Extension, “protrude”; retraction, “retract”; and static, “remain.” Percentage of pseudopods that protruded, retracted, or remained following an actin burst (within three frames) was a calculated per skin from at least eight bursts from four skins.
(K) Scatterplots showing pseudopod lifetime versus actin bursts. The line of best fit for Ctr has slope 7.2 ± 0.7, R2 = 0.55; for Cdc42 f/f, slope 10.8 ± 0.6, R2 = 0.77.
N = 5–6 embryos from four different litters, except (J), where N = 20 cells, over three experiments. Boxplots show mean, minimum, and maximum values. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, t test.
To determine whether a loss of actin dynamics might be responsible for the slow motility, we crossed Cdc42; Tyr::CreB+ with the Lifeact-GFP transgene [17] to observe filamentous actin dynamics. Bursts of actin occurred at the tips of both control and knockout pseudopods, driving them forward (Figure 4H; Movie S4). There was a small but significant increase in the time between actin bursts in knockout pseudopods from 9.0 ± 0.8 min SEM (Ctr) to 11.6 ± 0.7 min SEM (Cdc42 f/f) (Figure 4I), and cells appeared not to translocate as often or as rapidly following a burst (Movie S4). Actin bursts were usually accompanied by protrusion of the pseudopod, but this was impaired in Cdc42 nulls (Figure 4J). There was a linear correlation between the lifetime of a pseudopod and the number of actin bursts (Figure 4K). Thus, the frequency of actin bursting was not greatly affected by the loss of Cdc42, but the coupling with protrusion was lost.
Cdc42 Knockout Melanocytes Fail to Coordinate Actin Dynamics, Myosin Localization, and Migration
All cultured melanocyte lines were confirmed to be over 90% pure by staining with the melanocyte lineage factor MITF (Figure S4A shows EW7 as an example). Control cells formed large lamellipodia on fibronectin surfaces, with a length-to-width ratio close to 1, and moved at an average speed of 0.4 μm/min. Following deletion of Cdc42 (OHT), cells were almost stationary, with an elongated spindle shape and an average speed of 0.014 μm/min (Figures 5A–5C). As in skin explants, these thin, long protrusions were very stable, with the majority lasting over 600 min (Figure 5D). The increased length-to-width ratio was rescued through transient expression of Cdc42-YFP in knockout cells (Figures S4B and S4C). Thus, cultured melanocytes behaved similar in vitro to live skin explants and to Cdc42-depleted fibroblast cell lines [13].
Figure 5.

Primary Melanocytes Require Cdc42 for Efficient Pseudopod Extension and Ruffling but Not for Rac1 Activation
All panels show EW7 Cdc42 f/f; ROSA26::Cre-ERT2; CDKN2−/− melanocytes treated with DMSO (control) or OHT (Cdc42−/−).
(A) Melanocytes on fibronectin. Scale bar, 30 μm.
(B) Length-to-width ratio (N = 59 cells per condition).
(C) Migration speed (N = 90 cells per condition).
(D) Pseudopod lifetime (N = 57 pseudopods per condition).
(E) Nuclei (DAPI, blue), GM130 (Golgi, green), and F-actin (Alexa Fluor 568-phalloidin, red) of migrating melanocytes. Scale bars, 10 μm. Arrows show example quantification segments.
(F and G) Percentage of cells with Golgi polarized (F) and percentage of Golgi spread (G) over one, two, or three segments. N = 353–386 cells; 15 regions per condition over three experiments.
(H and I) DMSO-treated (H) and OHT-treated (I) EW7 F-actin (phalloidin, green) and Arp2/3 complex (p34) or WAVE2 (red). Insets show leading-edge protrusions. Scale bar, 15 μm.
Graphs show mean ± SEM. ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, t test. See also Figure S4.
Cdc42 has a well-described role in mediating the polarization of the Golgi complex in front of the nucleus in a moving cell [18], so we tested whether Cdc42 null melanocytes retained this ability. When cells were allowed to migrate out of a crowded space (see Supplemental Experimental Procedures), around 80% of the control cells reoriented their Golgi to face the direction of migration. However, Golgi orientation was random in Cdc42 null cells (Figures 5E and 5F). Additionally, the Golgi appeared more diffuse, and frequently spread over multiple segments of a 120° divided mask placed over the cell (Figure 5G). Thus, Cdc42 regulates Golgi polarity during motility of melanocytes, as previously described in other cell types [13, 18].
In addition to their polarity defects, Cdc42 null melanoblasts resembled blebbistatin-treated cells in embryo skin explants [4] with blebby static pseudopods. Cdc42 is linked to myosin activation via the myotonic dystrophy kinase-related MRCK kinases (reviewed in [19]), suggesting that pseudopod retraction defects may be due to reduced activation of myosin light chain (MLC). However, levels of MLC phosphorylation at T18/S19 were unaltered in Cdc42 null melanocytes (Figures S4D and S4E). Despite this, localization of p-MLC was abnormally concentrated toward the cell body of Cdc42 null cells, rather than peripheral in the lamellar region, as in the controls (Figures S4F and S4G). We also performed a blebbistatin wash-out experiment to test the sensitivity of Cdc42 null cells to inhibition of myosin-II (Figures S4H and S4I). Control cells (Cdc42 wt [wild-type]) became elongated and resembled Cdc42 null cells before blebbistatin treatment. Cdc42 nulls, however, showed very little change in shape, indicating that their myosin-II was already not engaged. Thus, although overall levels of p-MLC were not dramatically altered in Cdc42 nulls, p-MLC was mislocalized and Cdc42 null cells resembled blebbistatin-treated cells and showed very little response to blebbistatin treatment.
We next investigated whether loss of Cdc42 affected the major actin organizers Scar/WAVE and Arp2/3 complex as well as the Cdc42 target formin FMNL2 [20]. Both Arp2/3 and WAVE2 localized in a broad band in lamellipodia of control cells (Figure 5H), but in Cdc42 null cells they formed small patches near the cell edge (Figure 5I), in protrusions (Figure 5I, insets). Because FMNL2 is a Cdc42 target regulating lamellipodial actin [20], we tested whether constitutively active FMNL2 might rescue the loss of Cdc42. Endogenous FMNL2 localized prominently in lamellipodia and filopodia of both normal and Cdc42 null melanocytes (Figure 6A). Cells expressing constitutively active FMNL2 showed increased spreading but also a more fenestrated appearance (Figures 6B–6E). In particular, mutant FMNL2-GFP constructs FMNL2-ΔDAD-GFP and FMNL2-A272E-GFP (see Supplemental Experimental Procedures), which both lack the autoinhibitory regulation provided by the diaphanous autoinhibitory domain (DAD) region, rescued the length-to-width ratio (Figure 6C). We scored cells in shape categories (see the legend of Figure 6). Expression of active FMNL2-GFP increased the proportion of cells with a wider lamellipodial shape and decreased the proportion classified as elongated (Figure 6D). Wild-type FMNL2-GFP [20] and both active constructs gave a partial rescue of roundness (Figure 6E). Thus, a major part of the cell shape change that we observe upon loss of Cdc42 appears to be due to the inability of cells to activate FMNL2.
Figure 6.

Cdc42 Null Cells Have Morphological Defects Involving the Formin FMNL2 and Focal Adhesions
All panels show EW7 Cdc42 f/f; ROSA26::Cre-ERT2; CDKN2−/− melanocytes treated with DMSO (control) or OHT (to delete Cdc42).
(A) Control and OHT-treated EW7 cells stained for endogenous FMNL2 and F-actin. White boxes denote zoom-in.
(B) EW7 cells expressing FMNL2 constructs as indicated.
(C) Length-to-width ratio.
(D and E) Proportion of cells in different shape categories (D) and roundness (E) was calculated; N = 31–37 cells per condition over three experiments; t tests compare data to the EW7 OHT GFP condition.
(F) Control and OHT-treated EW7 cells stained for vinculin and F-actin.
(G–I) Adhesion number per cell (G), adhesion area (H), and number of adhesions normalized to the cell area (I) were calculated using thresholded vinculin staining. N = 29–33 cells per condition over three experiments.
Graphs (C, E, G, H, and I) show mean ± SEM. ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, t test with Welch’s correction. Scale bars, 10 μm. See also Figures S5 and S6.
We next used fluorescence lifetime imaging microscopy (FLIM) imaging of a Rac1FLARE dual-chain (dc) biosensor to determine whether loss of Cdc42 decreased Rac1 activation signals. Lifetime heatmaps of control dTurquoise (donor alone) or Rac1FLARE.dc showed a decreased lifetime, demonstrating Förster resonance energy transfer (FRET) (Figure S4J). The percentage FRET efficiency in Cdc42 knockout cells was to our surprise higher, indicating an increase in Rac1 signaling activity (Figure S4J). We also performed a biochemical pull-down assay (Figures S4K and S4L), which revealed no significant difference in Rac1 signaling activity between control and Cdc42 null cells. The FRET assay is more sensitive, so this likely means that there is a small or localized increase in Rac1 activity detectable only by FRET. Thus, the pathways that regulate Rac1 activation were active in Cdc42 null cells, despite cells being unable to use their actin dynamics productively for migration.
Taken together, Cdc42 null cells still showed active Rac signaling but a failure to organize Arp2/3, Scar/WAVE, FMNL2, and active myosin-II in their protrusions; thus, the various motility systems seemed intact, but uncoupled from movement.
Cdc42 Coordinates Adhesion Dynamics for Effective Migration
Adhesion controls the shape and migratory properties of cells, as well as their growth state. Global RNA sequencing was performed on Cdc42 f/f; ROSA26:Cre-ERT2; CDKN2−/− melanocyte lines (EW2.1 and EW2.2, isolated from littermate pups) to probe pathways affected by Cdc42 loss (Figure S5A; Data S1). Biological replicates were consistent by principal component analysis (PCA) of the data (Figure S5B). DAVID (https://david.ncifcrf.gov/) gene ontology analysis of significantly (padjusted < 0.05) downregulated (290) or upregulated (343) genes identified lysosomal pathways as significantly upregulated, and hierarchical clustering identified top consistent hits (Figures S5C and S5D). Additionally, “pathways in cancer,” “focal adhesion,” and “regulation of the actin cytoskeleton” (Figure S5E) were significantly downregulated. Hierarchical clustering analysis of KEGG (Kyoto Encyclopedia of Genes and Genomes) “focal adhesion” genes showed consistency between EW2.1 and EW2.2 (Figure S5F). α4 integrin showed the largest fold change (−2.93 ± 0.5), which we confirmed at the protein level (Figures S5G and S5H). Additionally, β3 integrin was decreased at the protein level (Figures S5I and S5J), despite an increase at the RNA level (+2.01 ± 0.7, padjusted = 0.2; Data S1). The expression levels of classical Rho-GTPases remained relatively unchanged in Cdc42 knockout cells (Figure S5K).
As suggested by global RNA-sequencing analysis and cell-shape abnormalities, Cdc42 null cells showed adhesion defects. Control melanocytes displayed a belt of multiple large adhesions underneath their lamellipodia that were rich in vinculin (Figure 6F), phospho-paxillin (Figure S6A), and β1 integrin (Figure S6E), whereas knockout melanocytes displayed only a few small adhesions with the same components (Figure 6F; Figures S6A and S6E). Overall, knockout melanocytes had fewer adhesions per cell, a smaller average adhesion size, and fewer adhesions per cell area (Figures 6G–6I; Figures S6B–S6D).
Because FMNL2 partially rescued the cell-shape defects of Cdc42 null cells, we asked whether it affected focal adhesions. Indeed, active FMNL2 expression increased the number of adhesions per cell (Figure S6F) and area of adhesions (Figure S6G) but not the number of adhesions per cell area, as FMNL2 expression increased the spread area quite significantly (Figure S6H). These changes were modest and might have complex reasons, but expression of FMNL2 appeared to give a partial rescue.
Live imaging of GFP-paxillin or mApple-vinculin revealed that control melanocytes dynamically extended lamellipodia supported by nascent adhesions, whereas adhesions of Cdc42 nulls were smaller and less dynamic. Adhesions in knockout melanocytes showed a slower assembly rate with GFP-paxillin and a slower disassembly rate with both GFP-paxillin and mApple-vinculin, indicating reduced adhesion dynamics in Cdc42 knockout cells (Figures 7A–7E; Figure S7A). Re-expression of α4 integrin in Cdc42 null cells gave a partial rescue (Figure 7F). The length-to-width ratio and number of adhesions per cell did not change significantly, but the roundness and total cell area increased modestly (Figures 7G–7J). Thus, α4 integrin levels account for part of the phenotype of Cdc42 null melanocytes.
Figure 7.

Cdc42 Knockout Melanocytes Form Less Dynamic Adhesions and Show Spreading Defects
All panels show EW7 Cdc42 f/f; ROSA::Cre-ERT2; CDKN2−/− melanocytes treated with DMSO (control) or OHT (to delete Cdc42).
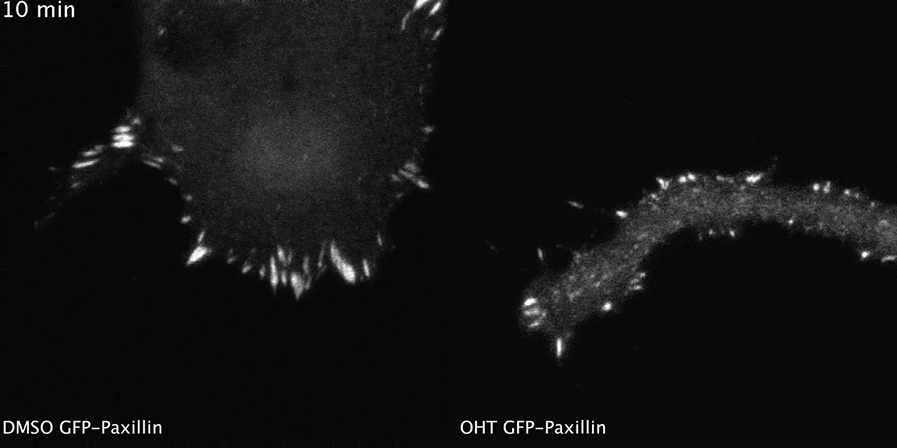
(A) Live confocal imaging sequence of control (top) versus OHT-treated cells (bottom) expressing GFP-paxillin, imaged every 4 min. The merge shows each frame in a unique color. Scale bar, 5 μm.
(B–E) Quantification of the rate of assembly and disassembly of adhesions marked with GFP-paxillin (B and C) or mApple-vinculin (D and E) over 30 min; N = 15 cells per condition over three experiments.
(F) EW7 cells treated with OHT transfected with α4 integrin and GFP and stained with anti-α4 integrin or anti-vinculin. Yellow boxes show zoom-in. Yellow arrows indicate α4 integrin-positive adhesions. Scale bars, 10 μm.
(G and H) Length-to-width ratio (G) and roundness (H) of EW7 +OHT cells transfected with pcDNA3.1 control vector or α4 integrin. N = 45 cells per condition over three experiments.
(I and J) Number (I) and size (J) of adhesion complexes in EW7 +OHT cells as in (G) and (H); N = 24–27 cells per condition over three experiments.
(K–M) Area of melanocyte spreading at the indicated times on fibronectin (K), laminin-111 (L), and concanavalin A (M); N = 89–156 cells over three experiments.
Graphs (B–E and G–J) show mean ± SEM. Boxplots (K–M) show mean with minimum and maximum values. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001, t test with Welch’s correction. See also Figure S7.
Cdc42 nulls also showed spreading defects on fibronectin or laminin but not concanavalin A, a lectin mediating integrin-independent adhesion [21] (Figures 7K–7M) and formation of fewer adhesions (Figures S7B, S7C, and S7E). Because filopodia-like protrusions during cell spreading are linked with nascent β1 integrin adhesions [22], we measured filopodia-like structures (FLSs) in cells spreading on fibronectin. Knockout melanocytes formed fewer, shorter FLSs (Figures S7D, S7F, and S7G) and showed a thicker band of cortical actin than controls (Figure S7D, white arrows), possibly reflecting less dynamic actin. Thus, Cdc42 null melanoblasts are defective in adhesion-dependent spreading, with fewer dynamic adhesions and misregulation of both localization and expression of focal adhesion components.
Discussion
Loss of Cdc42 in the melanocyte lineage gave a phenotype of similar severity to loss of Rac1, but with multiple unique features revealing Cdc42 as a complex organizer of motility systems. Both GTPases appear important for melanoblast proliferation and migration during embryogenesis, but Cdc42 null cells were exaggeratedly elongated in vivo, rather than rounded, like Rac1 nulls [4]. Cell shape has been proposed as a potential predictor of metastasis in melanoma [23, 24, 25, 26], but in the case of control by Rho-GTPases in the melanocyte lineage, we found no obvious correlation between shape and migration properties. Similar to Rac1, Cdc42 also plays a key role in promoting G1-to-S phase transition in melanocytic cells in vivo (Figure 3). Rac1 and Cdc42 have a well-established role in cell-cycle progression in cultured cells [27]. Cdc42 has a more direct role in cytokinesis than Rac, as it localizes on the spindle and kinetochores during mitosis [27]. Our observations that Cdc42 impacts on localization of phosphorylated myosin light chain suggest possible implications for cytokinesis. Curiously, proliferation and migration defects are generally coupled in melanoblasts in vivo [4, 5, 28], perhaps due to a metabolizable signal in the skin or coupling of the mitotic spindle to the cell cortex by motility proteins [29].
At the cellular level, loss of Cdc42 had a pleiotropic effect, causing defects in the organization and coordination of actin dynamics, contractile activity, and adhesion. Despite evidence of a hierarchy whereby Rac1 is activated downstream of Cdc42 in some systems, melanocytic Cdc42 null cells showed elevated Rac1 activation but only small lamellipodia. Their pseudopod extensions were not dynamic in vivo despite actin bursts visualized with Lifeact-GFP, and they failed to assemble broad lamellipodia driven by the formin FMNL2 [20], Scar/WAVE, and Arp2/3 in vitro. Myosin-II was still phosphorylated at normal levels, indicating signaling to activation, but it was not localized properly and cells resembled the blebbistatin-treated state. Previous studies connected Cdc42 and actin polymerization with phosphoinositides, p21-activated kinase (PAK) kinases, and N-WASP [30]. However, neither N-WASP nor PAK kinases affected pseudopod dynamics or migration of melanoblasts in skin explants [4].
Cdc42 null melanocytes showed adhesion defects, unlike Cdc42 null mouse embryonic fibroblasts (MEFs), which spread normally and had normal (β1 or β3, or α1, 2, 5, 6, or v) integrin subunit expression levels [13]. In contrast, Cdc42 null melanocytes showed reduced adhesion dynamics only partially rescued by re-expression of α4 integrin or FMNL2 active mutants. Despite adhesion and spreading defects in vitro, melanoblasts null for Cdc42 could still cross and interact with the epidermal basement membrane in vivo (Figure S2).
In addition to adhesion changes, several genes in a KEGG lysosomal pathway were upregulated at the RNA level in Cdc42 null melanocytes, including several subunits of ATP6vo, a vacuolar ATPase (V-ATPase) required for proton pumping in acidic organelles such as the lysosome (Figure S5). V-ATPase is linked to anoikis resistance of cancer cells [31] due to its ability to degrade internalized integrins. Thus, Cdc42 null cells may have increased ability to degrade internalized integrins, which would also contribute to diminished levels of α4 and β3 integrins.
Mouse melanocytes express Cdc42, Rac1, RhoA, and RhoC at high levels and RhoB, RhoG, RhoJ, and RhoQ at lower levels (Figure S5K; Data S1), which were not affected by deletion of Cdc42. RhoG has some overlapping functions with Rac1 and Cdc42 [32], but clearly it cannot compensate fully for the loss of Rac1 or Cdc42 in melanoblasts.
In summary, Cdc42 is an important regulator of cell migration and proliferation of melanocytic cells; its loss caused failure of multiple aspects of migration, which were not rescued by re-activation or expression of any single component.
Experimental Procedures
Reagents, Mice, and Genotyping
All animal experiments were performed according to UK Home Office regulations. Genotyping was performed by Transnetyx. All other mouse strains used were previously described, and all antibody and chemical reagents are detailed in Supplemental Experimental Procedures.
Melanocyte Isolation, Culture, and Transfection
Primary melanocytes were isolated and purified from the skin of 1-day-old pups and imaged according to methods previously described [4] and in Supplemental Experimental Procedures.
Author Contributions
E.F.W., N.R.P., and B.T. designed, performed, and analyzed experiments; H.J.S., M.R.S., K.S., E.G., and S.W.G.T. performed experiments; C.H.B. and L.L. provided reagents and advice; A.H. and W.C. performed RNA sequencing and analysis; F.K. constructed the FMNL2 mutants; D.J.M. and K.M.H. provided the Rac1FLARE.dc construct and advice; R.H.I. contributed to the experimental design and direction; and L.M.M. supervised the study and wrote the manuscript with E.F.W. and N.R.P.
Acknowledgments
We acknowledge funding from Cancer Research UK (to L.M.M. [A17196], R.H.I. [A19257], and S.W.G.T.) and NIH grants P01-GM103723 and P41-EB002025 (to K.M.H.). N.R.P. is supported by a Pancreatic Cancer Research Fund grant (to L.M.M.). We thank Prof. Klemens Rottner for advice and for sharing unpublished FMNL2 constructs, and acknowledge funding to Prof. Rottner by the Deutsche Forschungsgemeinschaft (grant RO2414/3-2). We thank Dr. Luke Tweedy for advice on image analysis and for writing a script to plot the phospho-MLC quantification data. We thank Dr. Matt Neilson for advice and help with data analysis methods and Dr. Ang Li for initial help with experiments. We thank Prof. Ian Jackson and Dr. Richard Mort for the DCT::LacZ mice and for helpful advice. We thank Margaret O’Prey, Dr. Kirsty Martin, and Tom Gilby for help with imaging and fluorescence-activated cell sorting training. We thank Drs. Mathieu Unbekandt and Mike Olson for providing reagents and protocols. We thank Dr. Jamie Whitelaw for help with the quantification of the cell-spreading experiments.
Published: February 23, 2017
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, seven figures, six movies, and one dataset and can be found with this article online at http://dx.doi.org/10.1016/j.cub.2017.01.033.
Supplemental Information
Movie showing cytokinesis of control (Ctl) and Cdc42 null (Cdc42 f/f) melanoblasts in skin explants from rounding up through to the separation of daughter cells. Images were taken every 5 min for 90 min; the movie plays at 8 frames/s.
{kind=link}
Time-lapse imaging showing melanocyte cytokinesis of control (Ctl) and Cdc42 null (Cdc42 f/f) from rounding up through to the separation of daughter cells. Images were taken every 15 min for 195 min; the movie plays at 2 frames/s.
{kind=link}
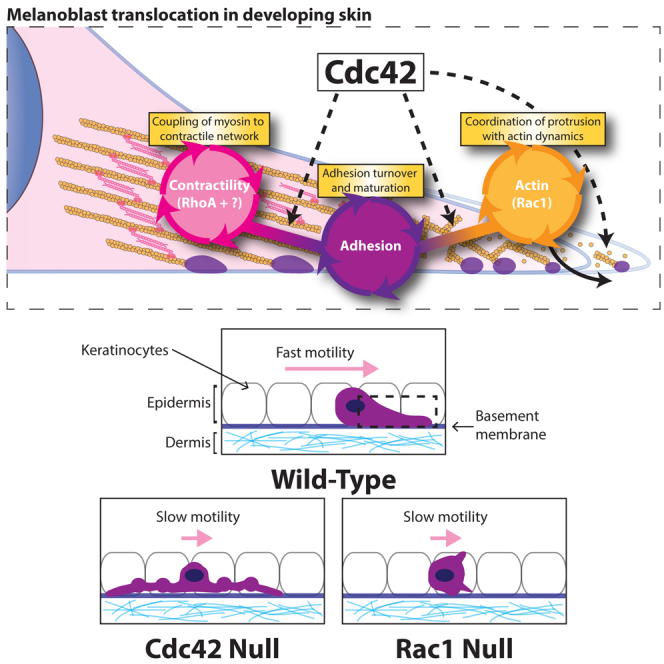
Movie of skin explant from Z/EG+/o-expressing embryos at E15.5. Confocal section of control and Cdc42 null (Cdc42 f/f) melanoblasts moving through the epidermal layer of the skin. Images were taken every 5 min for 240 min; the movie plays at 15 frames/s.
{kind=link}
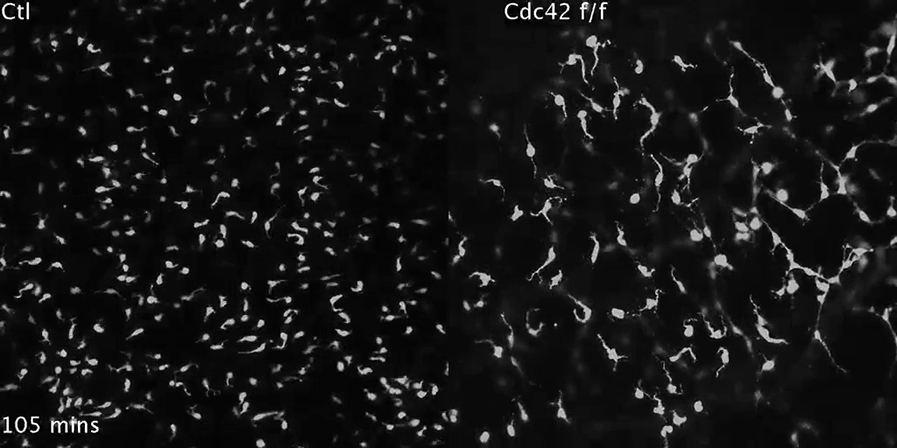
Movie of Lifeact-GFP-expressing melanoblasts moving through the embryo skin epidermis. Orange areas show bursts of actin. Images were taken every 1 min for 31 min; the movie plays at 8 frames/s.
{kind=link}
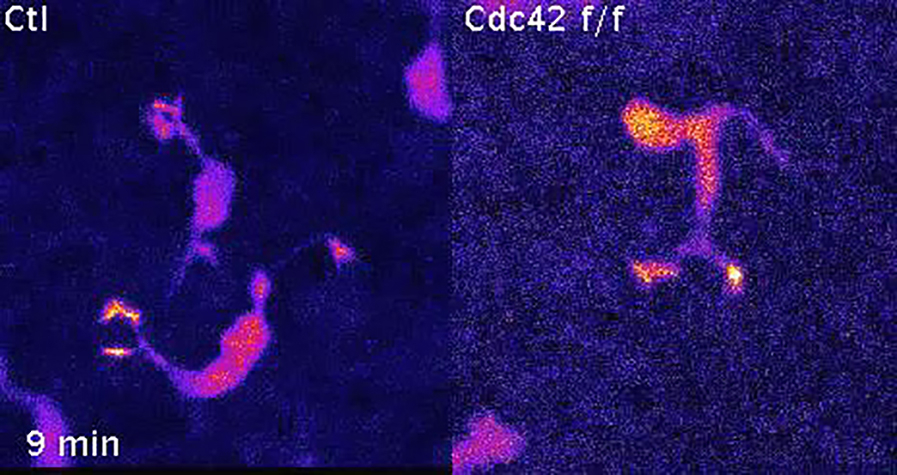
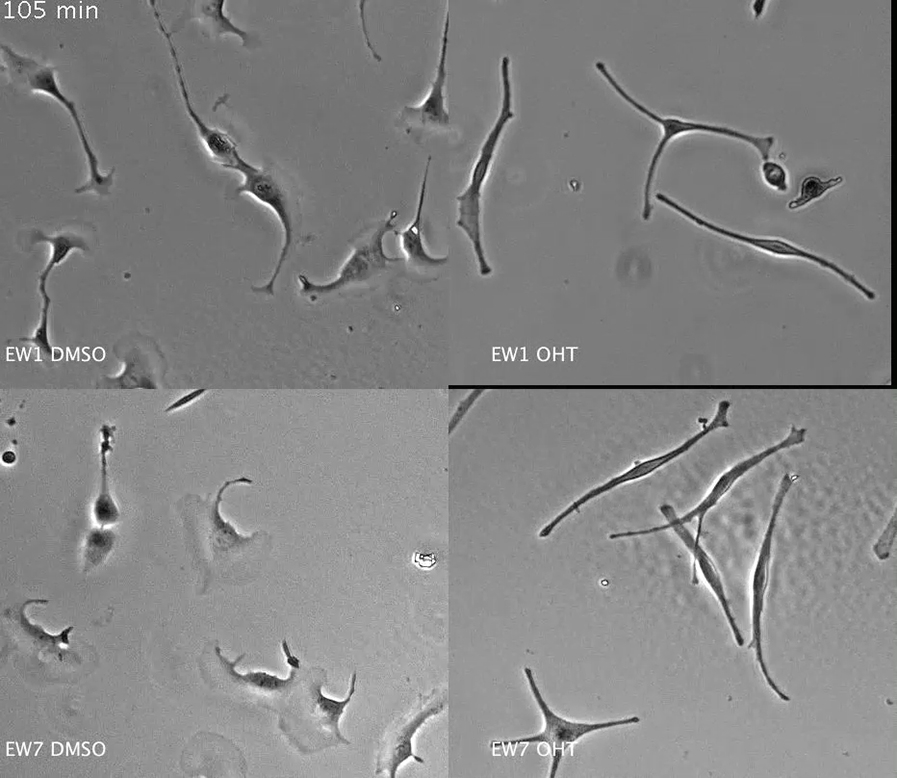
Time-lapse movies of immortalized melanocyte lines EW1 and EW7 migrating on fibronectin. Images were taken every 15 min; the movie plays at 8 frames/s.
{kind=link}
Confocal time-lapse imaging of DMSO- and OHT-treated EW7 melanocytes expressing GFP-paxillin. Cells were imaged every 2 min for 30 min; the movie plays at 5 frames/s.
{kind=link}
References
- 1.Luciani F., Champeval D., Herbette A., Denat L., Aylaj B., Martinozzi S., Ballotti R., Kemler R., Goding C.R., De Vuyst F. Biological and mathematical modeling of melanocyte development. Development. 2011;138:3943–3954. doi: 10.1242/dev.067447. [DOI] [PubMed] [Google Scholar]
- 2.Jordan S.A., Jackson I.J. MGF (KIT ligand) is a chemokinetic factor for melanoblast migration into hair follicles. Dev. Biol. 2000;225:424–436. doi: 10.1006/dbio.2000.9856. [DOI] [PubMed] [Google Scholar]
- 3.Thomas A.J., Erickson C.A. The making of a melanocyte: the specification of melanoblasts from the neural crest. Pigment Cell Melanoma Res. 2008;21:598–610. doi: 10.1111/j.1755-148X.2008.00506.x. [DOI] [PubMed] [Google Scholar]
- 4.Li A., Ma Y., Yu X., Mort R.L., Lindsay C.R., Stevenson D., Strathdee D., Insall R.H., Chernoff J., Snapper S.B. Rac1 drives melanoblast organization during mouse development by orchestrating pseudopod-driven motility and cell-cycle progression. Dev. Cell. 2011;21:722–734. doi: 10.1016/j.devcel.2011.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ma Y., Li A., Faller W.J., Libertini S., Fiorito F., Gillespie D.A., Sansom O.J., Yamashiro S., Machesky L.M. Fascin 1 is transiently expressed in mouse melanoblasts during development and promotes migration and proliferation. Development. 2013;140:2203–2211. doi: 10.1242/dev.089789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mort R.L., Ross R.J., Hainey K.J., Harrison O.J., Keighren M.A., Landini G., Baker R.E., Painter K.J., Jackson I.J., Yates C.A. Reconciling diverse mammalian pigmentation patterns with a fundamental mathematical model. Nat. Commun. 2016;7:10288. doi: 10.1038/ncomms10288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mort R.L., Jackson I.J., Patton E.E. The melanocyte lineage in development and disease. Development. 2015;142:620–632. doi: 10.1242/dev.106567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Genova J.L., Jong S., Camp J.T., Fehon R.G. Functional analysis of Cdc42 in actin filament assembly, epithelial morphogenesis, and cell signaling during Drosophila development. Dev. Biol. 2000;221:181–194. doi: 10.1006/dbio.2000.9671. [DOI] [PubMed] [Google Scholar]
- 9.Gotta M., Abraham M.C., Ahringer J. CDC-42 controls early cell polarity and spindle orientation in C. elegans. Curr. Biol. 2001;11:482–488. doi: 10.1016/s0960-9822(01)00142-7. [DOI] [PubMed] [Google Scholar]
- 10.Chen F., Ma L., Parrini M.C., Mao X., Lopez M., Wu C., Marks P.W., Davidson L., Kwiatkowski D.J., Kirchhausen T. Cdc42 is required for PIP(2)-induced actin polymerization and early development but not for cell viability. Curr. Biol. 2000;10:758–765. doi: 10.1016/s0960-9822(00)00571-6. [DOI] [PubMed] [Google Scholar]
- 11.Fuchs S., Herzog D., Sumara G., Büchmann-Møller S., Civenni G., Wu X., Chrostek-Grashoff A., Suter U., Ricci R., Relvas J.B. Stage-specific control of neural crest stem cell proliferation by the small rho GTPases Cdc42 and Rac1. Cell Stem Cell. 2009;4:236–247. doi: 10.1016/j.stem.2009.01.017. [DOI] [PubMed] [Google Scholar]
- 12.Liu Y., Jin Y., Li J., Seto E., Kuo E., Yu W., Schwartz R.J., Blazo M., Zhang S.L., Peng X. Inactivation of Cdc42 in neural crest cells causes craniofacial and cardiovascular morphogenesis defects. Dev. Biol. 2013;383:239–252. doi: 10.1016/j.ydbio.2013.09.013. [DOI] [PubMed] [Google Scholar]
- 13.Czuchra A., Wu X., Meyer H., van Hengel J., Schroeder T., Geffers R., Rottner K., Brakebusch C. Cdc42 is not essential for filopodium formation, directed migration, cell polarization, and mitosis in fibroblastoid cells. Mol. Biol. Cell. 2005;16:4473–4484. doi: 10.1091/mbc.E05-01-0061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Delmas V., Martinozzi S., Bourgeois Y., Holzenberger M., Larue L. Cre-mediated recombination in the skin melanocyte lineage. Genesis. 2003;36:73–80. doi: 10.1002/gene.10197. [DOI] [PubMed] [Google Scholar]
- 15.Delmas V., Beermann F., Martinozzi S., Carreira S., Ackermann J., Kumasaka M., Denat L., Goodall J., Luciani F., Viros A. Beta-catenin induces immortalization of melanocytes by suppressing p16INK4a expression and cooperates with N-Ras in melanoma development. Genes Dev. 2007;21:2923–2935. doi: 10.1101/gad.450107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Novak A., Guo C., Yang W., Nagy A., Lobe C.G. Z/EG, a double reporter mouse line that expresses enhanced green fluorescent protein upon Cre-mediated excision. Genesis. 2000;28:147–155. [PubMed] [Google Scholar]
- 17.Schachtner H., Li A., Stevenson D., Calaminus S.D., Thomas S.G., Watson S.P., Sixt M., Wedlich-Soldner R., Strathdee D., Machesky L.M. Tissue inducible Lifeact expression allows visualization of actin dynamics in vivo and ex vivo. Eur. J. Cell Biol. 2012;91:923–929. doi: 10.1016/j.ejcb.2012.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Etienne-Manneville S. Cdc42—the centre of polarity. J. Cell Sci. 2004;117:1291–1300. doi: 10.1242/jcs.01115. [DOI] [PubMed] [Google Scholar]
- 19.Zhao Z., Manser E. Myotonic dystrophy kinase-related Cdc42-binding kinases (MRCK), the ROCK-like effectors of Cdc42 and Rac1. Small GTPases. 2015;6:81–88. doi: 10.1080/21541248.2014.1000699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Block J., Breitsprecher D., Kühn S., Winterhoff M., Kage F., Geffers R., Duwe P., Rohn J.L., Baum B., Brakebusch C. FMNL2 drives actin-based protrusion and migration downstream of Cdc42. Curr. Biol. 2012;22:1005–1012. doi: 10.1016/j.cub.2012.03.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Burbelo P.D., Miyamoto S., Utani A., Brill S., Yamada K.M., Hall A., Yamada Y. p190-B, a new member of the Rho GAP family, and Rho are induced to cluster after integrin cross-linking. J. Biol. Chem. 1995;270:30919–30926. doi: 10.1074/jbc.270.52.30919. [DOI] [PubMed] [Google Scholar]
- 22.Shibue T., Brooks M.W., Inan M.F., Reinhardt F., Weinberg R.A. The outgrowth of micrometastases is enabled by the formation of filopodium-like protrusions. Cancer Discov. 2012;2:706–721. doi: 10.1158/2159-8290.CD-11-0239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bai S.W., Herrera-Abreu M.T., Rohn J.L., Racine V., Tajadura V., Suryavanshi N., Bechtel S., Wiemann S., Baum B., Ridley A.J. Identification and characterization of a set of conserved and new regulators of cytoskeletal organization, cell morphology and migration. BMC Biol. 2011;9:54. doi: 10.1186/1741-7007-9-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sanz-Moreno V., Marshall C.J. The plasticity of cytoskeletal dynamics underlying neoplastic cell migration. Curr. Opin. Cell Biol. 2010;22:690–696. doi: 10.1016/j.ceb.2010.08.020. [DOI] [PubMed] [Google Scholar]
- 25.Tyrrell B.J., Neilson M., Insall R.H., Machesky L.M. Predicting cell shapes in melanomas. Pigment Cell Melanoma Res. 2014;27:5–6. doi: 10.1111/pcmr.12176. [DOI] [PubMed] [Google Scholar]
- 26.Yin Z., Sadok A., Sailem H., McCarthy A., Xia X., Li F., Garcia M.A., Evans L., Barr A.R., Perrimon N. A screen for morphological complexity identifies regulators of switch-like transitions between discrete cell shapes. Nat. Cell Biol. 2013;15:860–871. doi: 10.1038/ncb2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chircop M. Rho GTPases as regulators of mitosis and cytokinesis in mammalian cells. Small GTPases. 2014;5:e29770. doi: 10.4161/sgtp.29770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lindsay C.R., Lawn S., Campbell A.D., Faller W.J., Rambow F., Mort R.L., Timpson P., Li A., Cammareri P., Ridgway R.A. P-Rex1 is required for efficient melanoblast migration and melanoma metastasis. Nat. Commun. 2011;2:555. doi: 10.1038/ncomms1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rosenblatt J., Cramer L.P., Baum B., McGee K.M. Myosin II-dependent cortical movement is required for centrosome separation and positioning during mitotic spindle assembly. Cell. 2004;117:361–372. doi: 10.1016/s0092-8674(04)00341-1. [DOI] [PubMed] [Google Scholar]
- 30.Ridley A.J. Rho GTPase signalling in cell migration. Curr. Opin. Cell Biol. 2015;36:103–112. doi: 10.1016/j.ceb.2015.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schempp C.M., von Schwarzenberg K., Schreiner L., Kubisch R., Müller R., Wagner E., Vollmar A.M. V-ATPase inhibition regulates anoikis resistance and metastasis of cancer cells. Mol. Cancer Ther. 2014;13:926–937. doi: 10.1158/1535-7163.MCT-13-0484. [DOI] [PubMed] [Google Scholar]
- 32.Wennerberg K., Ellerbroek S.M., Liu R.Y., Karnoub A.E., Burridge K., Der C.J. RhoG signals in parallel with Rac1 and Cdc42. J. Biol. Chem. 2002;277:47810–47817. doi: 10.1074/jbc.M203816200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie showing cytokinesis of control (Ctl) and Cdc42 null (Cdc42 f/f) melanoblasts in skin explants from rounding up through to the separation of daughter cells. Images were taken every 5 min for 90 min; the movie plays at 8 frames/s.
Time-lapse imaging showing melanocyte cytokinesis of control (Ctl) and Cdc42 null (Cdc42 f/f) from rounding up through to the separation of daughter cells. Images were taken every 15 min for 195 min; the movie plays at 2 frames/s.
Movie of skin explant from Z/EG+/o-expressing embryos at E15.5. Confocal section of control and Cdc42 null (Cdc42 f/f) melanoblasts moving through the epidermal layer of the skin. Images were taken every 5 min for 240 min; the movie plays at 15 frames/s.
Movie of Lifeact-GFP-expressing melanoblasts moving through the embryo skin epidermis. Orange areas show bursts of actin. Images were taken every 1 min for 31 min; the movie plays at 8 frames/s.
Time-lapse movies of immortalized melanocyte lines EW1 and EW7 migrating on fibronectin. Images were taken every 15 min; the movie plays at 8 frames/s.
Confocal time-lapse imaging of DMSO- and OHT-treated EW7 melanocytes expressing GFP-paxillin. Cells were imaged every 2 min for 30 min; the movie plays at 5 frames/s.