

Abstract
Atherosclerosis is associated with alterations in nitric oxide (NO)/cGMP signaling. In early stages of the disease, inflammatory and possibly other cells produce reactive oxygen species that scavenge vasoprotective NO. In addition to the oxidative stress, expression and activity of enzymes downstream to NO formation may also be affected. Here, we show in the aortas of chronically hypercholesterolemic rabbits (a model of late-stage atherosclerosis), both subunits and specific activity of the NO receptor soluble guanylyl cyclase (sGC) were significantly reduced, whereas overall NO synthase activity was unaffected. These changes were most prominent in the neointimal layer, wherein cGMP-dependent protein kinase I (cGK) levels also were reduced. Additionally, a protein (p38nt) that was constitutively tyrosine-nitrated was detected, and its expression was significantly reduced in atherosclerotic aorta. Phosphorylation of the cGK substrate vasodilator-stimulated phosphoprotein (VASP) at Ser-239, an established biochemical endpoint of NO/cGMP signaling, also was reduced. Thus, late-stage atherosclerosis is associated not only with enhanced NO breakdown but also with altered NO reception and cGMP signaling. Preferential down-regulation in neointima suggests a direct connection of these changes to neointimal proliferation and vascular dysfunction and provides a rationale for future pharmacotherapy using classical and novel sGC activators.
Keywords: hypercholesterolemia, soluble guanylyl cyclase, vasodilator-stimulated phosphoprotein, cGMP-dependent protein kinase
Atherosclerosis is major risk factor to cardiovascular diseases such as coronary heart disease and stroke. The pathophysiology of atherosclerosis is highly complex, multifactorial, and yet to be fully understood (1). A common feature observed is malperfusion of tissues as a result of luminally obstructive atherosclerotic plaques, augmented vascular tone, and reduced sensitivity to endothelium-dependent vasodilators in atherosclerotic arteries from humans (2) and cholesterol-fed animals (3).
Nitric oxide (NO) is an important antiatherosclerotic autocoid, which exerts antiproliferative and vasodilatory effect on vasculature and antiaggregatory effect on platelets. Vascular dysfunction in atherosclerosis is partly attributed to reduced tetrahydrobiopterin-dependent (4–7) production or bioavail-ability of endothelium-derived NO (8), which is supported by the observation of reduced activity and expression of endothelial NO synthase III (NOS-III) (9) in atherosclerotic vessels (10, 11). Additionally, the tissue half-life of NO is further reduced by reactive oxygen species (superoxide, 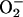 ) that scavenge NO and thereby impair its vasoprotective activity (12). Therefore, formation of O–2, by NADPH oxidase (13, 14) or xanthine oxidase (15) or uncoupled endothelial NOS (16) and/or its decomposition, by superoxide dismutase (SOD) (17–19), are thought to play important roles in the pathogenesis of atherosclerosis.
) that scavenge NO and thereby impair its vasoprotective activity (12). Therefore, formation of O–2, by NADPH oxidase (13, 14) or xanthine oxidase (15) or uncoupled endothelial NOS (16) and/or its decomposition, by superoxide dismutase (SOD) (17–19), are thought to play important roles in the pathogenesis of atherosclerosis.
Endothelium-derived NO stimulates the production of the second messenger cGMP by activation of soluble guanylyl cyclase (sGC) in vascular smooth muscle cells. cGMP alters the properties of several target proteins. For example, it activates cGMP-dependent protein kinase I (cGK), which also is essential for both vascular smooth muscle relaxation (20) and antiproliferative effects of NO (21). One of the best-characterized cGK substrates is the vasodilator-stimulated phosphoprotein (VASP) (22) originally described in platelets (23). In the vascular wall, phosphorylation of VASP at Ser-239 (24) (product denoted PSer-239-VASP) is regulated by NO and prostacyclin. Phosphorylation of VASP at Ser-239 is a sensitive monitor of defective vascular NO/cGMP signaling (25, 26), endothelial dysfunction (27, 28), and vascular oxidative stress (29).
Progression of atherosclerosis is accompanied by morphological and pathophysiological alterations, including the replacement of reversible early lesions (fatty streaks) by fibrous plaques (1). In later stages of the disease, endothelium-independent vasodilation caused by exogenous NO donors becomes increasingly attenuated (3). However, very little is known about the underlying molecular mechanisms such as the expression and activity of enzymes involved in NO-dependent signaling in atherosclerosis. Hence, we examined the possible alterations in sGC, cGK, and PSer-239-VASP levels in a rabbit model of late-stage atherosclerosis.
Materials and Methods
Animals. Rabbits were obtained from Charles River Laboratories; animal feed was from Ets Pietrements (Provin, France); cGMP enzyme immunoassay was from Biotrend (Cologne, Germany); tetrahydrobiopterin was from Schircks Laboratories (Jona, Switzerland); NADPH was from AppliChem (Darmstadt, Germany); Tris·HCl and acrylamide were from Roth (Karlsruhe, Germany); FAD, glutathione, leupeptin, and trypsin inhibitor were from Roche Molecular Biochemicals; [3H]arginine, nitrocellulose membrane, and the ECL Western blot detection kit were from Amersham Pharmacia; and FMN was from Fluka. All other chemicals and Kodak x-ray film were from Sigma. Water was deionized to 18 MΩ·cm by using the MilliQ system (Millipore).
Antibodies. Polyclonal antibodies against sGC subunits were raised by immunizing rabbits with synthetic peptides corresponding to conserved sequences of the α1 and β1 subunits of human sGC that had been affinity-purified as described in ref. 30. The anti-Cu,Zn-SOD antibody was from Calbiochem; the monoclonal anti-PSer-239-VASP antibody (31), the polyclonal anti-nitrotyrosine (anti-NT) antibody (32), the monoclonal antibody generated against tyrosine-nitrated albumin (33), and the cGK antiserum (34) were prepared as described. Secondary anti-rabbit and anti-mouse Ig antibodies were from Amersham Pharmacia; secondary anti-sheep Ig antibody was from Sigma.
Induction of Hypercholesterolemia. Rabbits were fed a cholesterol-enriched diet as described in refs. 3 and 35–37. Briefly, 8-week-old male New Zealand White rabbits (1.5–1.8 kg) were fed for 50 weeks on commercial rabbit feed (200 g per day) fortified with 0.3 ± 0.02% (wt/wt) cholesterol (hypercholesterolemic rabbits, n = 10). Normocholesterolemic rabbits fed on nonfortified commercial rabbit feed for the same period were used as controls (n = 7). All experiments were conducted in accordance with the institutional guidelines for animal studies at Julius-Maxmilians-Universität, Würzburg, Germany.
Preparation of Aortic Homogenates. Rabbits (3.45–5.10 kg) were euthanized with an i.v. dose of sodium pentobarbital (30 mg·kg–1), and blood samples were collected to determine total cholesterol, triglycerides, and high-density lipoprotein (HDL) as described in ref. 3. Subsequently, a 0.6-cm-long segment of the thoracic aorta was removed, rinsed with 0.9% NaCl, and carefully cleaned of fat and connective tissue. All of the steps were performed at 4°C. Each sample was weighed (wet) and immediately homogenized in a glass–glass Potter–Elvehjem homogenizer with 1 ml of homogenization buffer containing the following: 25 mM triethanolamine, pH 7.8; 1 mM EDTA disodium salt; 5 mM DTT; 1 μM leupeptin; and 0.5 mg·liter–1 trypsin inhibitor. Homogenates were centrifuged at 10,000 × g for 30 min. The supernatant was removed, and the pellet was resuspended in an equal volume of homogenization buffer and briefly sonicated. Supernatant and resuspended pellet fractions were quickly frozen in liquid nitrogen and stored at –80°C until further analysis. A further 0.6-cm-long segment of each aorta was microdissected into adventitia, media, and neointima, and each layer was processed as described for the whole aorta.
Protein Determination. Samples were solubilized with 1 M NaOH, and protein content was estimated as described by Bradford (38). In samples prepared for denaturing SDS/PAGE, protein was precipitated with trichloroacetic acid and quantified according to Lowry et al. (39).
NOS Activity Assay. NOS activity was assayed by the conversion of l-[3H]arginine to l-citrulline (40, 41). For the determination of total NOS activity, aortic homogenates (30 μl) were incubated for 15 min at 37°C in a total volume of 100 μl containing the following: 50 mM triethanolamine, pH 7.5; 10 μM calmodulin; 1 mM CaCl2; 250 μM 3-[(3-cholamidopropyl)dimethylammonio]-2-hydroxy-1-propanesulfonate (CHAPSO); 5 μM FAD; 5 μM FMN; 1 mM NADPH; 7 mM glutathione; 5 μM l-arginine; 7.4 kBq of l-[2,3,4,5-3H]arginine hydrochloride; and 10 μM (6R)-5,6,7,8-tetrahydrobiopterin. The reaction was terminated by adding 900 μl of stop buffer containing 25 mM acetic acid (pH 5.5) and 2 mM EDTA. The l-citrulline formed was separated from arginine substrate by cation-exchange chromatography and measured by liquid scintillation counting. To determine the Ca2+-independent NOS-II activity, CaCl2 was replaced by 5 mM EGTA. Activities are expressed in pmol of citrulline per mg of protein per min.
sGC Activity Assay. sGC activity was determined in the precleared (see under Western blotting) supernatant fractions of the tissue samples by the conversion of GTP to cGMP as described in ref. 30. Briefly, 10 μl of the samples was incubated for 10 min at 37°C in a total volume of 100 μl containing the following: 50 mM triethanolamine, pH 7.4; 1 mM 3-isobutyl-1-methylxantine; 3 mM MgCl2; 0.5 mM GTP; 3 mM DTT; 5 mM phosphocreatine; and 0.25 mg·ml–1 creatine kinase. The NO-stimulated sGC activity was measured in the presence of 100 μM sodium nitroprusside (SNP). The reaction was terminated by inactivation of sGC at 95°C for 10 min. cGMP was measured by using a commercially available cGMP enzyme-linked immunoassay (Biotrend) according to the manufacturer's instructions.
Phosphodiesterase (PDE) Activity Assay. To measure the activity of PDE, the cleavage of cGMP to GMP was determined as described in ref. 42. Briefly, 10 μl of sample was mixed in a total volume of 100 μl containing the following: 20 mM Tris·HCl, pH 7.5; 20 mM imidazole; 3 mM MgCl2;15 μM magnesium acetate; 0.25 μM calmodulin; and 0.25 μM CaCl2. After the addition of 10 μM cGMP, the mixture was incubated at 37°C, and aliquots were collected after 0, 5, and 10 min of incubation. The remaining cGMP was determined by enzyme-linked immunoassay as described above (sGC activity assay).
Western Blot Analysis. Rabbit aortic samples, particularly those of hypercholesterolemic rabbits, contained high levels of immunoglobulins (Ig) (43). Therefore, before Western blot analysis, 50 μl of supernatant fractions was precleared of rabbit Ig by incubating three times for 1 h with 50 μl of protein A-Sepharose (Sigma) at 4°C on an overhead shaker and intermittently pelleting the Sepharose beads. This procedure enabled the subsequent immunodetection of all proteins of interest, even when primary rabbit antibodies were used.
Samples were separated by SDS/PAGE on 8%, 10%, or 12% polyacrylamide gels (44) and electroblotted onto nitrocellulose membranes by using a semidry technique (45). Loading of equal amounts of protein was confirmed by reversible protein staining with Ponceau S. Membranes were thereafter blocked for 1 h at room temperature with 3% nonfat dry milk in TBST buffer, which contains 20 mM Tris·HCl (pH 7.5), 200 mM NaCl, and 0.1% Tween 20, and then probed overnight at 4°C with the respective antibodies diluted in the blocking buffer (dilutions: anti-sGCα1, 1:2000; anti-sGCβ1, 1:1,000; anti-PSer-239-VASP, 1:2,000; anti-Cu,Zn-SOD, 1:750; polyclonal anti-NT, 1:5,000 (32); monoclonal anti-NT, 1:4,000 (33); and anti-cGK, 1:4,000). After repeated washing and 1 h of incubation with the respective horseradish peroxidase-linked secondary antibody, immunoreactive protein was visualized on x-ray film by means of a commercially available enhanced chemiluminescence detection system (Amersham Pharmacia). Specificity of the signals of the anti-NT and anti-Cu,Zn-SOD antibodies was verified by preincubation of the diluted antibody with 50 μM 3-NT for 1 h or with 0.2 mg·ml–1 native bovine Cu,Zn-SOD for 6 h, respectively. For quantification of immunoreactivities, Western blots were scanned with a resolution of 300 dpi and subjected to densitometric analysis by using nih image software (National Institutes of Health, Bethesda, MD).
Statistical Analysis. Data are expressed as means ± SEM. Statistical significance (P < 0.05) was assessed by using a Student's unpaired two- or one-tailed t test.
Results
Morphology and Blood Lipid Levels. Rabbits fed a cholesterol-enriched diet had 50-fold higher total plasma cholesterol levels, compared with control animals, but there was no significant change in the levels of triglycerides and high-density lipoprotein (Fig. 1D). The body weight of the cholesterol-fed rabbits was significantly lower than that of the control animals (3.89 ± 0.11 kg vs. 4.66 ± 0.13 kg). Microscopically, it was observed that atherosclerotic plaques in the thoracic aorta contained foam cells, increased amounts of smooth muscle cells, and extracellular matrix (data not shown). In control aortas, α-actin-immunostained vascular smooth muscle cells were observed in the tunica media (M), whereas in atherosclerotic vessels, these cells also were observed in the neointima (N), where they formed a typical subendothelial fibromuscular cap, as described in ref. 37 (Fig. 1 A–C). Hence, this animal model represents a late stage of atherosclerosis in which the affected blood vessels show properties similar to those observed in human pathophysiology.
Fig. 1.

Phenotype of hypercholesterolemic rabbits. (A and B) Histology of thoracic aorta from control rabbits and hypercholesterolemic rabbit aorta. Sections of the thoracic aorta were immunohistochemically stained with antibodies against α-actin to identify vascular smooth muscle cells. M, media; N, neointima; L, lumen. (C) Ratio of total wet weight of adventitia, media, and (in the case of hypercholesterolemic animals) neointima expressed in percent of adventitia and media. (D) Total cholesterol, triglycerides, and HDL plasma levels in control (CON, n = 7) and hypercholesterolemic (HC, n = 9) rabbits. Data are represented as mean ± SEM (***, P < 0.001); n.s., not significant.
NOS Activity. Consistent with the inflammatory nature of atherosclerotic vascular disease, inducible NOS activity was higher in hypercholesterolemic animals, compared with controls, although this difference did not reach statistical significance (Fig. 2).
Fig. 2.

NOS activity measured in homogenates of intact aortas of control (CON, n = 3) and hypercholesterolemic (HC, n = 6) rabbits. Citrulline formation from arginine was determined in the presence (total NOS) and absence (inducible NOS, iNOS) of free Ca2+; constitutive NOS (cNOS) activity was calculated by subtracting iNOS from total activity for each individual rabbit. Data are represented as mean ± SEM; n.s., not significant.
Expression of Cu,Zn-SOD. The expression of Cu,Zn-SOD was reduced in aortic samples from hypercholesterolemic rabbits (Fig. 3), suggesting that antioxidant protection is diminished under late-stage atherosclerosis.
Fig. 3.

Western blot analysis of Cu,Zn-SOD in aortic homogenates. (A) Lanes 1–6, normocholesterolemic rabbits (CON, n = 6; 20 μg of protein per lane); lanes 7–12, hypercholesterolemic rabbits (HC, n = 6; 20 μg of protein per lane); lanes 13 and 14, 50 and 100 μg of bovine Cu,Zn-SOD, respectively. Signals were blocked by preincubation of the antibody with native Cu,Zn-SOD (data not shown). Molecular mass markers (in kDa) are given at the left side of the figure. (B) Densitometric analysis. Data are represented as mean ± SEM (*, P < 0.05; AU, arbitrary units).
Detection of Peroxynitrite (ONOO–) Formation by Protein Tyrosine Nitration. Superoxide rapidly reacts with NO, yielding ONOO– (33), which nitrates tyrosine residues in various proteins. By using the polyclonal antiserum, a single NT-positive protein with an apparent molecular mass of 38 kDa (p38nt) was detected that was specifically blocked by preincubation of antiserum with free 3-NT (data not shown). In the atherosclerotic aortas, this immunoreactivity was significantly decreased, compared with controls (Fig. 4 A and B). In contrast, the monoclonal antibody detected a 63-kDa band (p63nt) (Fig. 4 C and D) comigrating with BSA that was significantly increased in atherosclerotic rabbit aorta. The specificity of this signal is unclear, as it was not blocked by free 3-NT (data not shown).
Fig. 4.

Protein tyrosine nitration as analyzed by Western blot analysis. (A and C) Lanes 1–5, control aortas (CON, n = 5); lanes 6–11, atherosclerotic aortas (HC, n = 6); all samples contained 20 μg of protein. (A) Polyclonal antiserum against NT recognized one specific band at 38 kDa (p38nt), which was greatly diminished in the atherosclerotic samples. Preincubation of the antibody for 60 min with 50 μM free NT completely blocked formation of the band (data not shown). Rabbit IgG heavy chains (50 kDa) were recognized by the secondary antibody. (B) Densitometric analysis of bands from A; data are represented as mean ± SEM (***, P < 0.001; AU, arbitrary units). (C) Monoclonal anti-NO2-BSA antibody recognized a single but different protein band at 63 kDa (p63nt), which was significantly increased in the atherosclerotic samples but not readily blocked by preincubation with free NT. (D) Densitometric analysis of bands from C (**, P < 0.01).
Expression and Activity of sGC and PDEs. Both α1 (Fig. 5A) and β1 (Fig. 5C) subunits of sGC were detectable in hypercholesterolemic (lanes 8–13) and control (lanes 2–7) rabbits. In addition, the heavy chain of rabbit Ig was recognized in the atherosclerotic vessels by the secondary anti-rabbit antibody. The high level of Ig in atherosclerotic vessels was described in refs. 36 and 43. The expression levels of both α1 and β1 subunits of sGC were significantly lower in the atherosclerotic samples (P < 0.05; Fig. 5 B and D). Consistent with this observation, basal and NO-stimulated sGC activities were significantly lower in total homogenates of atherosclerotic aorta (Fig. 5E). In contrast, cGMP PDE activity was not significantly different between the groups (control, 45.1 ± 4.3 pmol·mg–1·min–1; and hypercholesterolemic, 35.3 ± 6.3 pmol·mg–1·min–1).
Fig. 5.

sGC immunoreactivity and activity in aortic homogenates. α1 subunits (A) and β1 subunits (C) (20 μg of protein per lane) were detected by using their respective antibodies. Lane 1, positive control (respective subunit expressed in Sf9 cells); lanes 2–7, control rabbits (CON, n = 6); lanes 8–13, hypercholesterolemic rabbits (HC, n = 6). (B and D) Quantitative determination of immunoreactivity of A and C by densitometry. Data are represented as mean ± SEM; AU, arbitrary units. sGC levels were significantly (*, P < 0.05) decreased in HC. (E) Specific sGC activity in aortic homogenates was determined in the absence (Basal, left y-axis) and presence of SNP (100 μM, right y-axis). Both basal and SNP-stimulated activities were significantly reduced in HC (n = 6) vs. CON (n = 6) (*, P < 0.05). (F) Specific sGC activity in the different layers of rabbit aorta (adventitia, n = 5; media, n = 5; neointima, n = 6). Whereas no significant difference in the adventitia and media could be determined between CON and HC, the neointimal layer revealed a markedly lower specific sGC activity (*, P < 0.05 vs. HC adventitia; †, P < 0.05 vs. CON adventitia or media).
In the hypercholesterolemic vessels, the newly formed neointimal layer represented 62.5 ± 12.5% wet weight, the media 12.5 ± 1.7%, and the adventitia 25.0 ± 4.2%. These different layers were homogenized and analyzed for sGC activity in the presence of 100 μM SNP (Fig. 5F). Whereas sGC activity in the media and adventitia revealed no significant difference between control and atherosclerotic samples, the level in the neointima, which represents the major part of the affected vessels, was markedly reduced.
cGK and PSer-239-VASP. In total homogenates of atherosclerotic vessels, the expression level of cGK was not significantly altered when compared with that of control vessels (Fig. 6 A and B). The expression of cGK was markedly lower in the neointima of atherosclerotic vessels, whereas the other layers contained nearly equal amounts of cGK immunoreactivity, compared with tissues from control rabbits (Fig. 6 C and D).
Fig. 6.

Western blot analysis of cGK expression and activity. (A) Expression of cGK-dependent protein kinase I in total aortic homogenates (20 μg of protein per lane); lanes 1–5, control rabbits (CON, n = 5); lanes 6–11, hypercholesterolemic rabbits (HC, n = 6). (B) Densitometric analysis of A with data expressed as mean ± SEM; AU, arbitrary units. (C) Expression of cGK in different aortic layers (20 μg of protein per lane). Lanes 1–3 and 4–6, adventitia of CON and HC, respectively (n = 3); lanes 7–9 and 10–12, media of CON and HC, respectively (n = 3); lanes 13–15, neointima (HC only, n = 3). Lane 16 contains platelet homogenate as a positive control. (D) Densitometry of the data in C (*, P < 0.05 vs. HC media; †, P < 0.05 vs. CON adventitia or media). (E) Western blot of PSer-239-VASP in aortic homogenates (20 μg of protein per lane) as a functional marker of the activity of cGMP-dependent protein kinases and the NO signaling pathway. Lanes 1–5, control (CON, n = 5); lanes 6–11, hypercholesterolemic samples (HC, n = 6); lane 12, PSer-239-VASP standard from human platelet homogenate treated for 10 min with 100 μM SNP. (F) Densitometric analysis of the results in E, with data expressed as mean ± SEM (***, P < 0.001).
PSer-239-VASP was quantified by Western blot analysis using a monoclonal antibody that binds VASP only in its Ser-239-phosphorylated state (31). The amount of PSer-239-VASP was markedly lower in the aorta of hypercholesterolemic rabbits (Fig. 6 E and F, lanes 6–11), which is consistent with the diminished NO/cGMP response in atherosclerotic blood vessels. However, it is possible that a concomitant down-regulation of total VASP contributed to this reduction in PSer-239-VASP.
Discussion
In atherosclerosis, several signal-transduction pathways are affected that are responsible for controlling vascular tone (46) and proliferation. Endothelium-derived NO, which inhibits platelet adhesion, vasocontraction, and proliferation of vascular smooth muscle cells, is one of the key protective mechanisms in the vasculature. The NO pathway is clearly affected in hypercholesterolemia, where increased O–2 levels scavenge NO and thereby reduce its bioavailability (14). Here, we show that PSer-239-VASP serves as a sensitive biochemical marker for this endothelial dysfunction. Moreover, we demonstrate alterations in signal-transduction components downstream of NO, among others the NO receptor sGC, particularly in neointima.
To investigate the activity and expression levels of the major protein components of the NO/cGMP pathway downstream of NO formation, we used a rabbit model that represents a relatively advanced stage of atherosclerosis. In contrast to studies in which rabbits are fed diets containing high levels of cholesterol for a relatively short period, we followed a feeding schedule for 1 year in this study while using only a moderately high cholesterol concentration in the diet. The atherosclerotic plaques in the thoracic aorta had properties similar to human lesions, as described in ref. 24.
Several years before endothelium-derived relaxing factor (EDRF) was identified as NO, various groups reported that superoxide radicals have a limiting effect on EDRF bioactivity (12, 47). Later, it was shown that  reacts effectively with NO, forming peroxynitrite (20), a short-lived anion with little or no vasodilatory potency. Atherosclerosis is an inflammatory process, and
reacts effectively with NO, forming peroxynitrite (20), a short-lived anion with little or no vasodilatory potency. Atherosclerosis is an inflammatory process, and 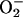 production in atherosclerotic plaques is increased (48). The free radical-scavenging enzyme SOD counteracts this by lowering
production in atherosclerotic plaques is increased (48). The free radical-scavenging enzyme SOD counteracts this by lowering 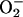 levels. Supporting this inhibition of SOD leads to greatly reduced NO activity in rats (49), whereas the administration of exogenous SOD partially restores the impaired vascular function in atherosclerotic vessels (17, 18, 50). Here, we observed a significant down-regulation of Cu,Zn-SOD in atherosclerotic aorta, suggesting an imbalance in vascular
levels. Supporting this inhibition of SOD leads to greatly reduced NO activity in rats (49), whereas the administration of exogenous SOD partially restores the impaired vascular function in atherosclerotic vessels (17, 18, 50). Here, we observed a significant down-regulation of Cu,Zn-SOD in atherosclerotic aorta, suggesting an imbalance in vascular  homeostasis.
homeostasis.
A rise in vascular wall 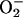 should result in an increased concentration of ONOO–, which then decomposes to nitrite and nitrate. ONOO– also oxidizes protein thiols and induces tyrosine nitration on different proteins, as observed in human patients with atherosclerosis (33). 3-NT immunoreactivity is considered to be a marker of concomitant formation of NO and reactive oxygen species (51) and can be detected by Western blot analysis. Here, we used a polyclonal antiserum generated against nitrated keyhole limpet hemocyanin (32) and a monoclonal antibody generated against nitrated BSA (NO2-BSA) (33). By using these antibodies, two different NT-positive proteins, p38nt and p63nt, were detected. The finding of proteins (p38nt and p63nt) that appear to be constitutively tyrosine nitrated, suggests the occurrence of selective ONOO–-mediated chemistry in healthy vasculature and formation of ONOO– under noninflammatory conditions. Thus, such proteins may have a role in the signaling mechanisms in vasculature under physiological conditions. Both proteins were detected previously by these antibodies, e.g., in heart (p38nt and p63nt), blood, and whole brain (unpublished data). Surprisingly, the degree of tyrosine nitration in the atherosclerotic model was enhanced for p63nt but decreased for p38nt. Our observation suggests that different anti-NT antibodies detect different protein epitopes, presumably depending on the neighboring amino acids of these nitrated proteins. Hence, we emphasize that a control with moderate concentration of authentic NT, as performed in the present study, should be routinely included in all such studies in future. It will be of great interest to identify p38nt and p63nt (presumably albumin) to draw conclusions about the functional significance of these modifications. These results also raise concerns about the applicability of protein tyrosine nitration as a general marker for peroxynitrite formation. As a possible explanation, ONOO– may be generated in different compartments of the blood vessel or the respective target proteins may be highly localized. The monoclonal antibody was raised against NO2-BSA, which was previously shown to be increased in atherosclerosis. The other protein, p38, may be a more abluminally expressed protein that is nitrated under physiological conditions. In atherosclerosis, it may be less nitrated, perhaps because the diffusion of ONOO– is diminished by neointimal formation or because NO or
should result in an increased concentration of ONOO–, which then decomposes to nitrite and nitrate. ONOO– also oxidizes protein thiols and induces tyrosine nitration on different proteins, as observed in human patients with atherosclerosis (33). 3-NT immunoreactivity is considered to be a marker of concomitant formation of NO and reactive oxygen species (51) and can be detected by Western blot analysis. Here, we used a polyclonal antiserum generated against nitrated keyhole limpet hemocyanin (32) and a monoclonal antibody generated against nitrated BSA (NO2-BSA) (33). By using these antibodies, two different NT-positive proteins, p38nt and p63nt, were detected. The finding of proteins (p38nt and p63nt) that appear to be constitutively tyrosine nitrated, suggests the occurrence of selective ONOO–-mediated chemistry in healthy vasculature and formation of ONOO– under noninflammatory conditions. Thus, such proteins may have a role in the signaling mechanisms in vasculature under physiological conditions. Both proteins were detected previously by these antibodies, e.g., in heart (p38nt and p63nt), blood, and whole brain (unpublished data). Surprisingly, the degree of tyrosine nitration in the atherosclerotic model was enhanced for p63nt but decreased for p38nt. Our observation suggests that different anti-NT antibodies detect different protein epitopes, presumably depending on the neighboring amino acids of these nitrated proteins. Hence, we emphasize that a control with moderate concentration of authentic NT, as performed in the present study, should be routinely included in all such studies in future. It will be of great interest to identify p38nt and p63nt (presumably albumin) to draw conclusions about the functional significance of these modifications. These results also raise concerns about the applicability of protein tyrosine nitration as a general marker for peroxynitrite formation. As a possible explanation, ONOO– may be generated in different compartments of the blood vessel or the respective target proteins may be highly localized. The monoclonal antibody was raised against NO2-BSA, which was previously shown to be increased in atherosclerosis. The other protein, p38, may be a more abluminally expressed protein that is nitrated under physiological conditions. In atherosclerosis, it may be less nitrated, perhaps because the diffusion of ONOO– is diminished by neointimal formation or because NO or  formation is reduced in the particular compartment. It should be noted that the validity of tyrosine nitration as an in vivo marker for ONOO– has been questioned recently, because no NT formation was observed under conditions where NO and
formation is reduced in the particular compartment. It should be noted that the validity of tyrosine nitration as an in vivo marker for ONOO– has been questioned recently, because no NT formation was observed under conditions where NO and 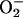 were generated simultaneously (52). Clearly, this issue should be readdressed by using more direct methods of NT quantification.
were generated simultaneously (52). Clearly, this issue should be readdressed by using more direct methods of NT quantification.
Our data suggest that down-regulation of heterodimeric sGC (53), an important target for cardiovascular therapeutics (42, 54), and cGK are involved in the underlying pathophysiology of atherosclerotic vascular disease. We considered the possibility that the accumulation of extracellular matrix proteins in atherosclerotic blood vessels led to “dilution” of the whole-tissue homogenates, resulting in an apparent “down-regulation” of cGMP pathway enzymes. It also could be possible that increase in NOS with the concomitant decrease in PDE activity may serve to offset the diminished sGC activity in hypercholesterolemia. However, NOS and PDE activities were unchanged in tissue homogenates, which argues against such a nonspecific effect. Previous investigations showed that the sensitivity of sGC to NO is reduced in atherosclerotic aorta, a reduction that may be mediated in part by oxidized lipoproteins (55–57). Indeed, our preliminary data indicate that oxidized LDL down-regulates endothelial cell sGC expression (unpublished data).
The neointima represented >60% of the aorta from hypercholesterolemic rabbits, wherein very low sGC activity was observed. This could clearly account for the reduced sGC activity or sensitivity observed in total tissue homogenates. cGMP modifies the properties of several effector proteins, including ion channels, PDEs, and cGK (58), which are essential for effective NO-induced vasodilation (20). As with sGC, expression of cGK in aorta from hypercholesterolemic rabbits was significantly lower in the neointima, whereas expression of cGK in the outer layers was identical to control tissue. In similar lines, cGK expression was reported to be unaltered in Watanabe heritable hyperlipidemic rabbits (59). The fact that smooth muscle cells have an attenuated NO/cGMP pathway may contribute to the reported hypercontractility in atherosclerotic disease, a situation similar to that in spontaneously hypertensive rats (23). Moreover, the ability of these cells to respond to antiproliferative stimuli could be affected, which may represent a key mechanism responsible for neointimal cell migration and proliferation. Importantly, adenoviral sGC gene transfer was reported to inhibit neointima formation (14), supporting the hypothesis that sGC activity has antiatherosclerotic effects.
PSer-239-VASP is an important in vivo marker for cGK activity (31) and a biochemical read-out for the activity of the vascular NO/cGMP pathway. The marked reduction in the levels of PSer-239-VASP illustrates the deficiency of NO/cGMP signaling in the late stage of atherosclerosis, which, as we have now shown, involves most of the protein components of this signaling cascade. This finding also should stimulate new therapeutic approaches. Our data are in agreement with other in vitro (21) and in vivo (60) studies wherein expression of protein components downstream of NO induced dramatic improvement in the vascular smooth muscle cell phenotype and prevented neointimal formation. As an alternative therapeutic approach, new experimental drugs that stimulate or sensitize sGC (e.g., YC-1) (61) or directly activate cGK may be beneficial in the later stages of atherosclerosis.
Acknowledgments
We thank Ulrich Walter, Suzanne Lohmann, and Albert Smolenski for providing antibodies and helpful discussions; and Manfred Bernhard, Birgit Wagner, and Monika Weeger for expert technical assistance. This study was supported by the Deutsche Forschungsgemeinschaft (SFB547/C7). V.O.M. is the recipient of the student award “Fortschritte in der Medizin, Studenten Forschen” and a Biomedical Sciences Exchange Program fellowship.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: cGK, cGMP-dependent protein kinase I; NOS, NO synthase; NT, nitrotyrosine; PDE, phosphodiesterase; sGC, soluble guanylyl cyclase; SNP, sodium nitroprusside; SOD, superoxide dismutase; VASP, vasodilator-stimulated phosphoprotein; PSer-239-VASP, VASP phosphorylated at Ser-239.
References
- 1.Ross, R. (1999) N. Engl. J. Med. 340, 115–126. [DOI] [PubMed] [Google Scholar]
- 2.Greager, M., Cooke, J. & Mendelsohn, M. (1990) J. Clin. Invest. 86, 228–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Verbeuren, T., Jordaens, F., Zonnekeyn, L., Van Hove, C., Coene, M. & Herman, A. (1986) Circ. Res. 58, 552–564. [DOI] [PubMed] [Google Scholar]
- 4.Reif, A., Zecca, L., Riederer, P., Feelisch, M. & Schmidt, H. H. H. W. (2001) Free Radical Biol. Med. 30, 803–808. [DOI] [PubMed] [Google Scholar]
- 5.Pantke, M. M., Reif, A., Valtschanoff, J. G., Shutenko, Z., Frey, A., Weinberg, R. J., Pfleiderer, W. & Schmidt, H. H. H. W. (2001) Biochem J. 356, 43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kotsonis, P., Frey, A., Frohlich, L. G., Hofmann, H., Reif, A., Wink, D. A., Feelisch, M. & Schmidt, H. H. H. W. (1999) Biochem J. 340, 745–752. [PMC free article] [PubMed] [Google Scholar]
- 7.Reif, A., Frohlich, L. G., Kotsonis, P., Frey, A., Bommel, H. M., Wink, D. A., Pfleiderer, W. & Schmidt, H. H. H. W. (1999) J. Biol. Chem. 274, 24921–24929. [DOI] [PubMed] [Google Scholar]
- 8.Verbeuren, T., Jordaens, F., Van Hove, C., Van Hoydonck, A. & Herman, A. (1990) Eur. J. Pharmacol. 191, 173–184. [DOI] [PubMed] [Google Scholar]
- 9.Butt, E., Bernhardt, M., Smolenski, A., Kotsonis, P., Frohlich, L. G., Sickmann, A., Meyer, H. E., Lohmann, S. M. & Schmidt, H. H. H. W. (2000) J. Biol. Chem. 275, 5179–5187. [DOI] [PubMed] [Google Scholar]
- 10.Buttery, L., Chester, A., Springall, D., Borland, J., Michel, T., Yacoub, M. & Polak, J. (1996) J. Pathol. 179, 197–203. [DOI] [PubMed] [Google Scholar]
- 11.Oemar, B., Tschudi, M., Godoy, N., Brovkovich, V., Malinski, T. & Lüscher, T. (1998) Circulation 97, 2494–2498. [DOI] [PubMed] [Google Scholar]
- 12.Gryglewski, R., Palmer, R. & Moncada, S. (1986) Nature 320, 454–456. [DOI] [PubMed] [Google Scholar]
- 13.Harrison, D. (1997) Clin. Cardiol. 20, 11–17. [PubMed] [Google Scholar]
- 14.Münzel, T. & Harrison, D. (1997) J. Mol. Med. 75, 891–900. [DOI] [PubMed] [Google Scholar]
- 15.Houston, M., Estevez, A., Chumley, P., Aslan, M., Marklund, S., Parks, D. & Freeman, B. (1999) J. Biol. Chem. 274, 4985–4994. [DOI] [PubMed] [Google Scholar]
- 16.Kuzkaya, N., Weissmann, N., Harrison, D. G. & Dikalov, S. (2003) J. Biol. Chem. 278, 22546–22554. [DOI] [PubMed] [Google Scholar]
- 17.Greenwald, R. (1990) Free Radical Biol. Med. 8, 201–209. [DOI] [PubMed] [Google Scholar]
- 18.Mügge, A., Elwell, J., Peterson, T., Hofmeyer, T., Heistad, D. & Harrison, D. (1991) Circ. Res. 69, 1293–1300. [DOI] [PubMed] [Google Scholar]
- 19.Luoma, J., Stralin, P., Marklund, S., Hiltunen, T., Sarkioja, T. & Yla-Herttuala, S. (1998) Arterioscler. Thromb. Vasc. Biol. 18, 157–167. [DOI] [PubMed] [Google Scholar]
- 20.Pfeifer, A., Klatt, P., Massberg, S., Ny, L., Sausbier, M., Hirneiss, C., Wang, G., Korth, M., Aszódi, A., Anderson, K., et al. (1998) EMBO J. 17, 3045–3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lincoln, T., Dey, N. B., Boerth, N., Cornwell, T. & Soff, G. (1998) Acta Physiol. Scand. 164, 507–515. [DOI] [PubMed] [Google Scholar]
- 22.Hauser, W., Knobeloch, K., Eigenthaler, M., Gambaryan, S., Krenn, V., Geiger, J., Glazova, M., Rohde, E., Horak, I., Walter, U. & Zimmer, M. (1999) Proc. Natl. Acad. Sci. USA 96, 8120–8125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rütten, H., Zabel, U., Linz, W. & Schmidt, H. (1999) Circ. Res. 85, 534–541. [DOI] [PubMed] [Google Scholar]
- 24.Ibarra-Alvarado, C., Galle, J., Melichar, V. O., Mameghani, A. & Schmidt, H. H. H. W. (2002) Mol. Pharmacol. 61, 312–319. [DOI] [PubMed] [Google Scholar]
- 25.Schulz, E., Tsilimingas, N., Rinze, R., Reiter, B., Wendt, M., Oelze, M., Woelken-Weckmuller, S., Walter, U., Reichenspurner, H., Meinertz, T. & Munzel, T. (2002) Circulation 105, 1170–1175. [DOI] [PubMed] [Google Scholar]
- 26.Chen, L., Daum, G., Chitaley, K., Coats, S. A., Bowen-Pope, D. F., Eigenthaler, M., Thumati, N. R., Walter, U. & Clowes, A. W. (2004) Arterioscler. Thromb. Vasc. Biol. 24, 1403–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oelze, M., Mollnau, H., Hoffmann, N., Warnholtz, A., Bodenschatz, M., Smolenski, A., Walter, U., Skatchkov, M., Meinertz, T. & Munzel, T. (2000) Circ. Res. 87, 999–1005. [DOI] [PubMed] [Google Scholar]
- 28.Mollnau, H., Wendt, M., Szocs, K., Lassegue, B., Schulz, E., Oelze, M., Li, H., Bodenschatz, M., August, M., Kleschyov, A. L., et al. (2002) Circ. Res. 90, E58–E65. [DOI] [PubMed] [Google Scholar]
- 29.Mulsch, A., Oelze, M., Kloss, S., Mollnau, H., Topfer, A., Smolenski, A., Walter, U., Stasch, J. P., Warnholtz, A., Hink, U., et al. (2001) Circulation 103, 2188–2194. [DOI] [PubMed] [Google Scholar]
- 30.Zabel, U., Weeger, M., La, M. & Schmidt, H. (1998) Biochem J. 335, 51–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smolenski, A., Bachmann, C., Reinhard, K., Honig-Liedl, P., Jarchau, T., Hoschuetzky, H. & Walter, U. (1998) J. Biol. Chem. 273, 20029–20035. [DOI] [PubMed] [Google Scholar]
- 32.Uttenthal, L., Alonso, D., Fernandez, A., Campbell, R., Moro, M., Leza, J., Lizasoain, I., Esteban, F., Barroso, J., Valderrama, R., et al. (1998) Microsc. Res. Tech. 43, 75–88. [DOI] [PubMed] [Google Scholar]
- 33.Beckmann, J., Ye, Y., Anderson, P., Chen, J., Accavitti, M., Tarpey, M. & White, C. (1994) Biol. Chem. Hoppe-Seyler 375, 81–88. [DOI] [PubMed] [Google Scholar]
- 34.Markert, T., Vaandrager, A., Gambaryan, S., Pöhler, D., Häusler, C., Walter, U., De Jonge, H., Jarchau, T. & Lohmann, S. (1995) J. Clin. Invest. 96, 822–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Simonet, S., Porro-de-Bailliemcourt, J., Descombes, J., Mennecier, P., Laubie, M. & Verbeuren, T. (1993) Circ. Res. 72, 616–630. [DOI] [PubMed] [Google Scholar]
- 36.Rupin, A., Behr, D. & Verbeuren, T. (1996) Br. J. Pharmacol. 119, 1233–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Behr, D., Rupin, A., Fabiani, J.-N. & Verbeuren, T. (1999) Atherosclerosis 142, 335–344. [DOI] [PubMed] [Google Scholar]
- 38.Bradford, M. (1976) Anal. Biochem. 72, 248–254. [DOI] [PubMed] [Google Scholar]
- 39.Lowry, O. H., Rosebrough, N. J., Farr, A. L. & Randall, R. J. (1951) J. Biol. Chem. 193, 265–275. [PubMed] [Google Scholar]
- 40.Bredt, D. & Snyder, S. (1990) Proc. Natl. Acad. Sci. USA 87, 682–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schmidt, H., Pollock, J., Nakane, M., Gorsky, L., Förstermann, U. & Murad, F. (1991) Proc. Natl. Acad. Sci. USA 88, 365–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Galle, J., Zabel, U., Hübner, U., Hatzelmann, A., Wagner, B., Wanner, C. & Schmidt, H. (1999) Br. J. Pharmacol. 127, 195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ylä-Herttuala, S., Palinski, W., Butler, S., Picard, S., Steinberg, D. & Witztum, J. (1994) Arterioscler. Thromb. 14, 32–40. [DOI] [PubMed] [Google Scholar]
- 44.Laemmli, U. K. (1970) Nature 227, 680–685. [DOI] [PubMed] [Google Scholar]
- 45.Kyhse-Andersen, J. (1994) J. Biochem. Biophys. Methods 10, 203–209. [DOI] [PubMed] [Google Scholar]
- 46.Verbeuren, T., Bonhomme, E., Laubie, M. & Simonet, S. (1993) J. Cardiovasc. Pharmacol. 21, 841–845. [DOI] [PubMed] [Google Scholar]
- 47.Rubanyi, G. & Vanhoutte, P. (1986) Am. J. Physiol. 250, H822–H827. [DOI] [PubMed] [Google Scholar]
- 48.Ohara, Y., Peterson, T. & Harrison, D. (1993) J. Clin. Invest. 91, 2546–2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lynch, S., Frei, B., Morrow, J., Robers, L., Xu, A., Jackson, T., Reyna, R., Klevay, L., Vita, J. & Keaney, J. (1997) Arterioscler. Thromb. Vasc. Biol. 17, 2975–2981. [DOI] [PubMed] [Google Scholar]
- 50.Hattori, Y., Kawasaki, H., Abe, K. & Kanno, M. (1991) Am. J. Physiol. 261, H1086–H1094. [DOI] [PubMed] [Google Scholar]
- 51.Beckman, J. & Koppenol, W. (1996) Am. J. Physiol. 271, C1424–C1437. [DOI] [PubMed] [Google Scholar]
- 52.Pfeiffer, S. & Mayer, B. (1998) J. Biol. Chem. 273, 27280–27285. [DOI] [PubMed] [Google Scholar]
- 53.Zabel, U., Häusler, C., Weeger, M. & Schmidt, H. (1999) J. Biol. Chem. 274, 18149–18152. [DOI] [PubMed] [Google Scholar]
- 54.Feelisch, M., Kotsonis, P., Siebe, J., Clement, B. & Schmidt, H. (1999) J. Pharmacol. Exp. Ther. 56, 243–254. [DOI] [PubMed] [Google Scholar]
- 55.Galle, J., Bauersachs, J., Busse, R. & Bassenge, E. (1992) Arterioscler. Thromb. 12, 180–186. [DOI] [PubMed] [Google Scholar]
- 56.Schmidt, K., Klatt, P., Graier, W., Kostner, G. & Kukovetz, W. (1992) Biochem. Biophys. Res. Commun. 182, 302–308. [DOI] [PubMed] [Google Scholar]
- 57.Schmidt, K., Klatt, P. & Mayer, B. (1993) Arterioscler. Thromb. 13, 1159–1163. [DOI] [PubMed] [Google Scholar]
- 58.Schmidt, H., Lohman, S. & Walter, U. (1993) Biochim. Biophys. Acta 1178, 153–175. [DOI] [PubMed] [Google Scholar]
- 59.Warnholtz, A., Mollnau, H., Heitzer, T., Kontush, A., Moller-Bertram, T., Lavall, D., Giaid, A., Beisiegel, U., Marklund, S. L., Walter, U., et al. (2002) J. Am. Coll. Cardiol. 40, 1356–1363. [DOI] [PubMed] [Google Scholar]
- 60.Sinnaeve, P., Chiche, J., Nong, Z., Varenne, O., Pelt, N., Gillijns, H., Collen, D., Bloch, K. & Jannssens, S. (1998) Circulation Res. 88, 103–109. [DOI] [PubMed] [Google Scholar]
- 61.Teng, C., Wu, C., Ko, F., Lee, F. & Kuo, S. (1997) Eur. J. Pharmacol. 320, 161–162. [DOI] [PubMed] [Google Scholar]