

Abstract
Cancer cells have epigenetic alterations that are known to drive cancer progression. The reversibility of the epigenetic posttranslational modifications on chromatin and DNA renders targeting these modifications an attractive means for cancer therapy. Cellular epigenetic status interacts with cell metabolism, and we are now beginning to understand the nature of how this interaction occurs and the biological contexts that mediate its function. Given the tremendous interest in understanding and targeting metabolic reprogramming in cancer, this nexus also provides opportunities for exploring the liabilities of cancers. This review summarizes recent developments in our understanding of the interaction of cancer metabolism and epigenetics.
Keywords: Acetyl-CoA, Serine, Methionine, One-carbon metabolism, S-adenosyl methionine, α-Ketoglutarate
1. Introduction
The term epigenetics has been used to describe heritable alterations of cellular phenotypes independent of mutations in the DNA sequence (Morgan et al., 2005). Now, it is also commonly referred to as a state of chromatin and DNA involving specific posttranslational modification of histones such as acetylation, methylation, ubiquitination, phosphorylation, crotonylation, and methylation, along with other modifications to DNA and RNA that affect gene expression (Dawson and Kouzarides, 2012; Tan et al., 2011). Changes in the epigenetic landscape have become evident in many pathophysiological conditions including cancer and diabetes (Baylin and Jones, 2011; Feinberg and Tycko, 2004; Suva et al., 2013). Cancer epigenetics was characterized in 1983 (Feinberg and Vogelstein, 1983; Gama-Sosa et al., 1983), when specific DNA methylation patterns of genes were found in human tumors in comparison to their normal tissue counterparts. It is now widely considered a significant contributor to cancer initiation and progression, along with genomic mutations (Sharma et al., 2010; Shen and Laird, 2013; Suva et al., 2013). In contrast to irreversible genetic mutations, the reversible property of epigenetic modifications along with the enzymatic nature of these modifications allows for therapeutic targeting through possible reversion of the epigenetic state associated with this modifications (Mosammaparast and Shi, 2010). To our knowledge, multiple drugs targeting epigenetic machinery have been approved by the Food and Drug Administration, in addition to a large variety of investigational compounds that are currently under avid clinical and laboratory investigation (Johnson et al., 2015; Yun et al., 2012). Despite advances in our understanding of cancer epigenetics over the past twenty years, what directly determines epigenetic status remains largely elusive. Recently, it has been proposed that metabolism interacts with epigenetic machinery (Carrer and Wellen, 2015; Gut and Verdin, 2013; Kaelin and McKnight, 2013) and that this interaction may have a substantial role in determining epigenetic state. Thus, there is an intriguing link between metabolism and epigenetics in cancer.
Metabolic reprogramming is a hallmark of cancer (Hanahan and Weinberg, 2011) with several common themes within this program that have emerged (Pavlova and Thompson, 2016). Now, cumulative evidence has reinforced this observation and revealed many additional metabolic characteristics in cancer cells, including alterations in the metabolism of glucose, amino acids, nucleotides and lipids (Boroughs and DeBerardinis, 2015; Pavlova and Thompson, 2016; Ward and Thompson, 2012). Furthermore, these metabolic features are heterogeneous and each cancer cell likely exhibits different metabolic features depending on its genetic, epigenetic, and environmental state.
In this review, we discuss the connection between cancer metabolism and epigenetics. We discuss metabolic pathways that can affect cellular epigenetics. We then highlight recent work on the interaction of metabolism and epigenetic modifications, focusing on methylation of DNA and histones, and acetylation of histones. In the end, we discuss the therapeutic potential of simultaneously targeting these connected processes. While this discussion due to space constraints is not comprehensive, it is our hope that it will give the reader an introduction to the key metabolic pathways known to affect epigenetics.
2. Serine, glycine, and one-carbon metabolism
3-phosphoglycerate dehydrogenase (PHGDH) encodes the enzyme that diverts glycolysis for serine synthesis. Quantitative characterizations of serine synthesis and the discovery amplifications in PHGDH, have reignited considerable interest in understanding the metabolic network downstream of this enzyme. This network, collectively referred to as serine, glycine, and one-carbon (SGOC) metabolism (Figure 1) encompasses a complex metabolic network involving the interconnected folate and methionine cycles (Locasale, 2013). In fact, modern cancer therapy partially arises from antagonizing folate metabolism that has been used in practice for over 60 years (Farber and Diamond, 1948; Locasale, 2013). SGOC metabolism integrates various nutrient inputs such as vitamins, glucose and amino acids, and generates substrates for the synthesis of macromolecules such as nucleotides and lipids, for the maintenance of cellular redox balance involving NADPH (nicotinamide adenine dinucleotide phosphate) and provides S-adenosyl methionine (SAM) for methylation reactions.
Figure 1. One-carbon metabolism.
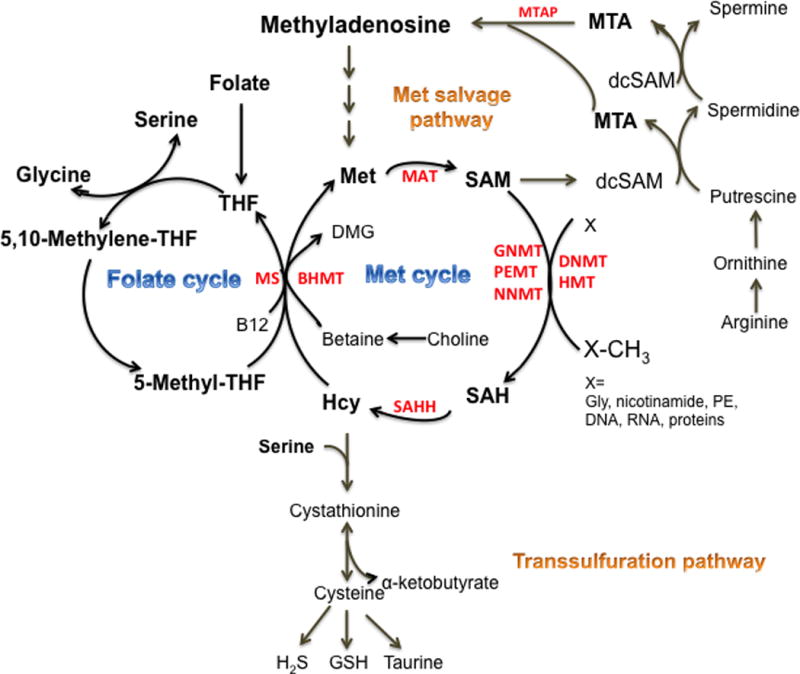
One-carbon metabolism is encompassed of a complex metabolic network centered by the folate and methionine cycles. Serine and glycine provide one-carbon unit to the folate cycle. The folate cycle is coupled with the methionine (Met) cycle for Met regeneration catalyzed by methionine synthase (MS). Met can also be regenerated through betaine-homocysteine (Hcy) methyltransferase (BHMT), using betaine as the one-carbon donor. Met provides the essential substrate for MAT (methionine S-adenosyltransferase)s, generating S-adenosylmethionine (SAM). SAM methylates a wide range of substrates DNA, RNA, lipids and proteins including histones, and generating S-adenosylhomocystine (SAH). SAH is catalyzed by SAH hydrolase to Hcy. One-carbon metabolism also interacts with the transsulfuration pathway and the methionine salvage pathway that is coupled with the polyamine synthesis. In the transsulfuration pathway, serine is required to divert Hcy for synthesis of cystathionine, and cystathionine is then catabolized into cysteine, the limiting factor for glutathione (GSH) production. Cysteine also provides substrate for the production of taurine and hydrogen sulfide (H2S). In the Met salvage pathway, SAM is decarboxylated to provide the supplaminopropyl groups to putrescine, supporting the polyamine synthesis. The byproduct 5′-methylthioadenosine (MTA) is salvaged back for SAM generation, with the initial step is catalyzed by MTA phosphorylase (MTAP). Other abbreviations: DNMT, DNA methyltransferase, HMT, histone methyltransferase, GNMT, glycine N-methyltransferase, PEMT, phosphatidylethanolamine N-methyltransferase, NNMT, nicotinamide N-methyltransferase.
Numerous studies have linked dietary changes and variation in the enzymes in the SGOC network to various types of cancer, especially leukemia and lymphoma where altered methylation is a critical contributor (Friso et al., 2002; Gemmati et al., 2004; Matsuo et al., 2001; Stern et al., 2000). For example, changes in dietary folate intake affect DNA methylation, as do genetic variants of methionine synthase (Anderson et al., 2012; Beaudin et al., 2012). Profiling the RNA levels of metabolic enzymes in a large cohort spanning multiple human cancer types reveals that methylene THF dehydrogenase 2 in the folate cycle is one of the top three metabolic enzymes most frequently overexpressed in cancer (Nilsson et al., 2014). In a wide range of cancers several elements of the network are highly variable, coordinately regulated, and overexpressed in certain cancer contexts (Mehrmohamadi et al., 2014).
In addition to glycine and serine, many cancer cells may also have a methionine dependency (Cavuoto and Fenech, 2012; Hoffman, 1982). It has recently been reported that methionine restriction alters the metabolism of cellular SAM that impacts gene expression reprogramming through histone methylation (Mentch et al., 2015). Nutritional manipulation of SAM by methionine is also physiologically sustainable in vivo (Mentch et al., 2015). Thus, modulation of SAM levels has physiological consequences. Supporting this hypothesis, some evidence shows that alterations of enzymes in the methionine cycle are also associated with cancer. Mice lacking MAT1A develop spontaneous liver tumors (Martinez-Chantar et al., 2002). Mice lacking glycine N-methyltransferase exhibit an accumulation of SAM in the liver and also develop hepatocellular carcinoma spontaneously (Martinez-Chantar et al., 2008). Overexpression of NNMT (nicotinamide N-methyltransferase) is also found in a variety of cancers such as the lung, liver, kidney, bladder and colon (Roessler et al., 2005; Ulanovskaya et al., 2013), where it might support tumor growth by diverting methyl groups away from other methylation processes (Ulanovskaya et al., 2013).
The methionine cycle also interacts with the transsulfuration pathway and methionine salvage pathway that is involved in polyamine synthesis (Lu and Mato, 2012). The transsulfuration pathway connects the methionine cycle to cysteine biosynthesis through Hcy. Hcy is converted to cystathionine by CBS (cystathionine β-synthase) that requires serine and vitamin B6. The resulting cystathionine is then cleaved into cysteine, the rate-limiting precursor for antioxidant GSH (glutathione) synthesis and α-ketobutyrate. In addition, CBS and cystathionase also catalyze reactions generating hydrogen sulfide (H2S) from cysteine and Hcy (Lu and Mato, 2012). Of interest, H2S released from the transsulfuration pathway may inhibit tumor growth (Kabil et al., 2014; Zhang et al., 2013).
Spermidine and spermine are two major polyamines derived from SAM. In polyamine synthesis, SAM is decarboxylated providing the supplaminopropyl groups to putrescine that derives from arginine. The byproduct 5′-methylthioadenosine (MTA) is salvaged back for SAM generation, and the initial step is catalyzed by MTA phosphorylase (MTAP), yielding adenine and 5-methylthioribose-1-phosphate. While polyamine synthesis has been associated with cancer (Thomas and Thomas, 2003), the connection with methionine salvage pathway is more recent. MTAP is ubiquitously expressed in normal tissues (Kryukov et al., 2016). However, due to its proximity to the tumor suppressor gene p16/CDKN2A, MTAP homozygous deletion occurs frequently in cancers including glioblastomas, pancreatic cancers, melanomas and others (Chen et al., 1996; Karikari et al., 2005; Kryukov et al., 2016). Recently, studies from two groups observed that MTAP-deleted cancer cells have specific vulnerabilities in their methylation reactions, notably the susceptibility to inhibition of an arginine methyltransferase PMRT5 (Figure 2) (Kryukov et al., 2016; Mavrakis et al., 2016).
Figure 2. Targeting One-carbon metabolism.
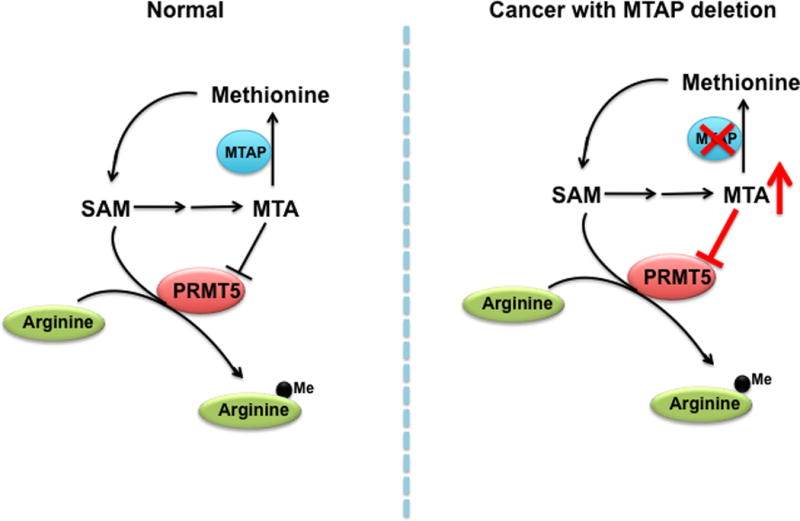
In the methionine salvage pathway, 5′-methylthioadenosine (MTA) functions as a competitive inhibitor of protein arginine methyltransferase 5 (PRMT5) that mediates methylation at arginine residues. Under normal conditions, cellular levels of S-adenosyl methionine (SAM) are quantitatively significant than MTA. In contrast, due to its proximity to the tumor suppressor gene p16, deletion of MTAP occurs frequently in a variety of cancers. The loss of MTAP leads to accumulation of MTA that competes with SAM for the binding of PRMT5. Recent studies have suggested that targeting PRMT5 could be effective for cancers with deletion of MTAP.
3. Methylation of DNA and histones by SAM
In cancer genomes, changes in DNA methylation are profound. Within a single cancer cell genome, 2000–3000 promoters can have aberrant methylation and most are hypermethylated, which typically represses gene expression (Plass et al., 2013). Promoter DNA methylation has been well studied. In mammals, the DNA methyl-group is typically transferred to the fifth position of cytosine within cytosine guanine (CpG) dinucleotides region that occupy 50–70% of human gene promoters (Sharma et al., 2010). In addition to promoter hypermethylation, the cancer genome can also be marked by genome-wide hypomethylation that takes place at various genomic sequences resulting in genomic instability (Yun et al., 2012). Patterns of histone methylation such as methylation of lysine 27 and lysine 9 on histone H3 (H3K9 and H3K27) have also been documented to correlate with aberrant gene silencing in a range of cancers (Yun et al., 2012).
One important property of DNA and histone methylation is its reversibility. DNA methylation is mediated by DNMT (DNA methyltransferase)s and reversed by several mechanisms including the removal of oxidized methylated bases by ten-eleven translocation (TET) proteins. TET proteins require Fe2+ and alpha-ketoglutarate (αKG) as co-factors to remove the methyl groups, producing succinate (Wu and Zhang, 2014). Similarly, histone methylation status is determined by histone methyltransferase (HMT)s and histone demethylases. While only three TET family members have been identified to date, over twenty histone demethylases are known (Kohli and Zhang, 2013; Mosammaparast and Shi, 2010). Histone demethylases are categorized into two families: LSD1, the flavin adenine dinucleotide-dependent oxidase, and the αKG- and Fe2+-dependent oxygenase, also known as JmjC-domain containing histone demethylase (JHDM) (Lu and Thompson, 2012). Up to three methyl-moieties can be attached to each lysine or arginine residue on histones. The outcome of methylation on gene expression depends on the specific site of histone as well as its valency, with trimethylation of H3K9 and H3K27 typical repressive marks and trimethylation of lysine 4 of histone H3 (H3K4me3) as a common active mark. We will first emphasize the interaction of metabolite SAM with the methylation of histone and DNA, and then highlight the importance of αKG in promoting both DNA and histone demethylation.
SAM provides a universal methyl-donor for a large variety of methyltransferases, including DNMTs and HMTs. Enzyme kinetics have revealed that the Michaelis constant (Km) values of many HMTs lies in the range of intracellular SAM levels (Mentch and Locasale, 2015). Of note, many methyltransferases can be inhibited by SAH, suggesting the ratio of SAM/SAH might also be relevant to histone and DNA methylation (Mentch and Locasale, 2015). Thus, histone and DNA methylation are affected by the levels of cellular SAM and by alterations in one-carbon metabolism that can modulate SAM levels. For example, it has been shown that H3K4 is often the most sensitive site to fluctuations of cellular SAM levels caused by nutritional availability of methionine (Mentch et al., 2015; Sadhu et al., 2013; Shiraki et al., 2014). This is the case of when lowering SAM by limited glycine input from threonine in mouse embryonic stem cells (Shyh-Chang et al., 2013). Similarly, C. elegans lacking SAM synthase sams-1 shows reduction in the levels of H3K4me3, which results in an impaired response to Pseudomonas infection (Ding et al., 2015). The relatively higher sensitivity of H3K4 methylation to cellular SAM is likely due to the relatively higher Km value of its corresponding methyltransferase. However, knockdown of SAM synthase sams-3 decreases methylation of H3K9, H3K27, and H3K36, but not H3K4me3 in embryos (Towbin et al., 2012), suggesting during embryonic development enzymes regulating H3K4 methylation are less sensitive to cellular SAM levels than those catalyzing other methylation sites. Furthermore, DNA methylation in osteoclasts also appears to be affected by SAM levels during differentiation (Nishikawa et al., 2015). Collectively, these data suggest hierarchy response of histone methylation to cellular fluctuation of SAM might be enzyme-dependent.
4. Demethylation of DNA and histones by α-ketoglutarate
In mammalian cells, αKG is produced from two different precursors: 1) from glutamate via GDH (glutamate dehydrogenase) and other transamination reactions and 2) from isocitrate via three non-redundant isocitrate dehydrogenase (IDH) enzymes: IDH1, IDH2, and IDH3. GDH is localized to the mitochondria and is either nicotinamide adenine dinucleotide (NAD)+ or NADP+ dependent (Li et al., 2012). In contrast, both IDH1 and IDH2 are NADP+ dependent enzymes localized to the cytosol and mitochondria, respectively, while IDH3 is NAD+ dependent and plays a major role in the TCA cycle (Dalziel, 1980). Gain-of-function mutations of IDH1 or IDH2 have been identified in gliomas, acute myeloid leukemia, cholangiocarcinoma and chondrosarcoma (Borger et al., 2012; Caramazza et al., 2010; Dang et al., 2010; Mardis et al., 2009; Rakheja et al., 2012; Yan et al., 2009). These heterozygous mutations convert αKG to (R)-2-hydroxyglutarate (2HG), sometimes causing rapid depletion of αKG and always causing millimolar level accumulation of 2HG (Dang et al., 2009; Losman and Kaelin, 2013). Although GDH mutations causing decreased αKG production have not been reported glutamine, the precursor of αKG, has been found to be depleted in the core region compared to the periphery of solid tumors (Reid et al., 2013).
Due to its essential role in DNA and histone demethylase activity, a loss in αKG levels may have profound effects on the epigenetic state of the cell, and results in a variety of physiological consequences. A CpG island hypermethylation is a phenotypic feature in IDH-driven gliomas, leukemias, and chondrosarcomas (Figueroa et al., 2010; Lu et al., 2013; Lu et al., 2012; Turcan et al., 2012). Studies by Noushmehr et. al. reveal that a subset of glioma patients displays a hypermethylated DNA phenotype that is highly associated with IDH1 mutations (Noushmehr et al., 2010). Ectopic expression of IDH1 and IDH2 mutants is sufficient to drive DNA hypermethylation via disruption of TET2 activity (Figure 3) (Figueroa et al., 2010). Similarly, introduction of mutant IDH1 and IDH2 leads to hypermethylation of histone residues, particularly on H3K9 and H3K27 (Lu et al., 2012). Interestingly, 2HG can cause histone methylation via inhibition of histone demethylase activity, although increasing levels of αKG reduces this effect (Lu et al., 2012). 2HG has been demonstrated to be a weak antagonist of αKG, and the combination of 2HG production and loss of αKG is required to induce histone and DNA hypermethylation (Xu et al., 2011). αKG-mediated histone and DNA demethylation has recently been shown to be critical for the maintenance of mouse embryonic stem cell pluripotency. Exogenous αKG decreases DNA methylation and histone methylation at H3K9 and H3K27, increases stem cell self-renewal, but suppresses cell differentiation (Carey et al., 2015). Conversely, ectopic expression of mutant IDH2 in 3T3-L1 preadipocytes results in αKG depletion and histone hypermethylation, and subsequent prevention of cell differentiation (Lu et al., 2012). Moreover, in acute myeloid leukemia patient-derived cells, mutant IDH2 promotes de-differentiation, and chemical inhibitors specific for IDH2 cause cell differentiation and increase their sensitivity to established chemotherapeutics (Wang et al., 2013). Similarly, inhibition of mutant IDH1 reduces cell growth, H3K9 hypermethylation, and promoted differentiation of glioma cells (Rohle et al., 2013). There are clear differences in the αKG–dependent effects between naïve embryonic stem cells and partially or terminally differentiated cells, therefore further studies are required to delineate the downstream effectors of cell fate in different physiological contexts.
Figure 3. Schematic representation of active vs. inactive histone and DNA demethylases.
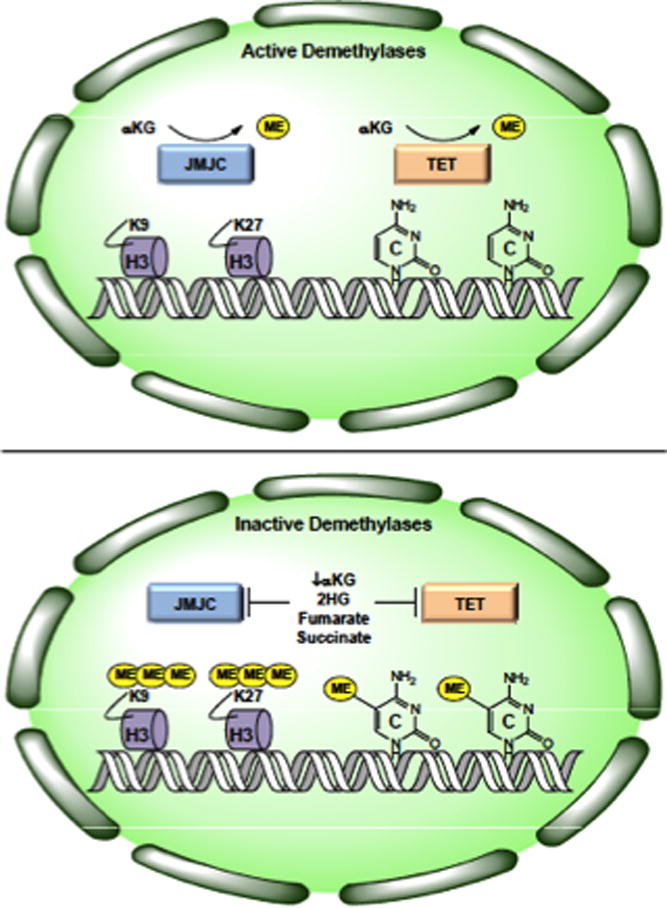
In the presence of αKG, JMJC-family histone demethylases and TET-family DNA demethylases remove methyl groups from histone H3 and cytosine, respectively (Top). When αKG is depleted or in the presence of excess 2HG, fumarate, or succinate, histone and DNA demethylase activity is inhibited resulting in broad genomic hypermethylation.
Besides 2HG, other metabolites reportedly inhibit demethylase activity. Recent evidence has demonstrated that succinate and fumarate can accumulate in high millimolar amounts due to loss-of-function mutations in genes coding for succinate dehydrogenase (SDH) and fumarate hydratase (FH) (Pollard et al., 2005). Tumors carrying these mutations share common characteristic features such as global DNA hypermethylation (Killian et al., 2013; Letouze et al., 2013; Xiao et al., 2012). Addition of succinate and fumarate is sufficient to inhibit the activity of αKG-dependent histone demethylase KDM4A, resulting in increased histone methylation, and knockdown of FH and SDH prevents TET1 and TET2-mediated DNA demethylation (Xiao et al., 2012). Fumarate has also been reported to inhibit JMJC-family histone demethylase KDM2B causing H3K36 hypermethylation, which promotes DNA double strand break repair (Jiang et al., 2015). Thus, it will be interesting to examine whether patients harboring FH mutations have less of a response to DNA-damaging therapeutics and whether supplementation with αKG may sensitize these tumors to treatment.
5. Histone acetylation and metabolites acetyl-CoA and NAD+
Histone acetylation is a dynamic and reversible process that involves the transfer of the acetyl groups from acetyl-CoA to the lysine residues, with half-life as short as ~2–3 min (Huang et al., 2015; Waterborg, 2002; Zheng et al., 2013). As acetylation removes positive charges of the lysine residues, it decreases the affinity between histones and DNA, and is considered a positive marker promoting gene expression (Huang et al., 2015; Yun et al., 2012). The reaction is driven by the compartmental abundance of acetyl-CoA and histone acetyltransferase (HAT)s. In the mitochondria, which contains high levels of acetyl-CoA (estimated to be 0.1–1.5 mM) and high pH, non-enzymatic acetylation may take place, accounting for substantial amounts of mitochondrial protein acetylation (Huang et al., 2015; Wagner and Payne, 2013). However, in the other cellular compartments where acetyl-CoA is lower, acetylation relies on HATs that have been divided into subfamilies: GCN5, MYST, and p300/CBP (Lu and Thompson, 2012; Yun et al., 2012). Several somatic mutations carried by HATs have been identified in human cancer (Dawson and Kouzarides, 2012). The acetyl groups from lysine residues can be removed by the histone deacetylase (HDAC)s (Lu and Thompson, 2012). HDACs are divided into classical HDACs (class I, II, or IV) that are Zn2+- and NAD+-dependent sirtuins (class III HDAC). Some evidence has shown that histone deacetylation is accelerated under low cellular pH, releasing the acetate anions to the extracellular environment to maintain the intracellular pH level (McBrian et al., 2013). This is intriguing as the Warburg effect, a common phenotype observed in cancer cells, leads to an acidic environment that may disrupt tissue architecture (Liberti and Locasale, 2016).
Compelling evidence support a physiological regulatory role for acetyl-CoA in histone acetylation (Cai et al., 2011; Cluntun et al., 2015; Henry et al., 2015; Takahashi et al., 2006; Wellen et al., 2009). Two landmark studies have been carried out with one in yeast where alterations of acetyl-CoA is enough to change histone acetylation and gene expression, and thereby affect cell growth (Cai et al., 2011). In mammals, cell metabolism has been linked to histone acetylation by ACLY (ATP-citrate lyase), an enzyme converting citrate into acetyl-CoA (Wellen et al., 2009). In fact, various HATs have Km values that fall within the range of estimated intracellular concentrations of acetyl-CoA (Cai et al., 2011; Langer et al., 2002). Some HATs are inhibited by the product CoA, therefore the ratio of acetyl-CoA:CoA may function as the physiological regulator of acetylation (Albaugh et al., 2011; Lee et al., 2014). In mammals, the nucleo-cytoplasmic acetyl-CoA derives from citrate or acetate by ACLY and acyl-CoA synthetase short-chain family member 2 (ACSS2) (Wellen and Thompson, 2012). ACLY-dependent acetyl-CoA production is positively regulated by AKT activation, and correlates with the status of histone acetylation in human prostate cancers and gliomas (Lee et al., 2014). The proto-oncogene MYC also promotes histone acetylation, presumably depending on citrate export from mitochondria and the nucleo-cytoplasmic ACLY activity (Edmunds et al., 2015). Another link to histone acetylation is the metabolic substrate acetate. ACSS2 catalyzes the conversion of acetate to acetyl-CoA. Expression of ACSS2 correlates with acetate uptake in tumors, whereas deletion of ACSS2 reduces tumor burden in mouse models (Comerford et al., 2014; Mashimo et al., 2014; Schug et al., 2015). In humans, the overexpression of ACSS2 is found in a wide range of cancers, and correlates with survival in triple-negative breast cancer patients (Comerford et al., 2014).
HDAC activity has been shown to be elevated in several types of cancers (Johnson et al., 2015). Sirtuins catalyze the deacetylation of histones into nicotinamide and 2′-O-acetyl-ADP-ribose, and this reaction requires NAD+ as a cofactor, which may affect sirtuin activity in physiology settings. The role of sirtuins in cancer is still inconclusive, and this might partially be due to their diverse distribution pattern among cellular compartments and tissues (Chalkiadaki and Guarente, 2015). Variations in the levels of cellular NAD+ and/or the NAD+/NADH ratio in response to changing metabolic environments and genetic alterations remain to be fully explored in cancer. Additionally, other Zn2+ dependent HDACs may also interact with metabolism. A group of metabolites, butyrate and its derivatives, inhibit HDAC enzyme activity (Shimazu et al., 2013). The physiological significance of this finding is that the ketone body β-hydroxybutryate, generated under nutrient-limiting conditions such as fasting, in the circulation falls in the range that inhibits HDACs (Huang et al., 2015; Shimazu et al., 2013). An additional line of evidence shows that butyrate, produced through bacterial fermentation, prevents colorectal tumorigenesis, suggesting a direct involvement of the gut microbiome (Donohoe et al., 2012; Donohoe et al., 2014).
6. Concluding remarks and future perspectives
Our understanding of metabolic reprogramming in cancer has grown substantially over the past ten years. In parallel, the possibility of starving cancers by caloric restriction or other nutritional interventions has come into focus (Meynet and Ricci, 2014). Glutamine, the most abundant free amino acid in human plasma (and tissue culture media), provides a link to epigenetics through demethylation reactions, yet this functions in cancer is still being understood, especially as it relates to the tumor microenvironment and corresponding nutrient limitations that exist (Davidson et al., 2016; Hensley et al., 2016). Acetate and fatty acids as nutritional sources in cancer has only recently been appreciated. Each of these macronutrients provides a source acetyl-CoA, the direct substrate for histone acetylation. Thus, acetyl-coA as it is centrally positioned in the network of carbon metabolism provides a gauge for the link between metabolism and chromatin status through reversible histone acetylation.
In the SGOC network, the variation of cellular SAM affects epigenetic status via histone and DNA methylation. In comparison to caloric restriction, dietary intervention through limiting the supply of amino acids could be an attractive cancer therapy. As an example, there is evidence that serum methionine levels in healthy individuals fall in the same order as that affecting histone methylation and its variability partially caused by nutritional intake (Mentch et al., 2015). Other nutrients that feed the methionine cycle such as choline, betaine and their derivative may also be attractive nutritional targets in cancer therapy. Furthermore, while epidemiological studies have revealed that insufficiency of SGOC-related nutrients such as folate and choline is positively associated with the risk of cancer, supplementing these nutrients are not exactly beneficial as in the case of folate supplementation studies (Kaelin and McKnight, 2013). Likely a U-shaped relationship exists where either low or high SAM levels will be harmful and thus there exists an optimal amount of these nutrients.
Recent evidence also supports cell metabolism as a key determinant of αKG-dependent demethylase activity. Whether producing 2HG or building up excess succinate and fumarate, tumor cell-selected mutations to key metabolic enzymes such as IDH, SDH, and FH directly inhibit DNA and histone demethylase activity (Figure 3). Although 2HG itself can inhibit demethylase activity, the loss of αKG may be even more effective in broad inhibition of histone and DNA demethylase activity and subsequent hypermethylation. Intriguingly, glutamine starvation has been shown to induce histone hypermethylation in mouse ES cells (Carey et al., 2015). Therefore, it will be interesting to examine the methylation state in glutamine depleted regions of solid tumors and other glutamine deprived environments.
All together, we are only beginning to learn the precise mechanisms of how cellular metabolic state communicates with chromatin to influence epigenetics. Therapeutically, combinations of metabolism- and epigenetics-related therapies are numerous and intriguing in many different context. Further development of our understanding will hopefully lead to the precise ways in which metabolism and epigenetics can be targeted in cancer.
Acknowledgments
The authors would like to thank support from R00CA168997 (JWL), R01CA193256 (JWL), R21 CA201963 (JWL), R01CA183989 (MK), Pew Scholar (MK), V Scholar in Cancer Research (MK) and Ralph M. Parsons foundation (MR). JWL thanks Katy Wellen and Ben Tu for helpful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Albaugh BN, Arnold KM, Denu JM. KAT(ching) metabolism by the tail: insight into the links between lysine acetyltransferases and metabolism. Chembiochem. 2011;12(2):290–298. doi: 10.1002/cbic.201000438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson OS, Sant KE, Dolinoy DC. Nutrition and epigenetics: an interplay of dietary methyl donors, one-carbon metabolism and DNA methylation. J Nutr Biochem. 2012;23(8):853–859. doi: 10.1016/j.jnutbio.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baylin SB, Jones PA. A decade of exploring the cancer epigenome – biological and translational implications. Nat Rev Cancer. 2011;11(10):726–734. doi: 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaudin AE, Abarinov EV, Malysheva O, Perry CA, Caudill M, Stover PJ. Dietary folate, but not choline, modifies neural tube defect risk in Shmt1 knockout mice. Am J Clin Nutr. 2012;95(1):109–114. doi: 10.3945/ajcn.111.020305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borger DR, Tanabe KK, Fan KC, Lopez HU, Fantin VR, Straley KS, Schenkein DP, Hezel AF, Ancukiewicz M, Liebman HM, Kwak EL, Clark JW, Ryan DP, Deshpande V, Dias-Santagata D, Ellisen LW, Zhu AX, Iafrate AJ. Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist. 2012;17(1):72–79. doi: 10.1634/theoncologist.2011-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boroughs LK, DeBerardinis RJ. Metabolic pathways promoting cancer cell survival and growth. Nat Cell Biol. 2015;17(4):351–359. doi: 10.1038/ncb3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai L, Sutter BM, Li B, Tu BP. Acetyl-CoA induces cell growth and proliferation by promoting the acetylation of histones at growth genes. Mol Cell. 2011;42(4):426–437. doi: 10.1016/j.molcel.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caramazza D, Lasho TL, Finke CM, Gangat N, Dingli D, Knudson RA, Siragusa S, Hanson CA, Pardanani A, Ketterling RP, Tefferi A. IDH mutations and trisomy 8 in myelodysplastic syndromes and acute myeloid leukemia. Leukemia. 2010;24(12):2120–2122. doi: 10.1038/leu.2010.213. [DOI] [PubMed] [Google Scholar]
- Carey BW, Finley LW, Cross JR, Allis CD, Thompson CB. Intracellular alpha-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature. 2015;518(7539):413–416. doi: 10.1038/nature13981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrer A, Wellen KE. Metabolism and epigenetics: a link cancer cells exploit. Curr Opin Biotechnol. 2015;34:23–29. doi: 10.1016/j.copbio.2014.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavuoto P, Fenech MF. A review of methionine dependency and the role of methionine restriction in cancer growth control and life-span extension. Cancer Treat Rev. 2012;38(6):726–736. doi: 10.1016/j.ctrv.2012.01.004. [DOI] [PubMed] [Google Scholar]
- Chalkiadaki A, Guarente L. The multifaceted functions of sirtuins in cancer. Nat Rev Cancer. 2015;15(10):608–624. doi: 10.1038/nrc3985. [DOI] [PubMed] [Google Scholar]
- Chen ZH, Zhang H, Savarese TM. Gene deletion chemoselectivity: codeletion of the genes for p16(INK4), methylthioadenosine phosphorylase, and the alpha- and beta-interferons in human pancreatic cell carcinoma lines and its implications for chemotherapy. Cancer Res. 1996;56(5):1083–1090. [PubMed] [Google Scholar]
- Cluntun AA, Huang H, Dai L, Liu X, Zhao Y, Locasale JW. The rate of glycolysis quantitatively mediates specific histone acetylation sites. Cancer Metab. 2015;3:10. doi: 10.1186/s40170-015-0135-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comerford SA, Huang Z, Du X, Wang Y, Cai L, Witkiewicz AK, Walters H, Tantawy MN, Fu A, Manning HC, Horton JD, Hammer RE, McKnight SL, Tu BP. Acetate dependence of tumors. Cell. 2014;159(7):1591–1602. doi: 10.1016/j.cell.2014.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalziel K. Isocitrate dehydrogenase and related oxidative decarboxylases. FEBS letters. 1980;117(Suppl):K45–55. doi: 10.1016/0014-5793(80)80569-2. [DOI] [PubMed] [Google Scholar]
- Dang L, Jin S, Su SM. IDH mutations in glioma and acute myeloid leukemia. Trends Mol Med. 2010;16(9):387–397. doi: 10.1016/j.molmed.2010.07.002. [DOI] [PubMed] [Google Scholar]
- Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, Marks KM, Prins RM, Ward PS, Yen KE, Liau LM, Rabinowitz JD, Cantley LC, Thompson CB, Vander Heiden MG, Su SM. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462(7274):739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson SM, Papagiannakopoulos T, Olenchock BA, Heyman JE, Keibler MA, Luengo A, Bauer MR, Jha AK, O’Brien JP, Pierce KA, Gui DY, Sullivan LB, Wasylenko TM, Subbaraj L, Chin CR, Stephanopolous G, Mott BT, Jacks T, Clish CB, Vander Heiden MG. Environment Impacts the Metabolic Dependencies of Ras-Driven Non-Small Cell Lung Cancer. Cell Metab. 2016;23(3):517–528. doi: 10.1016/j.cmet.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150(1):12–27. doi: 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]
- Ding W, Smulan LJ, Hou NS, Taubert S, Watts JL, Walker AK. s-Adenosylmethionine Levels Govern Innate Immunity through Distinct Methylation-Dependent Pathways. Cell Metab. 2015;22(4):633–645. doi: 10.1016/j.cmet.2015.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donohoe DR, Collins LB, Wali A, Bigler R, Sun W, Bultman SJ. The Warburg effect dictates the mechanism of butyrate-mediated histone acetylation and cell proliferation. Mol Cell. 2012;48(4):612–626. doi: 10.1016/j.molcel.2012.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donohoe DR, Holley D, Collins LB, Montgomery SA, Whitmore AC, Hillhouse A, Curry KP, Renner SW, Greenwalt A, Ryan EP, Godfrey V, Heise MT, Threadgill DS, Han A, Swenberg JA, Threadgill DW, Bultman SJ. A gnotobiotic mouse model demonstrates that dietary fiber protects against colorectal tumorigenesis in a microbiota- and butyrate-dependent manner. Cancer Discov. 2014;4(12):1387–1397. doi: 10.1158/2159-8290.CD-14-0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmunds LR, Sharma L, Kang A, Lu J, Vockley J, Basu S, Uppala R, Goetzman ES, Beck ME, Scott D, Prochownik EV. c-Myc programs fatty acid metabolism and dictates acetyl-CoA abundance and fate. J Biol Chem. 2015;290(33):20100. doi: 10.1074/jbc.A114.580662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farber S, Diamond LK. Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl-glutamic acid. N Engl J Med. 1948;238(23):787–793. doi: 10.1056/NEJM194806032382301. [DOI] [PubMed] [Google Scholar]
- Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer. 2004;4(2):143–153. doi: 10.1038/nrc1279. [DOI] [PubMed] [Google Scholar]
- Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature. 1983;301(5895):89–92. doi: 10.1038/301089a0. [DOI] [PubMed] [Google Scholar]
- Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, Tallman MS, Sun Z, Wolniak K, Peeters JK, Liu W, Choe SE, Fantin VR, Paietta E, Lowenberg B, Licht JD, Godley LA, Delwel R, Valk PJ, Thompson CB, Levine RL, Melnick A. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18(6):553–567. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friso S, Choi SW, Girelli D, Mason JB, Dolnikowski GG, Bagley PJ, Olivieri O, Jacques PF, Rosenberg IH, Corrocher R, Selhub J. A common mutation in the 5,10-methylenetetrahydrofolate reductase gene affects genomic DNA methylation through an interaction with folate status. Proc Natl Acad Sci U S A. 2002;99(8):5606–5611. doi: 10.1073/pnas.062066299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gama-Sosa MA, Slagel VA, Trewyn RW, Oxenhandler R, Kuo KC, Gehrke CW, Ehrlich M. The 5-methylcytosine content of DNA from human tumors. Nucleic Acids Res. 1983;11(19):6883–6894. doi: 10.1093/nar/11.19.6883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemmati D, Ongaro A, Scapoli GL, Della Porta M, Tognazzo S, Serino ML, Di Bona E, Rodeghiero F, Gilli G, Reverberi R, Caruso A, Pasello M, Pellati A, De Mattei M. Common gene polymorphisms in the metabolic folate and methylation pathway and the risk of acute lymphoblastic leukemia and non-Hodgkin’s lymphoma in adults. Cancer Epidemiol Biomarkers Prev. 2004;13(5):787–794. [PubMed] [Google Scholar]
- Gut P, Verdin E. The nexus of chromatin regulation and intermediary metabolism. Nature. 2013;502(7472):489–498. doi: 10.1038/nature12752. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Henry RA, Kuo YM, Bhattacharjee V, Yen TJ, Andrews AJ. Changing the selectivity of p300 by acetyl-CoA modulation of histone acetylation. ACS Chem Biol. 2015;10(1):146–156. doi: 10.1021/cb500726b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensley CT, Faubert B, Yuan Q, Lev-Cohain N, Jin E, Kim J, Jiang L, Ko B, Skelton R, Loudat L, Wodzak M, Klimko C, McMillan E, Butt Y, Ni M, Oliver D, Torrealba J, Malloy CR, Kernstine K, Lenkinski RE, DeBerardinis RJ. Metabolic Heterogeneity in Human Lung Tumors. Cell. 2016;164(4):681–694. doi: 10.1016/j.cell.2015.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman RM. Methionine dependence in cancer cells – a review. In Vitro. 1982;18(5):421–428. doi: 10.1007/BF02796468. [DOI] [PubMed] [Google Scholar]
- Huang Z, Cai L, Tu BP. Dietary control of chromatin. Curr Opin Cell Biol. 2015;34:69–74. doi: 10.1016/j.ceb.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Qian X, Shen J, Wang Y, Li X, Liu R, Xia Y, Chen Q, Peng G, Lin SY, Lu Z. Local generation of fumarate promotes DNA repair through inhibition of histone H3 demethylation. Nature cell biology. 2015;17(9):1158–1168. doi: 10.1038/ncb3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson C, Warmoes MO, Shen X, Locasale JW. Epigenetics and cancer metabolism. Cancer Lett. 2015;356(2 Pt A):309–314. doi: 10.1016/j.canlet.2013.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabil O, Vitvitsky V, Banerjee R. Sulfur as a signaling nutrient through hydrogen sulfide. Annu Rev Nutr. 2014;34:171–205. doi: 10.1146/annurev-nutr-071813-105654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaelin WG, Jr, McKnight SL. Influence of metabolism on epigenetics and disease. Cell. 2013;153(1):56–69. doi: 10.1016/j.cell.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karikari CA, Mullendore M, Eshleman JR, Argani P, Leoni LM, Chattopadhyay S, Hidalgo M, Maitra A. Homozygous deletions of methylthioadenosine phosphorylase in human biliary tract cancers. Mol Cancer Ther. 2005;4(12):1860–1866. doi: 10.1158/1535-7163.MCT-05-0103. [DOI] [PubMed] [Google Scholar]
- Killian JK, Kim SY, Miettinen M, Smith C, Merino M, Tsokos M, Quezado M, Smith WI, Jr, Jahromi MS, Xekouki P, Szarek E, Walker RL, Lasota J, Raffeld M, Klotzle B, Wang Z, Jones L, Zhu Y, Wang Y, Waterfall JJ, O’Sullivan MJ, Bibikova M, Pacak K, Stratakis C, Janeway KA, Schiffman JD, Fan JB, Helman L, Meltzer PS. Succinate dehydrogenase mutation underlies global epigenomic divergence in gastrointestinal stromal tumor. Cancer Discov. 2013;3(6):648–657. doi: 10.1158/2159-8290.CD-13-0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohli RM, Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature. 2013;502(7472):472–479. doi: 10.1038/nature12750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryukov GV, Wilson FH, Ruth JR, Paulk J, Tsherniak A, Marlow SE, Vazquez F, Weir BA, Fitzgerald ME, Tanaka M, Bielski CM, Scott JM, Dennis C, Cowley GS, Boehm JS, Root DE, Golub TR, Clish CB, Bradner JE, Hahn WC, Garraway LA. MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells. Science. 2016 doi: 10.1126/science.aad5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer MR, Fry CJ, Peterson CL, Denu JM. Modulating acetyl-CoA binding in the GCN5 family of histone acetyltransferases. J Biol Chem. 2002;277(30):27337–27344. doi: 10.1074/jbc.M203251200. [DOI] [PubMed] [Google Scholar]
- Lee JV, Carrer A, Shah S, Snyder NW, Wei S, Venneti S, Worth AJ, Yuan ZF, Lim HW, Liu S, Jackson E, Aiello NM, Haas NB, Rebbeck TR, Judkins A, Won KJ, Chodosh LA, Garcia BA, Stanger BZ, Feldman MD, Blair IA, Wellen KE. Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell Metab. 2014;20(2):306–319. doi: 10.1016/j.cmet.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letouze E, Martinelli C, Loriot C, Burnichon N, Abermil N, Ottolenghi C, Janin M, Menara M, Nguyen AT, Benit P, Buffet A, Marcaillou C, Bertherat J, Amar L, Rustin P, De Reynies A, Gimenez-Roqueplo AP, Favier J. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell. 2013;23(6):739–752. doi: 10.1016/j.ccr.2013.04.018. [DOI] [PubMed] [Google Scholar]
- Li M, Li C, Allen A, Stanley CA, Smith TJ. The structure and allosteric regulation of mammalian glutamate dehydrogenase. Archives of biochemistry and biophysics. 2012;519(2):69–80. doi: 10.1016/j.abb.2011.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberti MV, Locasale JW. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem Sci. 2016;41(3):211–218. doi: 10.1016/j.tibs.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locasale JW. Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat Rev Cancer. 2013;13(8):572–583. doi: 10.1038/nrc3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losman JA, Kaelin WG., Jr What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. 2013;27(8):836–852. doi: 10.1101/gad.217406.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Thompson CB. Metabolic regulation of epigenetics. Cell Metab. 2012;16(1):9–17. doi: 10.1016/j.cmet.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Venneti S, Akalin A, Fang F, Ward PS, Dematteo RG, Intlekofer AM, Chen C, Ye J, Hameed M, Nafa K, Agaram NP, Cross JR, Khanin R, Mason CE, Healey JH, Lowe SW, Schwartz GK, Melnick A, Thompson CB. Induction of sarcomas by mutant IDH2. Genes Dev. 2013;27(18):1986–1998. doi: 10.1101/gad.226753.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A, Wellen KE, O’Rourke DM, Berger SL, Chan TA, Levine RL, Mellinghoff IK, Thompson CB. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483(7390):474–478. doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu SC, Mato JM. S-adenosylmethionine in liver health, injury, and cancer. Physiol Rev. 2012;92(4):1515–1542. doi: 10.1152/physrev.00047.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, Koboldt DC, Fulton RS, Delehaunty KD, McGrath SD, Fulton LA, Locke DP, Magrini VJ, Abbott RM, Vickery TL, Reed JS, Robinson JS, Wylie T, Smith SM, Carmichael L, Eldred JM, Harris CC, Walker J, Peck JB, Du F, Dukes AF, Sanderson GE, Brummett AM, Clark E, McMichael JF, Meyer RJ, Schindler JK, Pohl CS, Wallis JW, Shi X, Lin L, Schmidt H, Tang Y, Haipek C, Wiechert ME, Ivy JV, Kalicki J, Elliott G, Ries RE, Payton JE, Westervelt P, Tomasson MH, Watson MA, Baty J, Heath S, Shannon WD, Nagarajan R, Link DC, Walter MJ, Graubert TA, DiPersio JF, Wilson RK, Ley TJ. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361(11):1058–1066. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Chantar ML, Corrales FJ, Martinez-Cruz LA, Garcia-Trevijano ER, Huang ZZ, Chen L, Kanel G, Avila MA, Mato JM, Lu SC. Spontaneous oxidative stress and liver tumors in mice lacking methionine adenosyltransferase 1A. FASEB J. 2002;16(10):1292–1294. doi: 10.1096/fj.02-0078fje. [DOI] [PubMed] [Google Scholar]
- Martinez-Chantar ML, Vazquez-Chantada M, Ariz U, Martinez N, Varela M, Luka Z, Capdevila A, Rodriguez J, Aransay AM, Matthiesen R, Yang H, Calvisi DF, Esteller M, Fraga M, Lu SC, Wagner C, Mato JM. Loss of the glycine N-methyltransferase gene leads to steatosis and hepatocellular carcinoma in mice. Hepatology. 2008;47(4):1191–1199. doi: 10.1002/hep.22159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashimo T, Pichumani K, Vemireddy V, Hatanpaa KJ, Singh DK, Sirasanagandla S, Nannepaga S, Piccirillo SG, Kovacs Z, Foong C, Huang Z, Barnett S, Mickey BE, DeBerardinis RJ, Tu BP, Maher EA, Bachoo RM. Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell. 2014;159(7):1603–1614. doi: 10.1016/j.cell.2014.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo K, Suzuki R, Hamajima N, Ogura M, Kagami Y, Taji H, Kondoh E, Maeda S, Asakura S, Kaba S, Nakamura S, Seto M, Morishima Y, Tajima K. Association between polymorphisms of folate- and methionine-metabolizing enzymes and susceptibility to malignant lymphoma. Blood. 2001;97(10):3205–3209. doi: 10.1182/blood.v97.10.3205. [DOI] [PubMed] [Google Scholar]
- Mavrakis KJ, McDonald ER, 3rd, Schlabach MR, Billy E, Hoffman GR, deWeck A, Ruddy DA, Venkatesan K, Yu J, McAllister G, Stump M, deBeaumont R, Ho S, Yue Y, Liu Y, Yan-Neale Y, Yang G, Lin F, Yin H, Gao H, Kipp DR, Zhao S, McNamara JT, Sprague ER, Zheng B, Lin Y, Cho YS, Gu J, Crawford K, Ciccone D, Vitari AC, Lai A, Capka V, Hurov K, Porter JA, Tallarico J, Mickanin C, Lees E, Pagliarini R, Keen N, Schmelzle T, Hofmann F, Stegmeier F, Sellers WR. Disordered methionine metabolism in MTAP/CDKN2A deleted cancers leads to dependence on PRMT5. Science. 2016 doi: 10.1126/science.aad5944. [DOI] [PubMed] [Google Scholar]
- McBrian MA, Behbahan IS, Ferrari R, Su T, Huang TW, Li K, Hong CS, Christofk HR, Vogelauer M, Seligson DB, Kurdistani SK. Histone acetylation regulates intracellular pH. Mol Cell. 2013;49(2):310–321. doi: 10.1016/j.molcel.2012.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehrmohamadi M, Liu X, Shestov AA, Locasale JW. Characterization of the usage of the serine metabolic network in human cancer. Cell Rep. 2014;9(4):1507–1519. doi: 10.1016/j.celrep.2014.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mentch SJ, Locasale JW. One-carbon metabolism and epigenetics: understanding the specificity. Ann N Y Acad Sci. 2015 doi: 10.1111/nyas.12956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mentch SJ, Mehrmohamadi M, Huang L, Liu X, Gupta D, Mattocks D, Gomez Padilla P, Ables G, Bamman MM, Thalacker-Mercer AE, Nichenametla SN, Locasale JW. Histone Methylation Dynamics and Gene Regulation Occur through the Sensing of One-Carbon Metabolism. Cell Metab. 2015;22(5):861–873. doi: 10.1016/j.cmet.2015.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meynet O, Ricci JE. Caloric restriction and cancer: molecular mechanisms and clinical implications. Trends Mol Med. 2014;20(8):419–427. doi: 10.1016/j.molmed.2014.05.001. [DOI] [PubMed] [Google Scholar]
- Morgan HD, Santos F, Green K, Dean W, Reik W. Epigenetic reprogramming in mammals. Hum Mol Genet. 2005;14(Spec No 1):R47–58. doi: 10.1093/hmg/ddi114. [DOI] [PubMed] [Google Scholar]
- Mosammaparast N, Shi Y. Reversal of histone methylation: biochemical and molecular mechanisms of histone demethylases. Annu Rev Biochem. 2010;79:155–179. doi: 10.1146/annurev.biochem.78.070907.103946. [DOI] [PubMed] [Google Scholar]
- Nilsson R, Jain M, Madhusudhan N, Sheppard NG, Strittmatter L, Kampf C, Huang J, Asplund A, Mootha VK. Metabolic enzyme expression highlights a key role for MTHFD2 and the mitochondrial folate pathway in cancer. Nat Commun. 2014;5:3128. doi: 10.1038/ncomms4128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa K, Iwamoto Y, Kobayashi Y, Katsuoka F, Kawaguchi S, Tsujita T, Nakamura T, Kato S, Yamamoto M, Takayanagi H, Ishii M. DNA methyltransferase 3a regulates osteoclast differentiation by coupling to an S-adenosylmethionine-producing metabolic pathway. Nat Med. 2015;21(3):281–287. doi: 10.1038/nm.3774. [DOI] [PubMed] [Google Scholar]
- Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, Verhaak RG, Hoadley KA, Hayes DN, Perou CM, Schmidt HK, Ding L, Wilson RK, Van Den Berg D, Shen H, Bengtsson H, Neuvial P, Cope LM, Buckley J, Herman JG, Baylin SB, Laird PW, Aldape K, Cancer Genome Atlas Research N Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer cell. 2010;17(5):510–522. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlova NN, Thompson CB. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016;23(1):27–47. doi: 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plass C, Pfister SM, Lindroth AM, Bogatyrova O, Claus R, Lichter P. Mutations in regulators of the epigenome and their connections to global chromatin patterns in cancer. Nat Rev Genet. 2013;14(11):765–780. doi: 10.1038/nrg3554. [DOI] [PubMed] [Google Scholar]
- Pollard PJ, Briere JJ, Alam NA, Barwell J, Barclay E, Wortham NC, Hunt T, Mitchell M, Olpin S, Moat SJ, Hargreaves IP, Heales SJ, Chung YL, Griffiths JR, Dalgleish A, McGrath JA, Gleeson MJ, Hodgson SV, Poulsom R, Rustin P, Tomlinson IP. Accumulation of Krebs cycle intermediates and overexpression of HIF1alpha in tumours which result from germline FH and SDH mutations. Human molecular genetics. 2005;14(15):2231–2239. doi: 10.1093/hmg/ddi227. [DOI] [PubMed] [Google Scholar]
- Rakheja D, Konoplev S, Medeiros LJ, Chen W. IDH mutations in acute myeloid leukemia. Hum Pathol. 2012;43(10):1541–1551. doi: 10.1016/j.humpath.2012.05.003. [DOI] [PubMed] [Google Scholar]
- Reid MA, Wang WI, Rosales KR, Welliver MX, Pan M, Kong M. The B55alpha subunit of PP2A drives a p53-dependent metabolic adaptation to glutamine deprivation. Molecular cell. 2013;50(2):200–211. doi: 10.1016/j.molcel.2013.02.008. [DOI] [PubMed] [Google Scholar]
- Roessler M, Rollinger W, Palme S, Hagmann ML, Berndt P, Engel AM, Schneidinger B, Pfeffer M, Andres H, Karl J, Bodenmuller H, Ruschoff J, Henkel T, Rohr G, Rossol S, Rosch W, Langen H, Zolg W, Tacke M. Identification of nicotinamide N-methyltransferase as a novel serum tumor marker for colorectal cancer. Clin Cancer Res. 2005;11(18):6550–6557. doi: 10.1158/1078-0432.CCR-05-0983. [DOI] [PubMed] [Google Scholar]
- Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, Tsoi J, Clark O, Oldrini B, Komisopoulou E, Kunii K, Pedraza A, Schalm S, Silverman L, Miller A, Wang F, Yang H, Chen Y, Kernytsky A, Rosenblum MK, Liu W, Biller SA, Su SM, Brennan CW, Chan TA, Graeber TG, Yen KE, Mellinghoff IK. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science. 2013;340(6132):626–630. doi: 10.1126/science.1236062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadhu MJ, Guan Q, Li F, Sales-Lee J, Iavarone AT, Hammond MC, Cande WZ, Rine J. Nutritional control of epigenetic processes in yeast and human cells. Genetics. 2013;195(3):831–844. doi: 10.1534/genetics.113.153981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schug ZT, Peck B, Jones DT, Zhang Q, Grosskurth S, Alam IS, Goodwin LM, Smethurst E, Mason S, Blyth K, McGarry L, James D, Shanks E, Kalna G, Saunders RE, Jiang M, Howell M, Lassailly F, Thin MZ, Spencer-Dene B, Stamp G, van den Broek NJ, Mackay G, Bulusu V, Kamphorst JJ, Tardito S, Strachan D, Harris AL, Aboagye EO, Critchlow SE, Wakelam MJ, Schulze A, Gottlieb E. Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell. 2015;27(1):57–71. doi: 10.1016/j.ccell.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010;31(1):27–36. doi: 10.1093/carcin/bgp220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, Laird PW. Interplay between the cancer genome and epigenome. Cell. 2013;153(1):38–55. doi: 10.1016/j.cell.2013.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimazu T, Hirschey MD, Newman J, He W, Shirakawa K, Le Moan N, Grueter CA, Lim H, Saunders LR, Stevens RD, Newgard CB, Farese RV, Jr, de Cabo R, Ulrich S, Akassoglou K, Verdin E. Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science. 2013;339(6116):211–214. doi: 10.1126/science.1227166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiraki N, Shiraki Y, Tsuyama T, Obata F, Miura M, Nagae G, Aburatani H, Kume K, Endo F, Kume S. Methionine metabolism regulates maintenance and differentiation of human pluripotent stem cells. Cell Metab. 2014;19(5):780–794. doi: 10.1016/j.cmet.2014.03.017. [DOI] [PubMed] [Google Scholar]
- Shyh-Chang N, Locasale JW, Lyssiotis CA, Zheng Y, Teo RY, Ratanasirintrawoot S, Zhang J, Onder T, Unternaehrer JJ, Zhu H, Asara JM, Daley GQ, Cantley LC. Influence of threonine metabolism on S-adenosylmethionine and histone methylation. Science. 2013;339(6116):222–226. doi: 10.1126/science.1226603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern LL, Mason JB, Selhub J, Choi SW. Genomic DNA hypomethylation, a characteristic of most cancers, is present in peripheral leukocytes of individuals who are homozygous for the C677T polymorphism in the methylenetetrahydrofolate reductase gene. Cancer Epidemiol Biomarkers Prev. 2000;9(8):849–853. [PubMed] [Google Scholar]
- Suva ML, Riggi N, Bernstein BE. Epigenetic reprogramming in cancer. Science. 2013;339(6127):1567–1570. doi: 10.1126/science.1230184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi H, McCaffery JM, Irizarry RA, Boeke JD. Nucleocytosolic acetyl-coenzyme a synthetase is required for histone acetylation and global transcription. Mol Cell. 2006;23(2):207–217. doi: 10.1016/j.molcel.2006.05.040. [DOI] [PubMed] [Google Scholar]
- Tan M, Luo H, Lee S, Jin F, Yang JS, Montellier E, Buchou T, Cheng Z, Rousseaux S, Rajagopal N, Lu Z, Ye Z, Zhu Q, Wysocka J, Ye Y, Khochbin S, Ren B, Zhao Y. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell. 2011;146(6):1016–1028. doi: 10.1016/j.cell.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas T, Thomas TJ. Polyamine metabolism and cancer. J Cell Mol Med. 2003;7(2):113–126. doi: 10.1111/j.1582-4934.2003.tb00210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towbin BD, Gonzalez-Aguilera C, Sack R, Gaidatzis D, Kalck V, Meister P, Askjaer P, Gasser SM. Step-wise methylation of histone H3K9 positions heterochromatin at the nuclear periphery. Cell. 2012;150(5):934–947. doi: 10.1016/j.cell.2012.06.051. [DOI] [PubMed] [Google Scholar]
- Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, Campos C, Fabius AW, Lu C, Ward PS, Thompson CB, Kaufman A, Guryanova O, Levine R, Heguy A, Viale A, Morris LG, Huse JT, Mellinghoff IK, Chan TA. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483(7390):479–483. doi: 10.1038/nature10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulanovskaya OA, Zuhl AM, Cravatt BF. NNMT promotes epigenetic remodeling in cancer by creating a metabolic methylation sink. Nat Chem Biol. 2013;9(5):300–306. doi: 10.1038/nchembio.1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner GR, Payne RM. Widespread and enzyme-independent Nepsilon-acetylation and Nepsilon-succinylation of proteins in the chemical conditions of the mitochondrial matrix. J Biol Chem. 2013;288(40):29036–29045. doi: 10.1074/jbc.M113.486753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Travins J, DeLaBarre B, Penard-Lacronique V, Schalm S, Hansen E, Straley K, Kernytsky A, Liu W, Gliser C, Yang H, Gross S, Artin E, Saada V, Mylonas E, Quivoron C, Popovici-Muller J, Saunders JO, Salituro FG, Yan S, Murray S, Wei W, Gao Y, Dang L, Dorsch M, Agresta S, Schenkein DP, Biller SA, Su SM, de Botton S, Yen KE. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science. 2013;340(6132):622–626. doi: 10.1126/science.1234769. [DOI] [PubMed] [Google Scholar]
- Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. 2012;21(3):297–308. doi: 10.1016/j.ccr.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterborg JH. Dynamics of histone acetylation in vivo. A function for acetylation turnover? Biochem Cell Biol. 2002;80(3):363–378. doi: 10.1139/o02-080. [DOI] [PubMed] [Google Scholar]
- Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324(5930):1076–1080. doi: 10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellen KE, Thompson CB. A two-way street: reciprocal regulation of metabolism and signalling. Nat Rev Mol Cell Biol. 2012;13(4):270–276. doi: 10.1038/nrm3305. [DOI] [PubMed] [Google Scholar]
- Wu H, Zhang Y. Reversing DNA methylation: mechanisms, genomics, and biological functions. Cell. 2014;156(1–2):45–68. doi: 10.1016/j.cell.2013.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, Liu L, Liu Y, Yang C, Xu Y, Zhao S, Ye D, Xiong Y, Guan KL. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012;26(12):1326–1338. doi: 10.1101/gad.191056.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Wang P, Xiao MT, Liu LX, Jiang WQ, Liu J, Zhang JY, Wang B, Frye S, Zhang Y, Xu YH, Lei QY, Guan KL, Zhao SM, Xiong Y. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer cell. 2011;19(1):17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, Friedman H, Friedman A, Reardon D, Herndon J, Kinzler KW, Velculescu VE, Vogelstein B, Bigner DD. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360(8):765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun J, Johnson JL, Hanigan CL, Locasale JW. Interactions between epigenetics and metabolism in cancers. Front Oncol. 2012;2:163. doi: 10.3389/fonc.2012.00163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Tang ZH, Ren Z, Qu SL, Liu MH, Liu LS, Jiang ZS. Hydrogen sulfide, the next potent preventive and therapeutic agent in aging and age-associated diseases. Mol Cell Biol. 2013;33(6):1104–1113. doi: 10.1128/MCB.01215-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Thomas PM, Kelleher NL. Measurement of acetylation turnover at distinct lysines in human histones identifies long-lived acetylation sites. Nat Commun. 2013;4:2203. doi: 10.1038/ncomms3203. [DOI] [PMC free article] [PubMed] [Google Scholar]