

The previous view that cellular senescence represented a stress-responsive tumor suppressor mechanism has been significantly revised recently with recognition of the profound non-autonomous functionality of senescent cells. Senescent cells interact with surrounding normal and transformed cells, in addition to immune cells, achieving outcomes that are sometimes tumor suppressive and sometimes pro-oncogenic. These different outcomes had been linked to paracrine signaling through the senescence-associated secretory phenotype (SASP), where senescent cells produce a cocktail of factors including cytokines, growth factors and matrix-modifying enzymes, transcriptionally regulated through the transcription factors, NF-κB and C/EBPβ.1 Previous studies linked different functions to individual SASP components, such as IL6-mediated autocrine reinforcement of senescence or TGF-β1-mediated paracrine senescence.
However, it was unclear how a SASP of static or stereotyped composition could underpin different functions in diverse target cell populations. How could individual SASP components, with distinct and sometimes contrasting functions, be regulated within a collective whole? We have recently discovered a critical role for NOTCH1 as both a master regulator of SASP composition and also regulator of spatially-restricted juxtacrine signaling in the context of RAS-induced senescence (RIS).2
The NOTCH signaling pathway relies on ligand-dependent activation and subsequent cleavage to liberate the NOTCH1 intracellular domain (N1ICD). This enters the nucleus to bind a multi-molecular complex, containing Mastermind-like 1 (MAML1) to drive a conserved transcriptional program. Notch has been studied in embryonic development, cell fate determination, and cancer. More recently, it has been linked with the development of senescence, where ectopic N1ICD was shown to induce senescence.3,4
In our study, while cell surface expression of NOTCH1 progressively increases as cells transitioned from growing to RIS, the level of downstream signaling is both dynamically regulated and temporally associated, either positively or negatively, with expression of TGF-β ligands or pro-inflammatory cytokines (such as IL1/6/8), respectively. Pharmacological and genetic manipulation of NOTCH activity in the context of RIS demonstrates that NOTCH drives a TGF-β- and growth factor-rich secretome, while simultaneously suppressing the canonical pro-inflammatory SASP through repression of C/EBPβ (Fig. 1). At ‘full senescence’ NOTCH activity is down-regulated, de-repressing C/EBPβ and permitting expression of the pro-inflammatory SASP. Importantly, our data places NOTCH-modulated C/EBPβ upstream of IL1A, previously thought of as a master regulator of the SASP. Expression profiling demonstrated that NOTCH-mediated secretome regulation is not limited to small numbers of factors, but a wholesale reprogramming of the output of the senescent cell between two functionally distinct secretomes. How NOTCH activity is downregulated at later stages of senescence, despite the high levels of NOTCH1 mRNA and total protein remains unclear.
Figure 1.
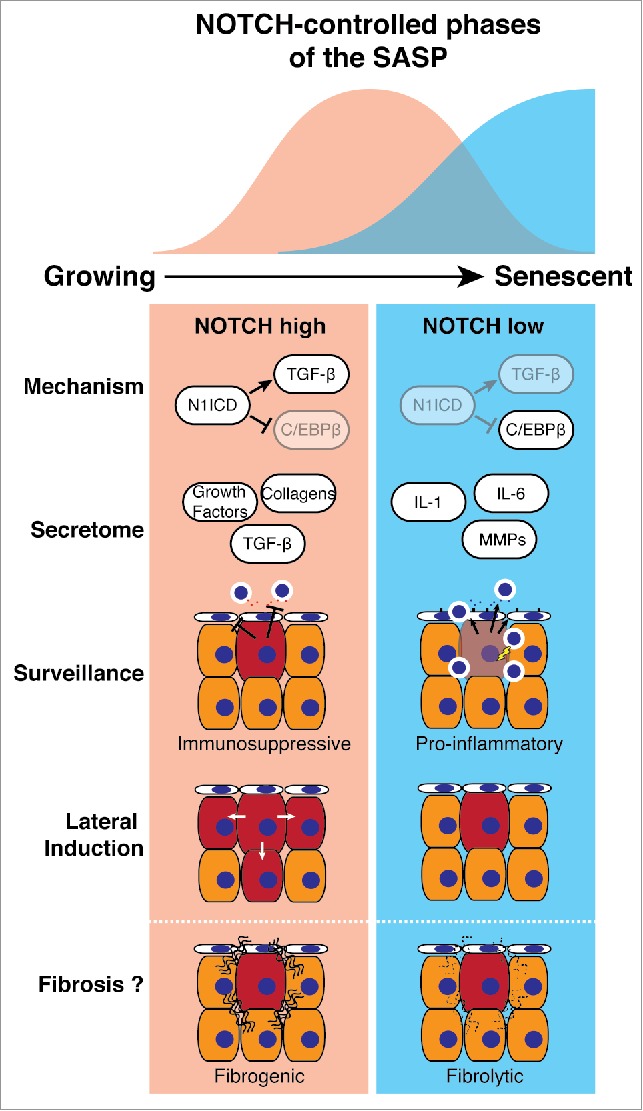
NOTCH-regulated 2-state model of the SASP. During senescence transient NOTCH activity drives a TGF-β-rich secretome (red), while inhibiting the pro-inflammatory secretome (blue), through repression of C/EBPβ. Functionally, the NOTCH-high phase of senescence is immunosuppressive and drives the juxtacrine, spatially-restricted lateral induction of NOTCH signaling. The 2-state SASP model could underpin physiological wound healing and pathological tissue fibrosis.
Utilizing an NRAS-induced murine hepatocyte senescence model we confirmed that Notch1 is upregulated during RIS in vivo; genetic inhibition of Notch activity is associated with an accelerated influx of CD3+ T-lymphocytes and immune-mediated clearance of senescent hepatocytes, presumably through promotion of the pro-inflammatory SASP. Furthermore, different SASPs, derived from senescent cells induced either by RAS, NOTCH, or both, radically alter the ability of human liver sinusoidal endothelial cells to trap lymphocytes on their surface, the first stage of immune trafficking into tissues. The SASP derived from RIS cells enhances lymphocyte trapping, an effect inhibited by NOTCH expression. Therefore, NOTCH represents a potential target to increase senescence surveillance, potentially involving non-autonomous modulation of endothelial cells by senescent cells.
Our data indicate that NOTCH autonomously controls the SASP, previously thought to be the major source of non-autonomous activities of senescent cells. However, we found that NOTCH directly induces a novel form of non-autonomous signaling in senescence. RIS cells are able to transmit a juxtacrine, cell-contact-dependent spatial expansion of Notch activation in vitro and in vivo. Interestingly, specific upregulation of the NOTCH ligand JAG1 occurred in both NOTCH-induced senescent (NIS) and ‘NOTCH-phase’ RIS fibroblasts. This NOTCH/JAG-mediated ‘lateral induction’ was originally identified in some embryological settings. Our data suggest that NOTCH is both a temporal and spatial regulator of the non-autonomous behavior of senescent cells.
What is the physiological role of this NOTCH-mediated switch between 2 different SASPs? Potential evidence emerges from studies of acute and chronic tissue injury and subsequent regeneration. Senescent cells are transiently detected in murine skin after acute wounding, before their elimination.5 Depletion of senescent cells delays wound closure due to loss of SASP-specific secretion of the growth factor PDGFA and its effect upon skin-resident myofibroblasts. In this model the expression of PDGFA peaked several days earlier than inflammatory cytokines such as IL6, potentially partly responsible for the later immune-mediated clearance of the senescent cells. Interestingly, whether or not NOTCH is involved in this dynamic SASP expression in this model remains to be tested; in our study PDGFA is strongly upregulated in NIS, but not in RIS. Such a mechanism may operate elsewhere. Notch1 and down-stream signaling are activated after liver injury, where hepatic stellate cells (HSC) become activated, releasing a pro-fibrotic secretome.6 Pharmacological Notch inhibition reduces liver fibrosis after injury. Post-activation senescence of HSCs is associated with a switch to an anti-fibrotic and pro-inflammatory secretome, limiting tissue fibrosis.7 This supports the idea of a 2 state SASP: a pro-fibrotic and growth factor-rich secretome promoting tissue regeneration, before a pro-inflammatory SASP drives the immune-mediated clearance of these ‘damage-management’ cells and restoration of tissue homeostasis.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1]. Coppé J-P, Desprez P-Y, Krtolica A, Campisi J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu Rev Pathol Mech Dis 2010; 5:99-118; http://dx.doi.org/ 10.1146/annurev-pathol-121808-102144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2]. Hoare M, Ito Y, Kang T-W, Weekes MP, Matheson NJ, Patten DA, Shetty S, Parry AJ, Menon S, Salama R, et al. NOTCH1 mediates a switch between two distinct secretomes during senescence. Nat Cell Biol 2016; 18:979-92; PMID:27525720; http://dx.doi.org/ 10.1038/ncb3397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3]. Liu Z-J, Tan Y, Beecham GW, Seo DM, Tian R, Li Y, Vazquez-Padron RI, Pericak-Vance M, Vance JM, Goldschmidt-Clermont PJ, et al. Notch activation induces endothelial cell senescence and pro-inflammatory response: Implication of Notch signaling in atherosclerosis. Atherosclerosis 2012; 225:296-303; PMID:23078884; http://dx.doi.org/ 10.1016/j.atherosclerosis.2012.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4]. Kagawa S, Natsuizaka M, Whelan KA, Facompre N, Naganuma S, Ohashi S, Kinugasa H, Egloff AM, Basu D, Gimotty PA, et al. Cellular senescence checkpoint function determines differential Notch1-dependent oncogenic and tumor-suppressor activities. Oncogene 2015; 34:2347-59; PMID:24931169; http://dx.doi.org/ 10.1038/onc.2014.169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5]. Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR, Laberge R-M, Vijg J, Van Steeg H, Dollé MET, et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell 2014; 31:722-33; PMID:25499914; http://dx.doi.org/ 10.1016/j.devcel.2014.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6]. Bansal R, van Baarlen J, Storm G, Prakash J. The interplay of the Notch signaling in hepatic stellate cells and macrophages determines the fate of liver fibrogenesis. Sci Rep 2015; 5:18272; PMID:26658360; http://dx.doi.org/ 10.1038/srep18272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7]. Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, Lee H, Zender L, Lowe SW. Senescence of activated stellate cells limits liver fibrosis. Cell 2008; 134:657-67; PMID:18724938; http://dx.doi.org/ 10.1016/j.cell.2008.06.049 [DOI] [PMC free article] [PubMed] [Google Scholar]