

Abstract
Background
Short linear motifs in host organisms proteins can be mimicked by viruses to create protein-protein interactions that disable or control metabolic pathways. Given that viral linear motif instances of host motif regular expressions can be found by chance, it is necessary to develop filtering methods of functional linear motifs. We conduct a systematic comparison of linear motifs filtering methods to develop a computational approach for predicting motif-mediated protein-protein interactions between human and the human immunodeficiency virus 1 (HIV-1).
Results
We implemented three filtering methods to obtain linear motif sets: 1) conserved in viral proteins (C), 2) located in disordered regions (D) and 3) rare or scarce in a set of randomized viral sequences (R). The sets C,D,R are united and intersected. The resulting sets are compared by the number of protein-protein interactions correctly inferred with them – with experimental validation. The comparison is done with HIV-1 sequences and interactions from the National Institute of Allergy and Infectious Diseases (NIAID).
The number of correctly inferred interactions allows to rank the interactions by the sets used to deduce them: D∪R and C. The ordering of the sets is descending on the probability of capturing functional interactions.
With respect to HIV-1, the sets C∪R, D∪R, C∪D∪R infer all known interactions between HIV1 and human proteins mediated by linear motifs. We found that the majority of conserved linear motifs in the virus are located in disordered regions.
Conclusion
We have developed a method for predicting protein-protein interactions mediated by linear motifs between HIV-1 and human proteins. The method only use protein sequences as inputs. We can extend the software developed to any other eukaryotic virus and host in order to find and rank candidate interactions. In future works we will use it to explore possible viral attack mechanisms based on linear motif mimicry.
Electronic supplementary material
The online version of this article (doi:10.1186/s12859-017-1570-7) contains supplementary material, which is available to authorized users.
Keywords: Virus, Host, Eukaryotes, Protein, Interaction, Prediction, Short, Linear, Motif, Disorder
Background
Virus-host Protein-protein interactions (VHPPIs) are essential to understand viral attack mechanisms. VHPPIs are used by viruses to disrupt or modulate host pathways in order to achieve goals like the evasion of the complement system [1], modulation of the cytokine system [2] and abrogation of apoptosis [3]. Some of these PPIs are based on mimicry: a viral protein mimicking a host protein might interact with the host protein binding partners. The mimicry is achieved through protein sequence or structural similarity [4]. We focus our study on predicting a subset of PPIs, the ones mediated by mimicked short linear motifs (SLiMs). SLiM-mediated PPI predictions, conveniently ranked, might help researchers to postulate hypothesis to elucidate viral attack mechanisms, design antivirals and vaccines [5–8].
A Short linear motif (SLiM) (also called linear motif, minimotif, ELM, LM) is a short region of a protein, 3 to 12 residues long, with functions like controlling the assembly of protein complexes, marking proteolytic cleavage, tagging protein localization and enzyme recruiting [9, 10]. SLiMs are structurally compact and participate in transitory low-affinity interactions [11, 12]. SLiMs in eukaryotic proteins are curated in the ELM database [13, 14].
SLiMs might evolve rapidly in viral disordered regions through insertions, deletions and mutations [15]. The new SLiMs can change the PPI networks creating new advantageous PPIs that can alter the cell cycle [16], form protein complexes and mediate conformational changes [17]. A recent analysis of the experimentally inferred human-virus PPIs concludes that human proteins interacting with viruses are enriched in SLiMs and binding interfaces [18].
Viruses use VHPPIs mediated by host-mimicked SLiMs to hijack cell regulation [19] and execute their viral cycle [20]. An example of this strategy is the set of SLiM-mediated interactions of human papilloma virus (HPV) protein E6 with members of the 14-3-3 protein family and proteins containing PDZ domains [21].
Experimental determination of VHPPIs is expensive since the number of proteins for some host organisms is large, more than 30.000 in humans. There are many viral protein sequences available but few corresponding three-dimensional structures resolved to use structure-based interaction prediction methods. These are reasons for developing a general method for predicting mimetic host-virus PPIs based solely in sequence data. A bioinformatic approach to predict SLiM-mediated VHPPIs might be an inexpensive alternative to experimentation or can guide experimental design.
SLiMs are represented computationally as regular expressions. A SLiM instance is a protein subsequence that matches the regular expression. For instance, a SLiM represented by the regular expression R.[RK]R. have several instances like RVRRE in Ebola virus [22] and RKRRF in Human respiratory syncytial virus A2 [23]. An algorithm for predicting virus-host PPIs consist in finding viral instances of SLiMs located in host proteins. The viral instances found need to be filtered by some criteria that increase the probability of inferring real interactions.
If a SLiM is conserved in a small viral genome it probably could be used to interact with a host protein. Evans et al. find that common SLiMs between HIV-1 and humans are significantly conserved in HIV-1 proteins [24]. They propose a criterion to filter SLiMs if they are conserved above a 70% in the available viral sequences.
Viral genomes have high mutation rates and are not too thermodynamically stable. This seems to favor protein structures with a small number of inter-residue interactions and a high number of polar residues that account for the abundance of disordered protein regions [25]. SLiMs occur more frequently in viral protein disordered regions [26], in different amounts between viral families [27]. Viral hubs, proteins that have many interactions with host proteins, tend to have more disordered regions [28]. With these antecedents Hagai et al. propose a criterion to filter SLiMs based on location in protein disordered regions [26].
Hagai et al. also propose another criterion to filter SLiMs based on rarity in a big set of randomized proteins [29]. A SLiM is judged as rare, or hard to form by pure chance, if it is counted in less than a percentage of the sequences in the set of randomized proteins, e.g. 1% of the sequences. Hagai et al. find that rare SLiMs located in disordered regions have a significant enrichment in functional SLiMs i.e. with experimental evidence for interaction with host proteins [29].
To our knowledge, there is no comparison of SLiM filtering methods in the literature. For that reason, we implement and compare the three criteria introduced above for SLiM filtering: conservation above a threshold of the available viral sequences, localization in a protein disordered region and rarity, or difficulty to form by pure chance. Each filtering method produces a set of SLiMs – conserved (C), disordered (D) and rare (R). With sets C,D,R we form derived union and intersection sets: C∪D, C∪R, D∪R, C∪D∪R and C∩D, C∩R, D∩R, C∩R∩D. Each of these sets allow us to predict interactions between the viral protein containing the SLiM and the host proteins that interact with the SLiM.
All the sets generated are compared by filtering strength. They also are compared by the number of VHPPIs derived from the set that have supporting evidence in a database –i.e. correctly predicted. The comparison by number of VHPPIs correctly predicted by set allow us to rank the VHPPIs partially.
To conduct the comparison of the sets we choose the Human immunodeficiency virus (HIV-1). It is the virus with more bioinformatic data available, with the NIAID databases for sequences and alignments [30] and for interactions with human proteins [31]. We also use the HIV-1-human PPIs mediated by SLiMs as reported in the LMPID database [32].
Methods
Disorder prediction
Protein preprocessing
We download alignments for HIV-1 proteins env, gag, nef, pol, rev, vif, vpr, tat, vpu for the year 2014 and an alignment of Gag-Pol DNA sequences with years previous to 2015 from the NIAID HIV-1 sequence database [30]. Gag-Pol sequences were translated following reference [33]: of 3648 sequences, 3626 containing the slippery subsequence TTTTTTA were used to perform a computational translation considering the frameshift at the given subsequence.
We filter all protein sequences by HIV-1 subtypes B and C for their worldwide dominance and computationally cleave some of the alignments in the following manner: env into gp120, gp41, pol into pr, rt, rtp51, in and gag into ma, ca, p2, nc, p1, p6 [31]. After the cleavage we eliminate the gaps and asterisks in the resulting alignments in order to reinterpret the files as sets of sequences, Fig. 1, Disorder panel. The number of sequences per HIV-1 protein is in Additional file 1: Table S1.
Fig. 1.
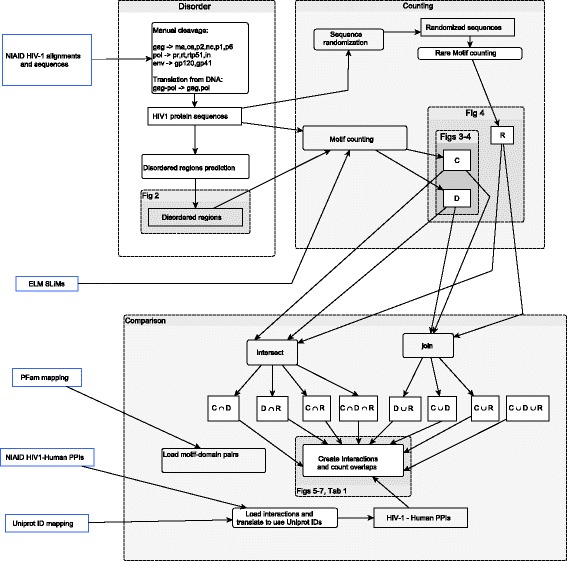
Methodology. The methods are divided in three parts: 1) Disorder: sequence preprocessing and prediction of disordered regions, 2) Counting: counting of SLiM patterns and instances, and 3) Comparison: analysis of the overlap between predicted interactions against interactions in NIAID and LMPID databases
Protein disorder prediction with IUPred
Among several disorder prediction algorithms for proteins [34] we use IUPred [35]. This predictor implements a physical model based on force fields between residues statistically calibrated with a set of globular proteins in PDB [35]. Its performance is comparable to other predictors [36] and can be installed locally.
IUPred is enhanced with a sliding window addition proposed by Hagai et al. that allows to define disordered regions [37]. Residues with IUPred computed values higher that 0.4 are considered disordered. For each residue an average disorder value is computed considering the IUPred values for surrounding residues in a window of size 10. This averaging is justified because the disorder tendency of the neighbors of a residue influence its disorder tendency. Residue windows with average disorder value higher than 0.4 are considered as disordered.
As IUPred receives as input a Fasta file with only one sequence, we split Fasta files with multiple sequences, call IUPred on every split sequence-file, compute the sliding window based average values and give as output a list of disordered regions per protein sequence id. We set the parameter long when calling IUPred, see Fig. 1, Disorder panel.
Protein randomization
We randomize the HIV-1 proteins to create a big data set. For each sequence in a protein file we create 1000 shuffled versions randomizing the residues located in disordered regions of the sequence, as computed with IUPred. All disordered residues in a protein are joined together in a temporary list, shuffled with the modern Fisher–Yates algorithm, and put back in the disordered regions, leaving the ordered residues intact.
SLiM counting
We download all the SLiMs, instances and interactions from the ELM database [14] and create an in-memory ELM data structure with each SLiM identifier, its regular expression, its instances and its interactions with protein domains. We wrote scripts to compute: the number of sequences with a given SLiM, the number of SLiM instances per protein, the number of SLiMs conserved above a percentage of sequences (set C) and the number of SLiMs in disordered regions (set D).
After randomizing as described above, we count the rare (scarce) SLiMs in these shuffled data set, i.e. the SLiMs that are found in 1% of the randomized sequences or less (set R).
Based on C, D, R we create the union sets C∪D, C∪R, D∪R, C∪D∪R and intersection sets C∩D, C∩R, D∩R, C∩R∩D. See Fig. 1, panels Counting and Comparison.
Prediction of protein-protein interactions
We download the NIAID human–HIV-1 PPI database [31]. As the proteins in the database are identified by RefSeq records and the SLiM-domain interactions given by the ELM database are given by UniProt records, we map RefSeq to UniProt identifiers for human proteins using UniProt id mappings. We also download the LMPID database that curates virus-host ELM-mediated interactions [32].
For each SLiM set (C,D,R,…) obtained per HIV-1 protein we create VHPPIs based on the ELM database interactions and interacting domains. For each interaction reported in ELM we add the human protein interacting with the SLiM located in the viral protein. We also add the proteins that contain the domains listed as interacting with the SLiM. To map domains to human proteins we used the domain-protein mapping for the human proteome in the PFAM ftp server [38]. Figure 1, Comparison panel.
Comparison of filtering methods
To validate a prediction we use two sets: the NIAID HIV-1-human interactions and the set of ELM mediated HIV-1-human interactions, as identified in LMPID [32]. We count the number of correctly predicted interactions, when an interaction deduced with one of the SLiM sets is in the NIAID database.
For all the SLiM sets obtained, and all the HIV-1 proteins, we analyze the overlap between the set of predicted human proteins interacting with HIV-1 and the set human proteins in NIAID interactions. We compute p-values for this overlap using the hyper-geometric distribution from the scipy python library, Table 1. The total number of human proteins was estimated as 30,057 from reference [39].
Table 1.
P-values for the overlap between predicted interactions and NIAID PPIs
|
|
|||||||||
The p-value indicates the probability that the overlap between our sets of predicted PPIs and the PPIs with literature support in the NIAID database takes place under the null hypothesis, that our sets were formed by random sampling. Red values are not significant at a level of 0.05
Results and discussion
A general method to identify SLiM-mediated PPIs in eukaryotes
As SLiMs are computationally represented by regular expressions there is always a possibility of finding instances in viral sequences by pure chance. For this reason, it is important to develop SLiM filtering methods.
Three filtering methods are implemented and systematically compared: conservation, location in disordered regions and rarity. The combination of filters produces a method to predict virus-host PPIs and rank them. The comparison of filtering methods performance is conducted with the virus with more abundant data, HIV-1. In Fig. 1 there is an overview of the methods used.
The developed method only use protein sequences as input and do not depend on protein 3D structures, for this reason it can be used with any sequenced eukaryotic virus to infer candidate VHPPIs. The restriction to eukaryotic viruses is based on the higher number of SLiMs in eukaryotes and the use of the ELM database, because the ELM SLiM classes MOD (post-translational modification) and TRG (targeting sites) are less used in prokaryotes [29].
Candidate interactions
The lists of predicted human-HIV-1 interactions that are not in the NIAID database are in the [Additional file 1].
Disordered regions and SLiMs in HIV-1 proteins
The disordered regions for HIV-1 proteins are in the [Additional file 2: Table S2]. They are depicted in Fig. 2. Subfigures A to U show the predicted disordered content in HIV-1 proteins and polyproteins. Each protein sequence is represented as a yellow line and disordered regions are depicted as red segments.
Fig. 2.

Disordered regions for HIV-1 proteins. Each subfigure from A to U contains an HIV-1 protein or protein precursor. For all subfigures, each yellow line represents a protein sequence. The red segments represent disordered regions as deduced with IUPred with the sliding window addition explained in the “Methods” section
We find that predicted disordered regions for HIV-1 proteins are relatively conserved. Perhaps the virus must keep flexibility in their proteins in order to interact with several partners.
In Fig. 3, we show the percentage of SLiMs conserved above a 70% of the input sequences that are also located inside a disordered region. Most of the conserved SLiMs in HIV-1 are located in protein disordered regions.
Fig. 3.
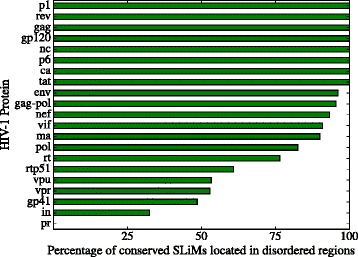
Percentage of conserved SLiMs that are located in disordered regions in HIV-1. We plot the percentage of conserved SLiMs, present in 70% or more of the input sequences, that are localized in a predicted disordered region
The proteins that deviate the most from this tendency are vpr, vpu, gp41, in, and pr, with a percentage of conserved motifs that are located in disordered regions of 53.3, 52.9, 48.5, 32.5 and 0% respectively. The reason for this discrepancy lies in the few disordered regions predicted in the five proteins. Indeed, pr, in and gp41 are considered mostly ordered, while vpr and vpu are considered moderately disordered [40].
A similar correlation between evolutionary conservation and location in disordered regions was found for the SLiMs that bind to SH2, SH3 and Ser/Thr Kinase domains [41].
We use IUPred as disorder predictor only because its performance finding the disordered regions of the VIF protein is outstanding compared to other 18 disorder predictors [36]. One procedure that could be used to avoid structured regions entirely is a BLAST query against HIV-1 proteins in the Protein Data Bank excluding hit regions. However, it seems that disorder is a viral strategy to buffer mutations and increase interactions with host proteins [42]. In this perspective, small disordered regions could be located inside structured protein regions to allow some interactions with the host, and not excluding the structured regions opens the possibility of finding these regions.
Analysis of SLiM sets obtained
A ranking of SLiM sets by filtering strength
The SLiM set sizes are in Additional file 3: Table S3 and the SLiM sets for HIV-1 proteins are in the [Additional file 3]. In Fig. 4 we plot the the number of SLiM regular expressions that were found in the HIV-1 proteins identified by set. The intersection SLiM sets C∩R (conserved and rare) and C∩D∩R (conserved, rare, located in disordered regions) were discarded for being almost empty for all proteins.
Fig. 4.
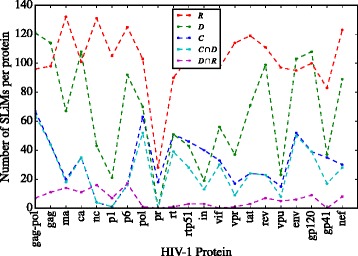
Number of SLiMs by set. We plot the number of SLiMs (regular expressions) that were found in HIV-1 proteins. The intersection SLiM sets C∩R (conserved and rare) and C∩D∩R (conserved, rare, located in disordered regions) were discarded for being almost null in all entries
Considering the sizes of SLiM sets we can rank them by the filtering strength; from low to high filtering. The obtained ranking is R,D,C,C∩D,D∩R. The criterion that filters the most is location in a disordered region and rarity. It is followed by location in a disordered region and conservation.
The sets D∩R (SLiMs hard to form by pure chance and located in protein disordered regions) studied by Hagai et al. [29], tend to have a smaller size than sets C∩D, of SLiMs conserved and located in protein disordered regions, Fig. 4. The intersection SLiM sets C∩R (conserved and rare) and C∩D∩R (conserved, rare, located in disordered regions) are almost empty so they can be discarded as useful filtering criteria –data in Additional file 3: Table S3.
Protein-protein interactions predicted with the SLiM sets are enriched in experimentally validated HIV-1-human protein-protein interactions
We validate against two virus-host PPIs databases: NIAID [31] and LMPID [32]. The NIAID contains 15074 PPIs at the moment of writing while LMPID contains 2203 PPIs between several viruses and hosts, with 6 PPIs between HIV-1 and human proteins.
The validation of the predicted PPIs with the NIAID database is not the best way to gauge the proportion of SLiM-based interactions. This database contains PPIs of all kinds, not only SLiM-mediated ones. However, it is the most complete virus-host PPI dataset.
A better validation set, conceptually, is constructed with pairs deemed to interact through a SLiM with the LMPID database. Nevertheless, this dataset is too small. We do the comparison with both databases, selecting the NIAID database to compare the sets prediction performance and check the statistical significance of the results.
Although we are suggesting a partial ranking of SLiM-based predicted PPIs, another addition would be to rank totally the interactions with a score representing the probability that the interaction takes place based on experimental data [43] or other techniques [44]. For the moment, a total ranking is difficult to achieve given the scarcity of data about SLiM-mediated PPIs [32, 45].
In the NIAID database
In Fig. 5 we plot the percentage of correctly predicted interactions, i.e. stored in the NIAID database and predicted with base on our SLiM sets. In Fig. 6 we plot the number of interactions predicted against the total number of interactions in the NIAID database per HIV-1 protein. The number of correctly predicted interactions is in Additional file 4: Table S4 and the number of novel interactions found with our method is in Additional file 4: Table S5.
Fig. 5.
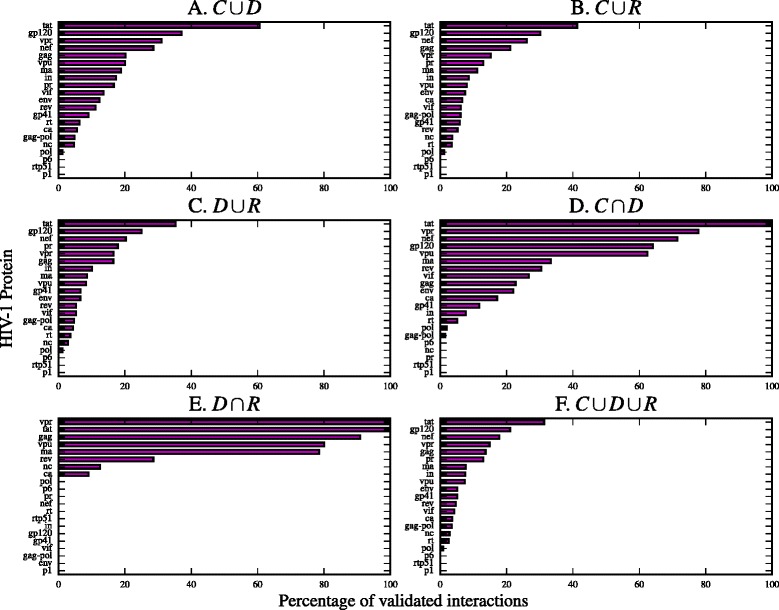
Percentage of validated interactions per SLiM set. Each subfigure plots the percentage of validated interactions with a SLiM set. Interactions were validated with the NIAID HIV-1 Human Interaction Database. The percentage of predicted interactions is represented with a magenta bar. The HIV-1 proteins are sorted by percentage in each subfigure
Fig. 6.
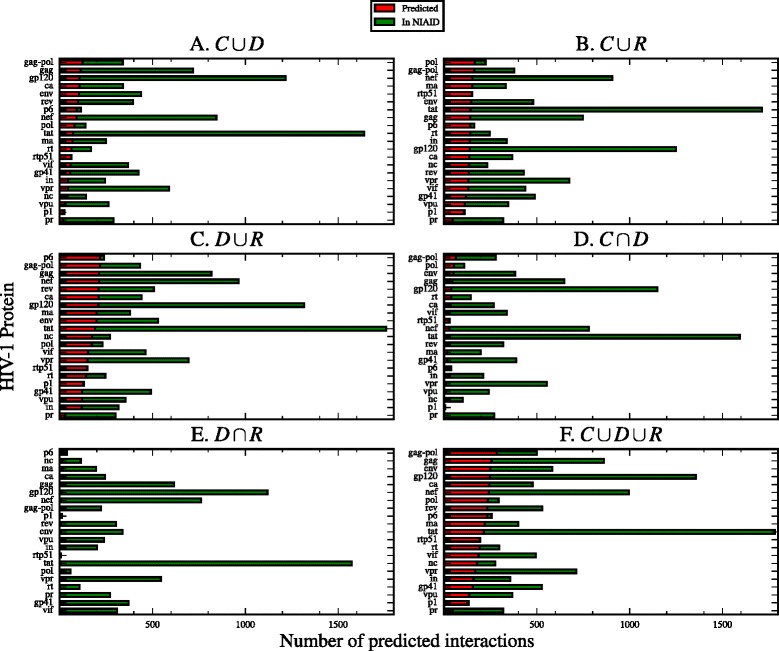
Number of predicted interactions per SLiM set. The number of predicted interactions by SLiM set, contrasted with the number of interactions in the NIAID HIV-1 Human Interaction Database. The number of predicted interactions is represented with a red bar. The green bar represents the total number of interactions in the NIAID database
We use the hyper-geometric distribution to measure the statistical significance of the sets of interactions we found. The p-values for the overlap between the PPIs predicted with base on each SLiM set and the PPIs in the NIAID database are in Table 1. The sensitivity and specificity for the SLiM sets as PPI predictors is in Additional file 4: Table S7 and Additional file 4: Table S8.
In the LMPID database
Using the literature curated LMPID database [32], we find that the motif sets C,C∩D,C∩R,C∩D∩R capture half of the interactions in LMPID, while the sets C∪D,C∪R,D∪R,C∪D∪R,D∪R allow to infer all of them. All the interactions between HIV-1 and human extracted from LMPID are in Additional file 4: Table S6.
The small number of human-HIV-1 interactions in this database (six), leaves open two possibilities: the number is really small, or the number is larger but few experiments have been performed to detect them. To estimate the number of human-HIV-1 SLiM-mediated PPIs more work is needed, perhaps an approach based on combining expert opinions [46].
PPIs correctly predicted serve as a ranking of filtering methods
In Fig. 7 we plot the number of predicted interactions correctly validated against the NIAID database identified by the SLiM set used to infer them. We find that the SLiM sets have an almost general tendency with respect to the number of PPIs correctly predicted across all HIV-1 proteins. For this reason we propose to rank the PPIs predicted according to the set used to deduce them.
Fig. 7.
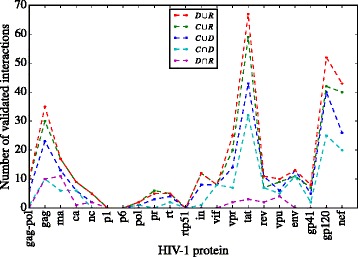
Number of validated interactions per set. The number of interactions as validated with the NIAID HIV-1 Human Interaction Database. Each set is represented by a dashed line with a different color
The ranking of the sets we found by its capacity to infer real interactions is: D∪R,C∪R,C∪D,C∩D,D∩R. This ranking allow to present the PPIs predicted to researchers in a partial order: first the set of interactions deduced with D∪R –SLiMs located in disordered regions or hard to form by pure chance, then the set deduced with C –conserved SLiMs.
Most used SLiMs in HIV-1 proteins suggest HIV-1 extensive use of human protein signaling and other processes
We consider the set C∩D of SLiMs conserved and located in disordered regions for their biological relevance to analyze their human counter domains. In Table 2 we include the most used SLiMs from this set, i.e the SLiMs that are present in 10 or more HIV-1 proteins, are conserved, and localize in a disordered region. In general, most of these SLiMs would interfere with host signaling. The most used counter domains are the Protein kinase domain (PF00069 in Pfam) that interact with 5 of the most used SLiMs and the Peptidase_S8 Subtilase family (PF00082 in Pfam) that interacts with 2 of the most used cleavage SLiMs. However, the list of counter domains in Table 2 suggest that HIV-1 SLiMs interfere with transcription regulation, autophagy, cell cycle control, apoptosis and cellular transport.
Table 2.
Most used SLiMs conserved and located in HIV-1 disordered regions
| SLiM | #HIV-proteins | Pfam domain | Domain name |
|---|---|---|---|
| LIG_WD40_WDR5_VDV_2 | 17 | IPR017986 | WD40-repeat-containing domain |
| DOC_USP7_1 | 17 | PF00917 | MATH domain |
| CLV_NRD_NRD_1 | 16 | PF00675 | Insulinase (Peptidase family M16) |
| CLV_PCSK_KEX2_1 | 15 | PF00082 | Peptidase_S8 Subtilase family |
| MOD_GSK3_1 | 15 | PF00069 | Protein kinase domain |
| MOD_PIKK_1 | 15 | PF00454 | Phosphatidylinositol 3- and 4-kinase |
| CLV_PCSK_SKI1_1 | 14 | PF00082 | Peptidase_S8 Subtilase family |
| LIG_SH3_3 | 14 | PF00018 | SH3 domain |
| MOD_NEK2_1 | 13 | PF00069 | Protein kinase domain |
| LIG_FHA_2 | 13 | PF00498 | FHA domain |
| MOD_CK1_1 | 13 | PF00069 | Protein kinase domain |
| LIG_FHA_1 | 12 | PF00498 | FHA domain |
| DOC_CYCLIN_1 | 12 | PF00134 | Dynein light chain type 1 |
| LIG_LIR_Nem_3 | 12 | PF02991 | Autophagy protein Atg8 ubiquitin like |
| DOC_WW_Pin1_4 | 12 | PF00397 | WW domain |
| MOD_ProDKin_1 | 12 | PF00069 | Protein kinase domain |
| MOD_PKA_2 | 12 | PF00069 | Protein kinase domain |
| MOD_CK2_1 | 12 | PF00069 | Protein kinase domain |
| TRG_ER_diArg_1 | 12 | PF00400 | WD domain, G-beta repeat |
| LIG_SH2_STAT5 | 11 | PF00017 | SH2 domain |
| LIG_LIR_Gen_1 | 11 | PF02991 | Autophagy protein Atg8 ubiquitin like |
| CLV_PCSK_PC1ET2_1 | 11 | PF00082 | Peptidase_S8 Subtilase family |
| MOD_GlcNHglycan | 11 | PF01048 | Phosphorylase superfamily |
| MOD_N-GLC_1 | 10 | PF02516 | Oligosaccharyl transferase STT3 subunit |
| TRG_ENDOCYTIC_2 | 10 | PF00928 | Adaptor complexes medium subunit family |
From the SLiMs that are conserved in more than 70% of the HIV-1 protein sequences, and are located in disordered regions too we counted how many HIV-1 proteins include them. In this table we report the SLiMs more commonly used in HIV-1 proteins, the ones that are included in 10 or more of the HIV-1 proteins. The table includes the counter domain for every SLiM
Conclusion
We develop a method to predict virus-host SLiM-mediated PPIs and rank them. It is applicable to any eukaryotic virus and host with available protein sequences. Using data for the most studied virus, HIV-1, we find a partial ordering of the PPIs obtained based on the set used to infer the interactions. This order is descending in the expected probability of inferring real interactions. We expect that the method gives interesting candidate interactions with other eukaryotic viruses and hosts. The call for using high-throughput methods to detect SLiM-mediated PPIs illustrates the benefits of a bioinformatic method that predicts SLiM-mediated PPIs and might guide experimental design [45].Although the number of SLiM-mediated PPIs might be small, there is evidence that these PPIs are used by several viruses, in contrast to virus-host domain-domain PPIs, that are virus-specific [18]. This kind of interactions can help to analyze common viral strategies for infection.
Indeed, in a previous work we used the method with the viruses in the NCBI virus variation resource to predict interactions with the proteins from the human protein synthesis machinery [47]. We found evidence that viruses interact with Eukaryotic initiation factors 3 and 4, and the Poly(A)-binding proteins using SLiMs. Even though the method developed is not a strong predictor, by using several viruses, interesting interactions with host subsystems can be uncovered. In a future work we want to scale the approach considering all the human proteome and more human viruses.
In future work we could also incorporate structural information in the prediction and analysis of SLiM-mediated VHPPIs in order to create otter SLiM filtering methods and compare them with the filters obtained in this work. One possibility is the study of fuzziness and SLiM flanking regions [48], another one is the use of disordered binding region prediction methods, like ANCHOR [49].
Acknowledgments
The authors would like to thank Dr. Aydin Tozeren for receiving AB in the Biomedical Engineering Department at Drexel university, and Drs. Irene Tischer and Angel García for their valuable input and comments at initial stages of the research.
Funding
This study was supported by a Colciencias scholarship granted to AB and the Escuela de Ingeniería de Sistemas y Computación de la Facultad de Ingeniería de la Universidad del Valle.
Availability of data and materials
The datasets generated during the current study are available in the figshare repository, with links in the Additional Files section.
Authors’ contributions
This study was conceived by AB with significant input to intellectual content at all stages from PM and VB. AB executed the model operations. All authors worked on and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Abbreviations
- ELM
Eukaryotic linear motifs
- HIV-1
Human immunodeficiency virus - 1
- LMPID
Linear motif mediated protein interaction database
- NIAID
National institute of allergy and infectious diseases
- PDB
Protein data bank
- Pfam
Protein families database
- PPI
Protein-protein interaction
- SLiM
Short linear motif
- VHPPI
Virus-host protein-protein interaction
Additional files
Table S1. Candidate interactions between human and HIV-1 (interactions.zip) available at https://figshare.com/articles/interactions_zip/4648714. (ZIP 2549.76 kb)
Table S2. Disordered regions for HIV-1 proteins (regions.zip), using filenames layout explained in Table S2 available at https://figshare.com/articles/Disordered_regions_predicted_in_HIV-1/4648729. (ZIP 708 kb)
Table S3. SLiM sets C,D,R and derived for HIV-1 proteins. (SLiM_sets.zip) available at https://figshare.com/articles/Short_Linear_Motif_Sets_common_to_human_and_HIV-1/4648732. (ZIP 145 kb)
Table S4. Supplement information describing the previous files. (Supplement - Prediction of Virus-Host Protein-Protein interactions based on Short Linear Motifs.pdf) available at https://figshare.com/articles/Supplement_-_Prediction_of_Virus-Host_Protein-Protein_interactions_based_on_Short_Linear_Motifs/4667461. (PDF 166 kb)
Contributor Information
Andrés Becerra, Email: andres.becerra.sandoval@correounivalle.edu.co.
Victor A. Bucheli, Email: victor.bucheli@correounivalle.edu.co
Pedro A. Moreno, Email: pedro.moreno@correounivalle.edu.co
References
- 1.Ahmad M, Pyaram K, Mullick J, Sahu A. Viral complement regulators: the expert mimicking swindlers. Indian J Biochem Biophys. 2007;44(5):331–43. [PubMed] [Google Scholar]
- 2.Alcami A. Viral mimicry of cytokines, chemokines and their receptors. Nat Rev Immunol. 2003;3(1):36–50. doi: 10.1038/nri980. [DOI] [PubMed] [Google Scholar]
- 3.Hasnain SE, Begum R, Ramaiah KV, Sahdev S, Shajil EM, Taneja TK, et al. Host-pathogen interactions during apoptosis. J Biosci. 2003;28(3):349–58. doi: 10.1007/BF02970153. [DOI] [PubMed] [Google Scholar]
- 4.Elde NC, Malik HS. The evolutionary conundrum of pathogen mimicry. Nat Rev Microbiol. 2009;7(11):787–97. doi: 10.1038/nrmicro2222. [DOI] [PubMed] [Google Scholar]
- 5.de Chassey B, Meyniel-Schicklin L, Aublin-Gex A, Andre P, Lotteau V. New horizons for antiviral drug discovery from virus-host protein interaction networks. Curr Opin Virol. 2012;2(5):606–13. doi: 10.1016/j.coviro.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 6.Zoraghi R, Reiner NE. Protein interaction networks as starting points to identify novel antimicrobial drug targets. Curr Opin Microbiol. 2013;16(5):566–72. doi: 10.1016/j.mib.2013.07.010. [DOI] [PubMed] [Google Scholar]
- 7.Dömling A, Mannhold R, Kubinyi H, Folkers G. Methods and Principles in Medicinal Chemistry. Weinheim: Wiley; 2013. Protein-Protein Interactions in Drug Discovery. [Google Scholar]
- 8.Ma-Lauer Y, Lei J, Hilgenfeld R, von Brunn A. Virus-host interactomes–antiviral drug discovery. Curr Opin Virol. 2012;2(5):614–21. doi: 10.1016/j.coviro.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Van Roey K, Uyar B, Weatheritt RJ, Dinkel H, Seiler M, Budd A, et al. Short linear motifs: ubiquitous and functionally diverse protein interaction modules directing cell regulation. Chem Rev. 2014;114(13):6733–78. doi: 10.1021/cr400585q. [DOI] [PubMed] [Google Scholar]
- 10.Diella F, Haslam N, Chica C, Budd A, Michael S, Brown NP, et al. Understanding eukaryotic linear motifs and their role in cell signaling and regulation. Front Biosci J Virtual Libr. 2008;13:6580–603. doi: 10.2741/3175. [DOI] [PubMed] [Google Scholar]
- 11.Davey NE, Van Roey K, Weatheritt RJ, Toedt G, Uyar B, Altenberg B, et al. Attributes of short linear motifs. Mol Biosyst. 2012;8(1):268–81. doi: 10.1039/C1MB05231D. [DOI] [PubMed] [Google Scholar]
- 12.Perkins JR, Diboun I, Dessailly BH, Lees JG, Orengo C. Transient protein-protein interactions: structural, functional, and network properties. Structure. 2010;18(10):1233–43. doi: 10.1016/j.str.2010.08.007. [DOI] [PubMed] [Google Scholar]
- 13.Dinkel H, Michael S, Weatheritt RJ, Davey NE, Van Roey K, Altenberg B, et al. ELM–the database of eukaryotic linear motifs. Nucleic Acids Res. 2012;40(Database issue):D242–251. doi: 10.1093/nar/gkr1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dinkel H, Van Roey K, Michael S, Davey NE, Weatheritt RJ, Born D, et al. The eukaryotic linear motif resource ELM: 10 years and counting. Nucleic Acids Res. 2014;42(Database issue):D259–266. doi: 10.1093/nar/gkt1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davey NE, Cyert MS, Moses AM. Short linear motifs - ex nihilo evolution of protein regulation. Cell Commun Signal. 2015;13(1):43. doi: 10.1186/s12964-015-0120-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berlow RB, Dyson HJ, Wright PE. Functional advantages of dynamic protein disorder. FEBS Lett. 2015;589(19 Pt A):2433–40. doi: 10.1016/j.febslet.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Latysheva NS, Flock T, Weatheritt RJ, Chavali S, Babu MM. How do disordered regions achieve comparable functions to structured domains? Protein Sci. 2015;24(6):909–22. doi: 10.1002/pro.2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Halehalli RR, Nagarajaram HA. Molecular principles of human virus protein-protein interactions. Bioinformatics. 2015;31(7):1025–33. doi: 10.1093/bioinformatics/btu763. [DOI] [PubMed] [Google Scholar]
- 19.Davey NE, Trave G, Gibson TJ. How viruses hijack cell regulation. Trends Biochem Sci. 2011;36(3):159–69. doi: 10.1016/j.tibs.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 20.Kadaveru K, Vyas J, Schiller MR. Viral infection and human disease–insights from minimotifs. Front Biosci. 2008;13:6455–471. doi: 10.2741/3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ganti K, Broniarczyk J, Manoubi W, Massimi P, Mittal S, Pim D, et al. The human Papillomavirus E6 PDZ binding motif: from life cycle to malignancy. Viruses. 2015;7(7):3530–51. doi: 10.3390/v7072785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Volchkova VA, Klenk HD, Volchkov VE. Delta-peptide is the carboxy-terminal cleavage fragment of the nonstructural small glycoprotein sGP of Ebola virus. Virology. 1999;265(1):164–71. doi: 10.1006/viro.1999.0034. [DOI] [PubMed] [Google Scholar]
- 23.Sugrue RJ, Brown C, Brown G, Aitken J, McL Rixon HW. Furin cleavage of the respiratory syncytial virus fusion protein is not a requirement for its transport to the surface of virus-infected cells. J Gen Virol. 2001;82(Pt 6):1375–86. doi: 10.1099/0022-1317-82-6-1375. [DOI] [PubMed] [Google Scholar]
- 24.Evans P, Dampier W, Ungar L, Tozeren A. Prediction of HIV-1 virus-host protein interactions using virus and host sequence motifs. BMC Med Genomics. 2009;2:27. doi: 10.1186/1755-8794-2-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xue B, Williams RW, Oldfield CJ, Goh GK, Dunker AK, Uversky VN. Viral disorder or disordered viruses: do viral proteins possess unique features? Protein Pept Lett. 2010;17(8):932–51. doi: 10.2174/092986610791498984. [DOI] [PubMed] [Google Scholar]
- 26.Fuxreiter M, Tompa P, Simon I. Local structural disorder imparts plasticity on linear motifs. Bioinformatics. 2007;23(8):950–6. doi: 10.1093/bioinformatics/btm035. [DOI] [PubMed] [Google Scholar]
- 27.Pushker R, Mooney C, Davey NE, Jacque JM, Shields DC. Marked variability in the extent of protein disorder within and between viral families. PLoS ONE. 2013;8(4):e60724. doi: 10.1371/journal.pone.0060724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meyniel-Schicklin L, de Chassey B, Andre P, Lotteau V. Viruses and interactomes in translation. Mol Cell Proteomics. 2012;11(7):M111.014738. doi: 10.1074/mcp.M111.014738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hagai T, Azia A, Babu MM, Andino R. Use of host-like peptide motifs in viral proteins is a prevalent strategy in host-virus interactions. Cell Rep. 2014;7(5):1729–39. doi: 10.1016/j.celrep.2014.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuiken C, Korber B, Shafer RW. HIV sequence databases. AIDS Rev. 2003;5(1):52–61. [PMC free article] [PubMed] [Google Scholar]
- 31.Fu W, Sanders-Beer BE, Katz KS, Maglott DR, Pruitt KD, Ptak RG. Human immunodeficiency virus type 1, human protein interaction database at NCBI. Nucleic Acids Res. 2009;37(Database issue):D417–422. doi: 10.1093/nar/gkn708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sarkar D, Jana T, Saha S. LMPID: a manually curated database of linear motifs mediating protein-protein interactions. Database (Oxford). 2015:2015. [DOI] [PMC free article] [PubMed]
- 33.Pettit SC, Gulnik S, Everitt L, Kaplan AH. The dimer interfaces of protease and extra-protease domains influence the activation of protease and the specificity of GagPol cleavage. J Virol. 2003;77(1):366–74. doi: 10.1128/JVI.77.1.366-374.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dosztanyi Z, Meszaros B, Simon I. Bioinformatical approaches to characterize intrinsically disordered/unstructured proteins. Brief Bioinforma. 2010;11(2):225–43. doi: 10.1093/bib/bbp061. [DOI] [PubMed] [Google Scholar]
- 35.Dosztanyi Z, Csizmok V, Tompa P, Simon I. The pairwise energy content estimated from amino acid composition discriminates between folded and intrinsically unstructured proteins. J Mol Biol. 2005;347(4):827–39. doi: 10.1016/j.jmb.2005.01.071. [DOI] [PubMed] [Google Scholar]
- 36.Reingewertz TH, Shalev DE, Friedler A. In: Making Order in the Intrinsically Disordered Regions of HIV-1 Vif Protein. Weinheim: John Wiley and Sons, Inc: 2011. p. 201–221. Available from: http://dx.doi.org/10.1002/9781118135570.ch8.
- 37.Hagai T, Azia A, Toth-Petroczy A, Levy Y. Intrinsic disorder in ubiquitination substrates. J Mol Biol. 2011;412(3):319–24. doi: 10.1016/j.jmb.2011.07.024. [DOI] [PubMed] [Google Scholar]
- 38.Uniprot. FTP server. 2016. Available from: ftp://ftp.uniprot.org/pub/databases/uniprot/current_release/knowledgebase/idmapping/. Accessed 14 Jan 2016.
- 39.Kim MS, Pinto SM, Getnet D, Nirujogi RS, Manda SS, Chaerkady R, et al. A draft map of the human proteome. Nature. 2014;509(7502):575–81. doi: 10.1038/nature13302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xue B, Mizianty MJ, Kurgan L, Uversky VN. Protein intrinsic disorder as a flexible armor and a weapon of HIV-1. Cell Mol Life Sci. 2012;69(8):1211–59. doi: 10.1007/s00018-011-0859-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ren S, Uversky VN, Chen Z, Dunker AK, Obradovic Z. Short linear motifs recognized by SH2, SH3 and Ser/Thr Kinase domains are conserved in disordered protein regions. BMC Genomics. 2008;9(Suppl 2):S26. doi: 10.1186/1471-2164-9-S2-S26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tokuriki N, Oldfield CJ, Uversky VN, Berezovsky IN, Tawfik DS. Do viral proteins possess unique biophysical features? Trends Biochem Sci. 2009;34(2):53–9. doi: 10.1016/j.tibs.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 43.Braun P, Tasan M, Dreze M, Barrios-Rodiles M, Lemmens I, Yu H, et al. An experimentally derived confidence score for binary protein-protein interactions. Nat Methods. 2009;6(1):91–7. doi: 10.1038/nmeth.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nouretdinov I, Gammerman A, Qi Y, Klein-Seetharaman J. Determining confidence of predicted interactions between HIV-1 and human proteins using conformal method. Pac Symp Biocomput. 2012;17:311–22. [PMC free article] [PubMed] [Google Scholar]
- 45.Blikstad C, Ivarsson Y. High-throughput methods for identification of protein-protein interactions involving short linear motifs. Cell Commun Signal. 2015;13:38. doi: 10.1186/s12964-015-0116-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tastan O, Qi Y, Carbonell JG, Klein-Seetharaman J. Refining literature curated protein interactions using expert opinions. Pac Symp Biocomput. 2015;20:318–29. [PubMed] [Google Scholar]
- 47.Becerra A, Moreno PA, Bucheli V. Computational analysis of the linear motif mediated subversion of the human protein synthesis machinery In: Pascal Lorenz SGS, editor. BIOTECHNO 2016, The Eighth International Conference on Bioinformatics, Biocomputational Systems and Biotechnologies. vol. 8. IARIA. PO Box 7827 Wilmington, DE 19803: ThinkMind(TM) Digital Library: 2016. p. 23–27.
- 48.Duro N, Miskei M, Fuxreiter M. Fuzziness endows viral motif-mimicry. Mol Biosyst. 2015;11(10):2821–9. doi: 10.1039/C5MB00301F. [DOI] [PubMed] [Google Scholar]
- 49.Meszaros B, Dosztanyi Z, Simon I. Disordered binding regions and linear motifs–bridging the gap between two models of molecular recognition. PLoS ONE. 2012;7(10):e46829. doi: 10.1371/journal.pone.0046829. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated during the current study are available in the figshare repository, with links in the Additional Files section.