

The discovery of superoxide dismutase by McCord and Fridovich (1) ushered in a new area of biology wherein free radicals had to be factored into the biology of animals and plants. The free radical species of oxygen, the superoxide anion radical, has become the focus of thousands of studies. In medicine, the number of disease processes that have been linked to overexposure to oxidants or a failure to defend against them is vast, including pathologies as diverse as neurodegeneration, sepsis, atherosclerosis, and arthritis. The NO free radical and its redox partners, the nitrosonium and nitroxide ions, have been similarly painted with broad negative brushes (2). More recently, NO has been recognized for its role in normal physiology, arising in part from its ability to act as a signal through regulation of guanylate cyclase (3) and to S-nitrosylate cysteinyl residues of proteins and peptides (3). In a recent issue of PNAS, we witnessed the emergence of a new paradigm wherein the interplay between different highly reactive species allows for complex and fast regulation of cellular processes whose disruption has potentially serious pathological consequences (4).
Research from the Hare laboratory (5) has shown that the different isoforms of NO synthase (NOS) are involved in controlling different aspects of cardiac contractility. However, Khan et al. (4) present simple and clear studies that describe a new aspect to the paradigm of NO control that has broad application beyond the realm of contractility. Khan et al. show that the neuronal isoform of NOS (nNOS) and the superoxide-generating enzyme xanthine oxidoreductase (XOR) are in physical proximity in the sarcoplasmic reticulum (SR) of the cardiac myocytes of mice. Earlier studies have shown that superoxide production within cardiac myocytes has a potentially important signaling role (6). Furthermore, disruption of this signaling is involved in the development of cardiac pathologies such as congestive heart failure. Other research has also elucidated a key role for nNOS-generated NO in controlling cardiac contractility through altered intracellular calcium storage (7), potentially through the formation of SNO on the ryanodine receptor (RyR) (8). Through the elegant use of knockout mice, Khan et al. (4) have connected these observations, thereby demonstrating that the regulation of XOR-generated superoxide represents another site of NO modulation of cardiac contractility. This is an immediately attractive proposal, because NO and superoxide are both free radical species that have long been known to react at nearly diffusion-limited rates (9). For background, it is of value to briefly summarize the XOR system. XOR is widely distributed and is implicated in apoptosis and pathophysiology (8). In vivo, the system exists in two forms: xanthine dehydrogenase (XDH) and xanthine oxidase (XO). Both forms convert hypoxanthine to xanthine or xanthine to uric acid. However, XDH utilizes NAD+ as the electron acceptor, whereas XO transfers electrons to molecular oxygen, forming superoxide (10). XDH can be converted to XO either irreversibly by proteolytic cleavage or reversibly by thiol modification (11, 12).
Nitric oxide synthase activity is very tightly regulated temporally, spatially, and quantitatively.
Importantly, XOR does not appear to associate with the other isoforms of NOS, particularly endothelial NOS (eNOS). Furthermore, within heart muscle both nNOS and XOR are found within the SR along with the calcium pump SERCA2a and the calcium channel RyR. Impressively, xanthine-mediated production of superoxide is significantly increased in nNOS knockout mice compared with wild-type and eNOS knockout animals. This enhanced superoxide production is not caused by altered XOR expression, nor is it caused by increased cleavage of XDH to XO. However, the centerpiece of the article by Khan et al. (4) is contained within figures 4 and 5; there the authors clearly demonstrate the effect of XOR-mediated superoxide production on contractility and the role of nNOS activity in controlling this activity. Measurements of the systolic calcium transient and re-uptake by the SR indicate that allopurinol in the nNOS knockout mice affects contractility without affecting calcium release or uptake. These data fit well with observations from stunned myocardium in which myofilament contractility is reduced without affecting release or uptake of calcium. Hence, giving allopurinol to the myocardia of nNOS knockout mice improves calcium-sensitive contractility by reducing superoxide production rather than by restoring NO. It will be interesting to see whether NO donors increase calcium release or re-uptake in these knockout mice. Interestingly, allopurinol did not alter calcium sequestration within wild-type and eNOS knockout animals. Because nNOS is functional within these myocytes and is involved in the regulation of calcium storage by the SR, one might have predicted that XOR inhibition would accentuate this control.
The novel findings of Khan et al. are highlighted by a number of key observations. The physical proximity of nNOS and XOR, as demonstrated by immunoprecipitation studies, extends the idea that NOS isoform expression and localization are designed to control NO function. Traditionally we have considered NO signaling in terms of its rapid diffusibility. These findings, along with previous work on the cardiac myocyte and the consistent observation that NOS isoform expression is tightly controlled in terms of cellular location, as well as expression (13), present us with the novel challenge of determining how NO's activity is so strictly controlled. Within the myocyte there are three direct reaction pathways for NO: reaction with a target protein, namely, RyR (8); consumption via reaction with superoxide produced by XOR (4); and reaction with myoglobin in a way that NO cannot traverse the cytosolic space (14). What Khan et al. (4) have made evident is that NOS activity is very tightly regulated temporally, spatially, and quantitatively. Another key aspect of their study is that NO may serve not only to regulate particular protein targets but also to control another reactive diffusible signaling species. This finding presents a newly described form of cellular regulation, one in which an intracellular messenger, NO, functions to interrupt another signaling molecule, superoxide, by mediating its consumption. An undiscussed aspect of this model is that the end product of the reaction is itself a reactive species, peroxynitrite. Peroxynitrite has been implicated in a number of pathophysiologies and may even play a role in heart failure (15). It is possible that peroxynitrite generated within this system becomes rapidly protonated and isomerizes to nitrate, an inert metabolite. However, it could also react with either CO2, becoming a potent nitrating agent (16), or with a thiol and generate SNO (17). The interplay between nNOS and XOR presents an intriguing model for the pathophysiology of cardiac disease (see Fig. 1), particularly in light of the potential for increased XO formation during chronic heart failure (18).
Fig. 1.

nNOS and XOR are expressed within close proximity within the SR. nNOS catalyzes the conversion of arginine to citruline with the concomitant production of the NO group. NO produced in this way is capable of nitrosylating a specific cysteine on the SR calcium channel, otherwise called RyR (19). SNO formation on the RyR increases channel opening probability and hence calcium release during excitation–contraction coupling. XOR, in the oxidase form, utilizes molecular oxygen as an electron acceptor for purine metabolism to produce superoxide (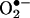 ). Superoxide is capable of diffusing out of the SR and inhibiting the calcium responsiveness of the contractile machinery, thus inhibiting contraction (6, 20). In addition to activating calcium release through the RyR, NO can increase contractility by inhibiting the release of superoxide from the SR by direct reaction (solid red arrow). Additionally, superoxide may be able to inhibit contractility by inhibiting NO-mediated activation of the RyR (dashed blue line).
). Superoxide is capable of diffusing out of the SR and inhibiting the calcium responsiveness of the contractile machinery, thus inhibiting contraction (6, 20). In addition to activating calcium release through the RyR, NO can increase contractility by inhibiting the release of superoxide from the SR by direct reaction (solid red arrow). Additionally, superoxide may be able to inhibit contractility by inhibiting NO-mediated activation of the RyR (dashed blue line).
It has been suggested that the production of XOR and the buildup of purines during ischemia prime the myocyte for superoxide production upon reperfusion. Clearly one can see that a positive consequence of such superoxide generation would be the inhibition of cardiac contraction, thereby protecting the energetically compromised cell. Khan et al. present another potential consequence: the limitation of NO to produce intracellular calcium storage, another energetically costly process. As the intracellular purine concentration falls with the restoration of normal cellular energy production, superoxide production decreases, allowing contractility and calcium storage to be reestablished. Conversely, the opposite condition can also be envisioned, in which NO production results in increased intracellular calcium metabolism and activated contractility by consuming superoxide. Thus, it is the stoichiometric balance between NO and superoxide, rather than the absolute concentrations of either, that is critical. Imbalance that may occur in diseases like congestive heart failure could have serious consequences for both contractility and myocyte viability.
One of the most important deities of Roman culture during the Augustinian era was Janus, the god of beginnings and endings, for whom January was named. NO has often been referred to as a Janus molecule because it has both positive and negative attributes. The research presented by Khan et al. (4) may now allow us to refer to NO as truly a Janus molecule, not in that it has negative and positive consequences, but rather that it represents both the opening and closing of doors. In this way it is a true molecular switch.
Acknowledgments
The National Institutes of Health Center of Biomedical Research Excellence Protein Research Center at the University of Puerto Rico is funded by National Institutes of Health Grant 5P20 RR016439.
See companion article on page 15944 in issue 45 of volume 101.
References
- 1.McCord, J. M. & Fridovich, I. (1969) J. Biol. Chem. 244, 6049–6055. [PubMed] [Google Scholar]
- 2.Beckman, J. S. & Koppenol, W. H. (1996) Am. J. Physiol. 271, C1424–C1437. [DOI] [PubMed] [Google Scholar]
- 3.Murad, F., Mittal, C. K., Arnold, W. P., Katsuki, S. & Kimura, H. (1978) Adv. Cyclic Nucleotide Res. 9, 145–158. [PubMed] [Google Scholar]
- 4.Khan, S. A., Lee, K., Minhas, K. M., Gonzalez, D. R., Raju, S. V. Y., Tejani, A. D., Li, D., Berkowitz, D. E. & Hare, J. M. (2004) Proc. Natl. Acad. Sci. USA 101, 15944–15948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khan, S. A., Skaf, M. W., Harrison, R. W., Lee, K., Minhas, K. M., Kumar, A., Fradley, M., Shoukas, A. A., Berkowitz, D. E. & Hare, J. M. (2003) Circ. Res. 92, 1322–1329. [DOI] [PubMed] [Google Scholar]
- 6.Perez, N. G., Gao, W. D. & Marban, E. (1998) Circ. Res. 83, 423–430. [DOI] [PubMed] [Google Scholar]
- 7.Barouch, L. A., Harrison, R. W., Skaf, M. W., Rosas, G. O., Cappola, T. P., Kobeissi, Z. A., Hobai, I. A., Lemmon, C. A., Burnett, A. L., O'Rourke, B., et al. (2002) Nature 416, 337–339. [DOI] [PubMed] [Google Scholar]
- 8.Eu, J. P., Sun, J., Xu, L., Stamler, J. S. & Meissner, G. (2000) Cell 102, 499–509. [DOI] [PubMed] [Google Scholar]
- 9.Koppenol, W. H., Moreno, J. J., Pryor, W. A., Ischiropoulos, H. & Beckman, J. S. (1992) Chem. Res. Toxicol. 5, 834–842. [DOI] [PubMed] [Google Scholar]
- 10.Waud, W. R. & Rajagopalan, K. V. (1976) Arch. Biochem. Biophys. 172, 354–364. [DOI] [PubMed] [Google Scholar]
- 11.Stirpe, F. & Della, C. E. (1969) J. Biol. Chem. 244, 3855–3863. [PubMed] [Google Scholar]
- 12.Waud, W. R. & Rajagopalan, K. V. (1976) Arch. Biochem. Biophys. 172, 365–379. [DOI] [PubMed] [Google Scholar]
- 13.Sowa, G., Pypaert, M. & Sessa, W. C. (2001) Proc. Natl. Acad. Sci. USA 98, 14072–14077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brunori, M. (2001) Trends Biochem. Sci. 26, 21–23. [DOI] [PubMed] [Google Scholar]
- 15.Mihm, M. J., Coyle, C. M., Schanbacher, B. L., Weinstein, D. M. & Bauer, J. A. (2001) Cardiovasc. Res. 49, 798–807. [DOI] [PubMed] [Google Scholar]
- 16.Gow, A., Duran, D., Thom, S. R. & Ischiropoulos, H. (1996) Arch. Biochem. Biophys. 333, 42–48. [DOI] [PubMed] [Google Scholar]
- 17.Schrammel, A., Gorren, A. C., Schmidt, K., Pfeiffer, S. & Mayer, B. (2003) Free Radical Biol. Med. 34, 1078–1088. [DOI] [PubMed] [Google Scholar]
- 18.Farquharson, C. A., Butler, R., Hill, A., Belch, J. J. & Struthers, A. D. (2002) Circulation 106, 221–226. [DOI] [PubMed] [Google Scholar]
- 19.Sun, J., Xin, C., Eu, J. P., Stamler, J. S. & Meissner, G. (2001) Proc. Natl. Acad. Sci. USA 98, 11158–11162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berry, C. E. & Hare, J. M. (2004) J. Physiol. 555, 589–606. [DOI] [PMC free article] [PubMed] [Google Scholar]