

Abstract
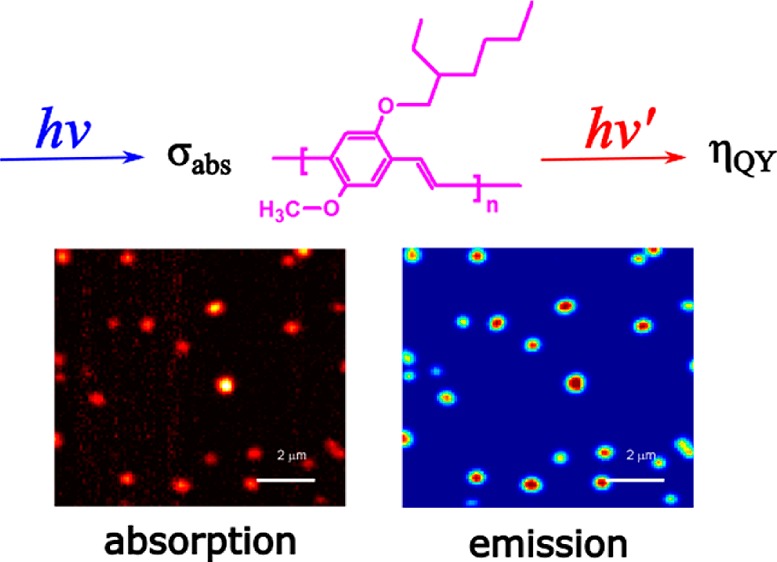
We simultaneously measured the absorption and emission of single conjugated polymer poly[2-methoxy-5-(2-ethylhexyloxy)-1,4-phenylenevinylene] (MEH-PPV) molecules in a poly(methyl methacrylate) (PMMA) matrix using near-critical xenon to enhance the photothermal contrast for direct absorption measurements. We directly measured the number of monomers and the quantum yield of single conjugated polymer molecules. Simultaneous absorption and emission measurements provided new insight into the photophysics of single conjugated polymers under optical excitation: quenching in larger molecules is more efficient than in smaller ones. Photoinduced traps and defects formed under prolonged illumination lead to decrease of both polymer fluorescence and absorption signals with the latter declining slower.
Keywords: Conjugated polymer, absorption, photothermal, quantum yield, quenching
Conjugated polymers represent an important class of organic materials widely used in applications such as organic light emitting devices (OLEDs),1 solar energy conversion,2 thin-film transistors, and chemical sensors.3,4 It is widely accepted that the optical and electrical properties of conjugated polymer chains strongly depend on their microscopic structure and on their local surroundings.5,6 Over the past decades, with the help of single-molecule fluorescence spectroscopy,7 extensive studies have been carried out aiming to clarify the optical properties of conjugated polymers.6 Barbara’s group first applied single-molecule spectroscopy to single conjugated polymers PPV–PPyV in polystyrene.8 Surprisingly, they found that a single conjugated polymer that consists of many chromophores can behave like a single emitter. After many factors were examined, they concluded that efficient intrachain energy transfer accounts for the single-emitter behavior. Since then, many approaches based on single-molecule fluorescence spectroscopy have been proposed and applied to conjugated polymers. A common approach is to measure the fluorescence time traces of single polymer chains9,10 under different experimental conditions such as different chemical structures, molecular weights, solvents, matrixes, and sample preparations.9,11−13 Another important approach is the fluorescent excitation and emission polarization anisotropy measurements on conjugated molecules, combined with computer simulations,5 to characterize the anisotropic conformation properties14,15 and energy transfer.16 Room temperature17−19 and low-temperature single-molecule spectroscopy11,20−22 measure the spectra of single conjugated polymer molecules to resolve the interaction between chromophores and provide a picture of the landscape of energy transfer. Time-resolved measurements can give information on the fast dynamics of excitons at different intermediate fluorescence intensity levels when fluorescence exhibits blinking behavior. These measurements indirectly yield insight into exciton migration along a single chain and the population of localized domains.23,24 Fluorescence correlation spectroscopy has been used to study the solvent effect on the conformation of conjugated polymers in solution,25,26 while dynamics of excitons under variable excitation power showed singlet–triplet interaction at the single chain level.27 By combining far-field and near-field microscopy, researchers proposed to measure the absorption properties of single conjugated polymers.28 By providing a few nm spatial resolution, atomic force microscopy (AFM) can be exploited to resolve the conformational structure of some conjugated polymer molecules.29
Most single-molecule spectroscopic studies on conjugated polymers were based on their fluorescence. It has been impossible so far to probe the absorption and nonradiative decay of excitons directly at the single-molecule level. Measurements of fluorescence polarization properties provide information on the anisotropy of the relative fluorescence excitation cross-section. Absolute value of the fluorescence excitation cross-section can be assessed by measuring of fluorescence brightness of single molecules.30,31 However, without knowing the fluorescence quantum yield, the absorption cross-section cannot be derived from fluorescence brightness. Therefore, direct absorption measurements are needed to provide complementary information for unraveling nonradiative decay channels of excitons and obtaining new insights into the quenching mechanisms and formation of dark states of conjugated polymer molecules. Such measurements may thus clarify some of the questions that have arisen in previous studies.30
Photothermal microscopy is a tool for probing the local energy dissipation at the nanoscale. Detection sensitivity has been demonstrated down to single 2.5 nm gold nanoparticles32 and single dye molecules.33 Conventional photothermal microscopy relies on high optical excitation powers to reach single-molecule sensitivity.33 However, such high powers limit the detection to those molecules which resist photobleaching at such harsh excitation conditions. Recently, we have experimentally shown that by using near-critical xenon as the transducing medium, the photothermal contrast of weak absorbers such as small gold nanoparticles can be greatly enhanced.34 This allows lowering of the excitation power to become more compatible with single-molecule measurements of light-sensitive compounds. In this work, we simultaneously measure the absorption and emission of single poly[2-methoxy-5-(2-ethylhexyloxy)-1,4-phenylenevinylene] (MEH-PPV) molecules in a poly(methyl methacrylate) (PMMA) matrix with an excitation power about 450 W/cm2 by using near-critical xenon as the transducing medium for the photothermal microscope. Our results yield the number of monomers and the quantum yield of single MEH-PPV molecules and provide valuable information on the radiative and nonradiative decays of excitons in conjugated polymer molecules.
Experimental Details
Simultaneous absorption and fluorescence measurements were performed on a home-built inverted confocal microscope. See Figure S1 in the Supporting Information for the scheme of the setup. A continuous-wave (CW) laser with a wavelength of 488 nm was intensity-modulated by an acousto-optic modulator at 53 kHz and used as the excitation/heating beam to excite the conjugated polymer molecules. A CW laser beam (probe beam) with a wavelength of 815 nm was spatially overlapped with the excitation/heating beam. The two beams were both expanded to fill the back aperture of an oil immersion objective (Olympus PLAPON, 60× , NA = 1.45) and were focused on the sample. The reflected probe beam was collected by a photodiode (Femto DHPCA-100-F), and the signal was demodulated by a lock-in amplifier (SR844, Stanford Research Systems). The demodulated voltage signal gives the photothermal contrast. The fluorescence signal of the polymer molecules was collected by a single-photon-counting avalanche photodetector (SPCM-AQR-16, PerkinElmer), and a set of optical filters was used to remove the excitation and probe beams. The excitation power used in the experiments was typically about 450 W/cm2 in focus at 488 nm. The probe power in focus was about 3.5 MW/cm2 at 815 nm, and the integration time was 30 ms. The gain of the photodiode was 10 000. All these parameters remain the same in our measurements unless noted otherwise.
Cover glasses were cleaned via sonication in acetone, ethanol, and Milli-Q water, separately. Then they were treated with ozone for about 30 min to remove all the possible organic contaminations. MEH-PPV polymers with average molecular weight by number Mn ≈ 200 kDa and polydispersity index ≈ 6 (measured by GPC) and PMMA (with molecular weight Mw ≈ 996 kDa) were bought from Sigma-Aldrich. PMMA was dissolved in toluene with concentration of about 4 g/L. MEH-PPV was dissolved in chloroform with a concentration of about 0.66 μg/L for typical single-molecule measurements. A droplet of PMMA solution (about 15 μL) was first spin-coated on the clean cover glass to obtain a film about 20 nm thick. Then a droplet of MEH-PPV solution was spin-coated on the PMMA film. Although part of the lower PMMA layer might be dissolved by the chloroform in the MEH-PPV drop-casting solution, we believe that at least some PMMA was still left on the glass because the fluorescence background was clearly different from the case where only MEH-PPV solution was spin-coated on glass. On top of the MEH-PPV film, we spin-coated a second PMMA layer with a thickness of about 20 nm to form a sandwich-structure sample (see the scheme in Figure 1). Each spin-coating was performed with a speed of 2000 rpm for 30 s. For the gold nanoparticle sample, we first spin-coated gold nanoparticles on the clean slide and then spin-coated a PMMA layer on the gold nanoparticles with a thickness of about 20 nm.
Figure 1.
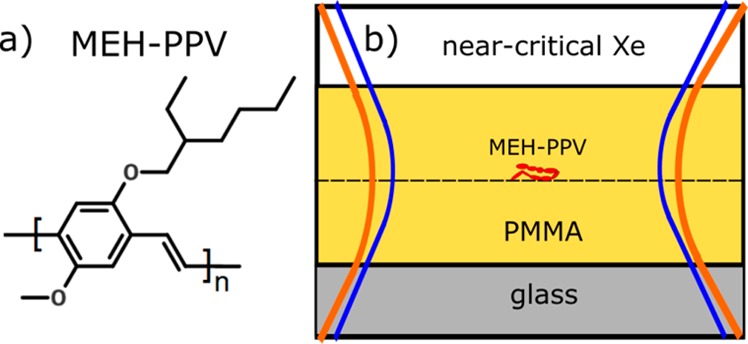
Sample. (a) Chemical structure of MEH-PPV. (b) Scheme of the sample: MEH-PPV molecules (in red) are imbedded in a PMMA matrix and undergo illumination by excitation (blue) and probe (orange) beams.
The cover glass coated with MEH-PPV/PMMA was glued to a home-built high-pressure cell, which left a clear aperture of about 0.75 mm diameter for optical access.34 The high pressure was applied to xenon gas through thin steel tubes and controlled with an auxiliary device. The temperature of the whole cell was also controlled with a feedback device (see ref (34) for more details). We varied the temperature and pressure of xenon close to its critical point to achieve optimal sensitivity, and we used 20 nm diameter gold nanoparticles to calibrate the photothermal signal (see Figure S1b and c in the Supporting Information).
Results
To find the optimal conditions for the photothermal detection of single conjugated polymer molecules, we carried out measurements on small gold nanoparticles (20 nm in diameter) in xenon under variable temperature and pressure. We found that the best photothermal signals arise when the pressure and temperature of the xenon cell are about 6.26 MPa and 288 K. Note that the optimal conditions may change slightly for different measurements, probably because the purity of xenon in the pressure cell may not be exactly the same every time, even though the cell is flushed with fresh xenon before each measurement. Figure S1b and c show a typical image and the associated histogram of the photothermal contrast for 20 nm gold nanoparticles imbedded in PMMA in xenon under circularly polarized 488 nm excitation, when the pressure and temperature were optimized. The mean photothermal signal (with respect to the background) over 40 particles was about 164 ± 27 μV. These data will be used for calibration to deduce the absorption cross-section of single conjugated polymers since the absorption cross-section of a gold nanosphere can be easily calculated using Mie theory (for a 20 nm gold sphere, the absorption cross-section is about 296 nm2 in PMMA at 488 nm).
We simultaneously measured the absorption and emission of single MEH-PPV molecules embedded in PMMA using the enhanced photothermal setup. Typical confocal images are shown in Figure 2a and b. The excitation beam at 488 nm was circularly polarized in all the measurements discussed below. Single conjugated polymers show both fluorescence and absorption signals, and the spots in the two images are well correlated. The excitation power at the sample is approximately 600 nW, which corresponds to a power density about 450 W/cm2. The photothermal contrast of these polymers under current excitation power is good enough, with an average signal-to-noise ratio (SNR) of about 29. We scanned different regions of the same sample, and we found 177 spots in the fluorescence image, from which 174 correlated spots were found in the photothermal image. The distributions of photothermal and fluorescence signals are shown in Figure 2d and e. Note that these histograms show only spots correlated in both images. The distributions reflect the molecular weight or conformational heterogeneity of the molecules. The distribution of fluorescence intensities, between 1.0 and 1.8 Mcps, appears somewhat narrower than the distribution of absorption strengths, between 10 and 30 μV. Moreover, the absorption distribution is narrower than that of molecular weight indicated by the manufacturer and obtained by gel permeation chromatography (GPC; polydispersity index, PDI = 6). The origin of this difference is unclear. It may arise from sample preparation. We correlated the absorption and fluorescence signals, and the result is shown in Figure 2c. As is clearly seen in spite of the dispersion of data points, the fluorescence brightness of single polymer molecules appears to scale sublinearly with their absorption signal. This sublinear dependence explains the different shape and the narrower width of the fluorescence distribution compared to the absorption distribution.
Figure 2.

Simultaneous absorption and emission measurements on single conjugated polymers embedded in PMMA in near-critical xenon. (a) Photothermal image of single conjugated MEH-PPV molecules; (b) confocal fluorescence image of the area in panel a, recorded simultaneously. The pressure and temperature of the sample cell were 6.26 MPa and 288 K, respectively. (c) Correlated scatter plot of fluorescence and photothermal signals. (d, e) Histograms of the photothermal signal and the fluorescence signal, respectively.
We can deduce the absorption cross-section of a single polymer and the number of monomers in each polymer based on the photothermal signal of polymer molecules and on the calibration data of 20 nm gold particles (see the Supporting Information). Figure 3a shows the histogram of absorption cross-sections of 174 conjugated polymers. The mean value is about 11.2 nm2 for the current sample under study. The wide distribution again reflects the size heterogeneity or aggregation of chains. Note that the histogram in Figure 3 may not fully reflect the distribution of the absorption cross-sections of these molecules simply because of the detection limit of our current photothermal setup. For a meaningful discussion of signal strength and distribution, the SNR normally should not be lower than 10.35 Then we estimate the minimum detectable absorption cross-section to be around 4 nm2 under our current experimental conditions. The photothermal contrast could be slightly improved by using longer integration times, provided a good stability of the optical microscope and the photostability of the conjugated molecules are ensured.
Figure 3.
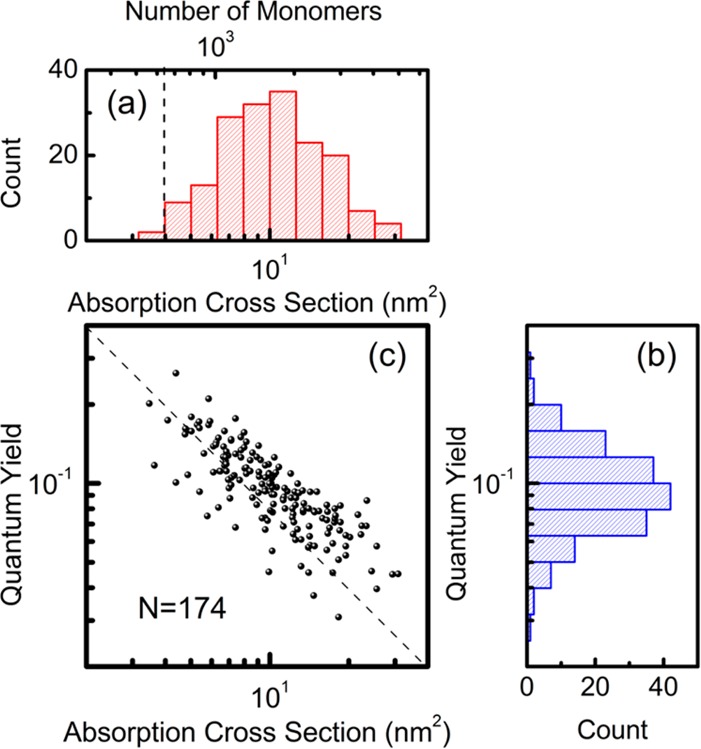
Absorption cross-section and quantum yield of single MEH-PPV molecules in PMMA matrix. (a) Histogram of the absorption cross-section (bottom axis) and the number of monomers (top axis) of single MEH-PPV molecules. The dashed line indicates the current photothermal detection limit. (b) Histogram of the quantum yield of single MEH-PPV polymers deduced from the simultaneous measurements. (c) Scatter plot of the quantum yield versus the absorption cross-section. The dashed line has slope −1 and indicates a decrease of the quantum yield as the inverse of the absorption cross-section.
To estimate the number of monomers in single polymer molecules by their experimentally measured absorption cross-section, we use the absorption cross-section of MEH-PPV per its monomer unit, that is, the molecular absorption cross-section normalized to the number of monomer units in the molecule. The cross-section per monomer unit of MEH-PPV is known from the literature to be 6.2 × 10–3 nm2 at 488 nm.36 The number of monomers per individual chain deduced from their absorption cross-section is shown on the top axis of Figure 3a. From the specification of our MEH-PPV sample by the manufacturer, the average molecular weight Mn is around 200 kDa. As the monomer molecular weight is 276 Da, the expected average number of monomers per chain is around 720, close to the smallest measurable number in our experiments. However, the absorption distribution of Figure 3a is considerably narrower than the large polydispersity (PDI = 6) indicated by the manufacturer. It is therefore reasonable to assume that sample preparation or selective effects in the detection has led us to a biased observation of the largest molecules. In any case, the number of monomers deduced from our measurements reasonably matches what is expected from the specification. To the best of our knowledge, our results are the first quantitative measurement of the monomer numbers in single conjugated polymers by photothermal microscopy. This useful information will help us to better estimate the fluorescence quantum yield of single MEH-PPV molecules.
The quantum yield (QY) of an emitter is the ratio between the number of photons emitted and the number of photons absorbed under optical excitation, as written in eq 1:
| 1 |
The total number of photons emitted Nemit is the fluorescent signal divided by the detection efficiency of the setup (taking into account the collection efficiency of all optics in the detection path and the efficiency of photon detectors), while the number of photons absorbed by a molecule Nabs is written as the ratio between absorbed power and incident photon energy:
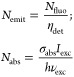 |
2 |
where Nfluo is the measured fluorescence signal from a single conjugated molecule (counts per second); ηdet is the detection efficiency of our current setup and is estimated to be about 13% (see the Supporting Information); σabs is the absorption cross-section of a single MEH-PPV molecule, deduced from the photothermal signal; Iexc and vexc are the excitation intensity at the sample and the excitation frequency; and h is the Planck constant.
In Figure 3b, we plot the histogram of quantum yields for 174 molecules with an excitation intensity of 450 W/cm2. We can see that the quantum yield of these conjugated molecules is around 10%. In this evaluation, we will neglect the small fraction of absorbed energy re-emitted as fluorescence. As the fluorescence quantum yield is about 10% for conjugated polymers, the error involved is at most 10–20%, much less than the experimental accuracy. The measured quantum yield is close to the ensemble results reported in ref (37).
We also had a look at the simultaneous transient traces of absorption and emission from individual MEH-PPV molecules. We measured simultaneous time traces of about 30 molecules and found most of them to show continuous bleaching in both fluorescence and absorption under continuous illumination. One example is shown in Figure 4. A few molecules prepared by spin coating a mixture of PMMA and MEH-PPV in toluene on glass, however, showed clear blinking behavior (see Figure S6 in the Supporting Information).
Figure 4.

Transient time traces of absorption and emission of a single MEH-PPV molecule recorded simultaneously. (a) Fluorescence; (b) absorption cross-section deduced from the photothermal signal. The dashed lines represent the background level. The integration time was 100 ms. (c) Scatter plot of fluorescence and absorption showing the decay of the two signals. Each point is a binned data point where ten successive points in the raw data (a, b) were summed up to reduce the noise. The arrow indicates the time evolution. The red line is the fitted line with slope 1.34.
Two main types of transient fluorescence traces have been reported in the literature.9,14,38 For the first type, single polymer molecules behave like fluorescent beads. They tend to have extended conformations. For these extended molecules, the energy transfer is inefficient, although some energy transfer can happen over a limited range of a few chromophores. Once a trap is formed, it can only quench the few chromophores available within this limited range (a simple scheme is shown in Figure S7 in the Supporting Information). In this case, the fluorescence time traces normally show continuous decrease with the illumination time because of photobleaching. The second type of conjugated polymer fluorescence is attributed to collapsed conformations and is characterized by strong intrachain interactions. Excitons created upon photon absorption by the chromophores can migrate over large distances along the conjugated chain and relax toward low-energy traps, which give rise to the “funneling” effect. In this second case, emission mostly takes place from a few low-energy sites,8 and time traces normally show blinking dynamics. It was also proposed previously that large parts of the collapsed molecule are completely quenched and behave as “dark matter”, while only a few emissive chromophores are left.30 The observed blinking behavior comes then from the blinking of these active chromophores due to energy funneling. The decays shown in Figure 4 seem to point to the first kind, extended conformations with limited energy transfer.
Discussion
In Figure 2, we showed that the absorption and emission of single conjugated polymers can be measured simultaneously in near-critical xenon with the enhanced photothermal microscope, with an excitation power of a few hundred W/cm2. On the basis of the absorption signal, for the first time, we obtained the number of monomers in single MEH-PPV molecules, and evaluated their quantum yields. As mentioned in the Experimental Details, the conjugated polymers present a large distribution in molecular weight. Thus, it is not surprising that the histograms of both the absorption cross-section and the quantum yield show broad maxima as shown in Figure 3. Because of the limited photothermal sensitivity, we could not detect the absorption of molecules with molecular weight smaller than 150 kDa. Figure 3a and b may only partially reflect the molecular weight heterogeneity in the sample. Although we cannot completely rule out that weakly emitting molecules may have been photobleached at the excitation power we used, or that molecules with dense collapsed conformations disappear more rapidly because of early quenching by photoinduced traps, the absence of any weak fluorescence spot in Figure 2b supports our belief that we observe all molecules in the sample. Because of the preparation of our sample, most of these molecules would be in extended conformations, with limited energy transfer only.
We noticed from Figure 2c that for different molecules, fluorescence correlates with absorption/photothermal signal in a sublinear way, even though the excitation power remained the same for all the molecules. Molecules with larger absorption tend to be relatively less florescent than the ones with smaller absorption. This trend becomes even clearer if we look at the scatter plot of the quantum yield against the molecular absorption cross-section, shown in Figure 3c. Each point represents one molecule in the simultaneous measurements. The quantum yield decreases significantly as the absorption cross-section increases. In other words, large molecules have lower fluorescence efficiency than small ones. We can propose two possible explanations for this observation. The first possibility is that molecules with large molecular weights have a larger concentration of chemical or physical defects such as kinks. Indeed, the probability of kinks increases for large molecules due to folding, topological constraints, and hydrodynamic forces they undergo during sample preparation and spin coating. These defects would then more efficiently quench the fluorescence of larger molecules. The second interpretation is that the reduced quantum yield is the result of exciton–exciton annihilation (singlet–singlet or singlet–triplet). Exciton–exciton collisions are obviously more likely in larger polymer molecules because of their larger absorption cross-section and of some limited energy transfer, which leads to a higher annihilation rate and to a lower quantum yield for larger molecules.27,38 Our current results alone do not allow us to distinguish which of these two mechanisms dominates. In principle, the role of exciton–exciton annihilation could be tested experimentally by varying the excitation power. However, such measurements are hampered by the very limited power range that can be used in single-molecule photothermal experiments because of conflicting requirements on signal level and photostability of molecules.
To propose a tentative answer to this question, we measured conjugated polymer films, where interactions between chains are supposed to be even stronger than in isolated molecules. Indeed, emission and absorption of conjugated polymer films are known to depend on excitation power. Before saturation, high excitation rates lead to nonlinear behavior such as exciton–exciton annihilation. We thus measured the fluorescence and photothermal signals with MEH-PPV films, which resist photobleaching better than isolated molecules, and which give good photothermal contrast at lower excitation power. Although the single-molecule and the film samples are very different, and the power dependence of their luminescence and adsorption may be different too, this comparison gives us a rough idea of the range over which absorption and fluorescence vary linearly with excitation power. Figure S3 of the Supporting Information shows a plot of fluorescence and photothermal signals of a MEH-PPV film as functions of the excitation power. Clearly, the power dependence of the two signals starts to deviate from a linear relationship at powers above 500 W/cm2. We attribute the deviation from linearity at higher excitation rates to exciton–exciton annihilation. These processes not only limit fluorescence,27 but also appear to influence the photothermal absorption signal slightly. On the basis of those film measurements, we limited the excitation intensity of single molecules to 500 W/cm2. Therefore, we think it unlikely that the deviation from linearity seen in Figures 2 and 3 is due to exciton–exciton annihilation, and we rather attribute it to conformational defects of the chains. This question will have to be clarified in future investigations.
We now turn to the evolution of absorption and fluorescence signals of single molecules under continuous illumination. As we see from Figure 4, both the fluorescence and the absorption of single conjugated polymer molecules decay with the illumination time. The absorption decay originates from the continuous bleaching of the absorbing units or monomers of single MEH-PPV molecules, and the fluorescence decay might have been expected to follow the absorption. However, fluorescence appears to decay faster than absorption, at least in the first 20 s or so. This is confirmed by the correlated plot of the fluorescence versus the absorption in Figure 4c. There, we binned the data to reduce noise in the traces, and we found a slope larger than 1 (1.34) in log–log scale for the fluorescence versus absorption dependency, that is, a superlinear behavior. Most of the molecules present a similar behavior, as shown in Figure S4 in the Supporting Information for another eight molecules. The superlinear relation between fluorescence and absorption found here differs strikingly from the sublinear one found on varying the molecular size. They must thus arise from very different physical mechanisms. We propose that the superlinear decay in the bleaching experiments results from fluorescence quenching by photoinduced traps. In this case, one trap formed by a photochemical reaction, although decreasing the absorption coefficient very little, can quench several nearby chromophores of the polymer chain by both static and dynamic mechanisms involving intrachain exciton migration,23,30,39 on a time scale shorter than the lifetime of excitons.23 That is why the absorption, being mainly due to all unbleached monomers, decays more slowly with illumination time than the fluorescence, which scales as the number of unquenched chromophores. At very long illumination times, the absorption signal eventually drops close to the background level. Because of the large noise and the inaccuracy of measurements, it is not clear whether the fluorescence and the absorption also decay in different ways for heavily bleached molecules.
Conclusions and Outlook
We measured the absorption and emission of individual conjugated MEH-PPV polymers simultaneously in near-critical xenon with an enhanced photothermal setup. For the first time, we showed direct measurements of the absorption cross-sections, the number of monomers, and the quantum yield of single conjugated polymer molecules. We observed that for larger MEH-PPV molecules, the quantum yield decreases. Complementary experiments on polymer films lead us to propose that conformational defects are mostly responsible for fluorescence quenching in larger molecules. In addition, the fluorescence and the absorption of single conjugated molecules showed different bleaching behaviors, that is, dependence on the illumination time. We attribute this behavior to photoinduced quenchers. The photophysics of conjugated polymers is strongly dependent on the conformation, host matrix, and sample preparation. Future simultaneous absorption and fluorescence transient traces and spectral measurements on different polymers and matrixes will provide more insight into the dependence of the optical properties of these complex systems on molecular conformation and host matrix.
Acknowledgments
We would like to acknowledge Harmen van der Meer and Tina Ding for their efforts in building the pressure cell used in the measurements. L.H. acknowledges the financial support of China Scholarship Council. S.A. and M.O. acknowledge Foundation for Fundamental Research on Matter (FOM), which is part of The Netherlands Organization for Scientific Research (NWO). I.G.S. and Y.T. thank Swedish Research council, Knut & Alice Wallenberg foundation, and Lund Laser Center for support. Y.T. acknowledges National Natural Science Foundation of China (NSFC Grant No. 21673114) and “Dengfeng Program of Nanjing University”.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.nanolett.6b04726.
Optical setup and supporting figures (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Burroughes J. H.; Bradley D. D. C.; Brown A. R.; Marks R. N.; Mackay K.; Friend R. H.; Burns P. L.; Holmes A. B. Nature 1990, 347 (6293), 539–541. 10.1038/347539a0. [DOI] [Google Scholar]
- Günes S.; Neugebauer H.; Sariciftci N. S. Chem. Rev. 2007, 107 (4), 1324–1338. 10.1021/cr050149z. [DOI] [PubMed] [Google Scholar]
- Hu D.; Yu J.; Barbara P. F. J. Am. Chem. Soc. 1999, 121 (29), 6936–6937. 10.1021/ja990139d. [DOI] [Google Scholar]
- Barbara P. F.; Gesquiere A. J.; Park S.-J.; Lee Y. J. Acc. Chem. Res. 2005, 38 (7), 602–610. 10.1021/ar040141w. [DOI] [PubMed] [Google Scholar]
- Hu D.; Yu J.; Wong K.; Bagchi B.; Rossky P. J.; Barbara P. F. Nature 2000, 405 (6790), 1030–1033. 10.1038/35016520. [DOI] [PubMed] [Google Scholar]
- Vacha M.; Habuchi S. NPG Asia Mater. 2010, 2, 134–142. 10.1038/asiamat.2010.135. [DOI] [Google Scholar]
- Orrit M.; Bernard J. Phys. Rev. Lett. 1990, 65 (21), 2716–2719. 10.1103/PhysRevLett.65.2716. [DOI] [PubMed] [Google Scholar]
- Bout D. A. V.; Yip W.-T.; Hu D.; Fu D.-K.; Swager T. M.; Barbara P. F. Science 1997, 277 (5329), 1074–1077. 10.1126/science.277.5329.1074. [DOI] [Google Scholar]
- Huser T.; Yan M.; Rothberg L. J. Proc. Natl. Acad. Sci. U. S. A. 2000, 97 (21), 11187–11191. 10.1073/pnas.97.21.11187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker K.; Lupton J. M. J. Am. Chem. Soc. 2006, 128 (19), 6468–6479. 10.1021/ja0609405. [DOI] [PubMed] [Google Scholar]
- Schindler F.; Lupton J. M.; Feldmann J.; Scherf U. Proc. Natl. Acad. Sci. U. S. A. 2004, 101 (41), 14695–14700. 10.1073/pnas.0403325101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grey J. K.; Kim D. Y.; Norris B. C.; Miller W. L.; Barbara P. F. J. Phys. Chem. B 2006, 110 (51), 25568–25572. 10.1021/jp065990a. [DOI] [PubMed] [Google Scholar]
- Lee Y. J.; Kim D. Y.; Barbara P. F. J. Phys. Chem. B 2006, 110 (20), 9739–9742. 10.1021/jp056727x. [DOI] [PubMed] [Google Scholar]
- Ebihara Y.; Vacha M. J. Phys. Chem. B 2008, 112 (40), 12575–12578. 10.1021/jp806963u. [DOI] [PubMed] [Google Scholar]
- Kobayashi H.; Onda S.; Furumaki S.; Habuchi S.; Vacha M. Chem. Phys. Lett. 2012, 528, 1–6. 10.1016/j.cplett.2011.11.064. [DOI] [Google Scholar]
- Camacho R.; Tubasum S.; Southall J.; Cogdell R. J.; Sforazzini G.; Anderson H. L.; Pullerits T.; Scheblykin I. G. Sci. Rep. 2015, 5, 15080. 10.1038/srep15080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J.; Hu D.; Barbara P. F. Science 2000, 289 (5483), 1327–1330. 10.1126/science.289.5483.1327. [DOI] [PubMed] [Google Scholar]
- Liang J.-J.; White J. D.; Chen Y. C.; Wang C. F.; Hsiang J. C.; Lim T. S.; Sun W. Y.; Hsu J. H.; Hsu C. P.; Hayashi M.; Fann W. S.; Peng K. Y.; Chen S. A. Phys. Rev. B: Condens. Matter Mater. Phys. 2006, 74 (8), 085209. 10.1103/PhysRevB.74.085209. [DOI] [Google Scholar]
- Sahoo D.; Tian Y.; Sforazzini G.; Anderson H. L.; Scheblykin I. G. J. Mater. Chem. C 2014, 2 (32), 6601–6608. 10.1039/C4TC00831F. [DOI] [Google Scholar]
- Müller J. G.; Lemmer U.; Raschke G.; Anni M.; Scherf U.; Lupton J. M.; Feldmann J. Phys. Rev. Lett. 2003, 91 (26), 267403. 10.1103/PhysRevLett.91.267403. [DOI] [PubMed] [Google Scholar]
- Feist F. A.; Basché T. J. Phys. Chem. B 2008, 112 (32), 9700–9708. 10.1021/jp802585m. [DOI] [PubMed] [Google Scholar]
- Feist F. A.; Zickler M. F.; Basché T. ChemPhysChem 2011, 12 (8), 1499–1508. 10.1002/cphc.201001010. [DOI] [PubMed] [Google Scholar]
- Lin H.; Tabaei S. R.; Thomsson D.; Mirzov O.; Larsson P.-O.; Scheblykin I. G. J. Am. Chem. Soc. 2008, 130 (22), 7042–7051. 10.1021/ja800153d. [DOI] [PubMed] [Google Scholar]
- Nguyen T.-Q.; Doan V.; Schwartz B. J. J. Chem. Phys. 1999, 110 (8), 4068–4078. 10.1063/1.478288. [DOI] [Google Scholar]
- Onda S.; Kobayashi H.; Hatano T.; Furumaki S.; Habuchi S.; Vacha M. J. Phys. Chem. Lett. 2011, 2 (21), 2827–2831. 10.1021/jz2012512. [DOI] [Google Scholar]
- Wang D.; Yuan Y.; Mardiyati Y.; Bubeck C.; Koynov K. Macromolecules 2013, 46 (15), 6217–6224. 10.1021/ma4011523. [DOI] [Google Scholar]
- Yu J.; Lammi R.; Gesquiere A. J.; Barbara P. F. J. Phys. Chem. B 2005, 109 (20), 10025–10034. 10.1021/jp0506742. [DOI] [PubMed] [Google Scholar]
- Ebihara Y.; Vacha M. J. Chem. Phys. 2005, 123 (24), 244710. 10.1063/1.2140706. [DOI] [PubMed] [Google Scholar]
- Shinohara K.-i.; Kitami T.; Nakamae K. J. Polym. Sci., Part A: Polym. Chem. 2004, 42 (16), 3930–3935. 10.1002/pola.20247. [DOI] [Google Scholar]
- Lin H.; Tian Y.; Zapadka K.; Persson G.; Thomsson D.; Mirzov O.; Larsson P.-O.; Widengren J.; Scheblykin I. G. Nano Lett. 2009, 9 (12), 4456–4461. 10.1021/nl9027473. [DOI] [PubMed] [Google Scholar]
- Tian Y.; Halle J.; Wojdyr M.; Sahoo D.; Scheblykin I. G. Methods Appl. Fluoresc. 2014, 2 (3), 035003. 10.1088/2050-6120/2/3/035003. [DOI] [PubMed] [Google Scholar]
- Boyer D.; Tamarat P.; Maali A.; Lounis B.; Orrit M. Science 2002, 297 (5584), 1160–1163. 10.1126/science.1073765. [DOI] [PubMed] [Google Scholar]
- Gaiduk A.; Yorulmaz M.; Ruijgrok P. V.; Orrit M. Science 2010, 330 (6002), 353–356. 10.1126/science.1195475. [DOI] [PubMed] [Google Scholar]
- Ding T. X.; Hou L.; Meer H. v. d.; Alivisatos A. P.; Orrit M. J. Phys. Chem. Lett. 2016, 7, 2524–2529. 10.1021/acs.jpclett.6b00964. [DOI] [PubMed] [Google Scholar]
- Gaiduk A.; Ruijgrok P. V.; Yorulmaz M.; Orrit M. Chem. Sci. 2010, 1 (3), 343–350. 10.1039/c0sc00210k. [DOI] [PubMed] [Google Scholar]
- Mirzov O.; Scheblykin I. G. Phys. Chem. Chem. Phys. 2006, 8 (47), 5569–5576. 10.1039/b612073c. [DOI] [PubMed] [Google Scholar]
- Wu C.; Bull B.; Szymanski C.; Christensen K.; McNeill J. ACS Nano 2008, 2 (11), 2415–2423. 10.1021/nn800590n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaiduk A.; Yorulmaz M.; Ishow E.; Orrit M. ChemPhysChem 2012, 13 (4), 946–951. 10.1002/cphc.201100788. [DOI] [PubMed] [Google Scholar]
- Camacho R.; Thomsson D.; Sforazzini G.; Anderson H. L.; Scheblykin I. G. J. Phys. Chem. Lett. 2013, 4 (6), 1053–1058. 10.1021/jz400142x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.