

Abstract
An enhanced thrombotic environment and premature atherosclerosis are key factors for the increased cardiovascular risk in diabetes. The occlusive vascular thrombus, formed secondary to interactions between platelets and coagulation proteins, is composed of a skeleton of fibrin fibres with cellular elements embedded in this network. Diabetes is characterised by quantitative and qualitative changes in coagulation proteins, which collectively increase resistance to fibrinolysis, consequently augmenting thrombosis risk. Current long-term therapies to prevent arterial occlusion in diabetes are focussed on anti-platelet agents, a strategy that fails to address the contribution of coagulation proteins to the enhanced thrombotic milieu. Moreover, antiplatelet treatment is associated with bleeding complications, particularly with newer agents and more aggressive combination therapies, questioning the safety of this approach. Therefore, to safely control thrombosis risk in diabetes, an alternative approach is required with the fibrin network representing a credible therapeutic target. In the current review, we address diabetes-specific mechanistic pathways responsible for hypofibrinolysis including the role of clot structure, defects in the fibrinolytic system and increased incorporation of anti-fibrinolytic proteins into the clot. Future anti-thrombotic therapeutic options are discussed with special emphasis on the potential advantages of modulating incorporation of the anti-fibrinolytic proteins into fibrin networks. This latter approach carries theoretical advantages, including specificity for diabetes, ability to target a particular protein with a possible favourable risk of bleeding. The development of alternative treatment strategies to better control residual thrombosis risk in diabetes will help to reduce vascular events, which remain the main cause of mortality in this condition.
Keywords: Fibrinolysis, Diabetes, Fibrinogen
Background
Diabetes is becoming the epidemic of the twenty first century with more than 1.5 million deaths worldwide directly attributed to this condition in 2012 (http://www.who.int). Despite advances in therapy, cardiovascular disease (CVD) remains the main cause of morbidity and mortality in individuals with diabetes. The risk of vascular complications is doubled in diabetes, resulting in significant reduction in life expectancy [1, 2]. Moreover, following a vascular event, the outcome in patients with diabetes is worse compared with individuals with normal glucose metabolism, regardless of the therapeutic strategy used in the acute stage [3–7]. There are two key reasons for the adverse vascular outcome in patients with diabetes. The first is related to more extensive vascular pathology and the second involves an enhanced thrombotic environment [8]. There are two main types of diabetes, one is characterised by insulin deficiency, termed type 1 diabetes (T1DM), and the other, type 2 diabetes (T2DM), arising mainly due to insulin resistance secondary to increased prevalence of obesity. However, the two conditions can overlap as a significant proportion of individuals with T1DM develop a phenotype seen in T2DM, making them fall into a new category termed double diabetes [9]. Equally, longer duration of T2DM can lead to insulin deficiency, making these individuals similar to T1DM patients.
Blood clot formation occurs secondary to interactions between the cellular and protein arms of coagulation, resulting in a network of fibrin fibres populated by various blood cells. Diabetes is characterised by enhanced activation of platelets as well as the formation of compact fibrin networks that are resistant to fibrinolysis [10–15], consequently increasing thrombosis risk.
Although the presence of a prothrombotic environment in diabetes is well documented, current anti-thrombotic treatment strategies aiming to reduce vascular risk are largely similar in those with and without diabetes. Moreover, therapies are mainly directed to control platelet activation and the fibrin network is not usually targeted unless there are additional pathologies such as atrial fibrillation or valvular heart disease. Therefore, in order to reduce residual vascular risk in diabetes, more effective anti-thrombotic treatment strategies are required.
This review will summarise current knowledge in fibrin network abnormalities in diabetes with special emphasis on fibrinolysis. The work will also explore diabetes-specific novel anti-thrombotic therapeutic targets aiming to reduce the unacceptably high risk of vascular disease in individuals with diabetes.
Clot formation and lysis
The formation of a fibrin clot is the final step in the atherothrombotic process, involving complex interactions between platelets and plasma coagulation proteins. After rupture of an atherosclerotic plaque, platelets adhere to the site of injury and become partially activated. Exposed tissue factor (TF) binds factor VII (FVII), promoting proteolysis and activation to FVIIa. The TF/FVIIa complex activates FIX and FX resulting in the generation of FIXa and FXa, and the latter subsequently associates with cofactor FVa to form a prothrombinase complex, which converts prothrombin into thrombin [16]. The generated thrombin converts soluble fibrinogen into insoluble fibrin fibres. Thrombin also activates FXIII, a transglutaminase that crosslinks neighbouring fibrin fibres resulting in the formation of a branched fibrin clot structure, which is more resistant to lysis [17]. Activated FXIII (FXIIIa) crosslinks other proteins into the fibrin network, including plasmin inhibitor (PI) [18], thrombin activatable fibrinolysis inhibitor (TAFI) [19] and plasminogen activator inhibitor-2 (PAI-2) [20], further increasing resistance of the clot to lysis. The coagulation cascade results in the formation of a fibrin mesh, with embedded erythrocytes and other cellular blood elements, which can occlude the vascular lumen, ultimately resulting in the acute complications of CVD, including myocardial infarction, stroke and critical limb ischaemia.
Normal physiology ensures a balance between fibrin clot formation and lysis in order to prevent widespread vascular occlusion or excessive bleeding. Plasmin is the enzyme responsible for fibrin breakdown (fibrinolysis) and is generated from plasminogen through the action of the serine protease tissue plasminogen activator (tPA), a product of endothelial cells [21]. The binding of tPA to fibrin increases the catalytic conversion of plasminogen to plasmin, while also localising the generation of plasmin to the site of thrombus formation. Plasmin cleavage of fibrin generates new C-terminal lysine residues (additional tPA/plasminogen binding sites) within the fibrin network [22].
Several proteins prevent unregulated plasmin or plasminogen activator activity, thereby inhibiting excessive clot lysis. Plasminogen activator inhibitor (PAI-1) is produced by endothelial cells, platelets and adipose tissue. Like other serpins (serine protease inhibitors), PAI-1 forms a stable 1:1 complex with tPA, thereby inhibiting its action [23]. Plasmin inhibitor (PI), another member of the serpin family, is the primary physiological inhibitor of plasmin. PI forms a stable inactive complex with plasmin, and can also be covalently cross-linked to fibrin making fibrin more resistant to lysis [24, 25]. Importantly, the fibrinolytic inhibitory properties of PI are significantly enhanced once the protein is cross-linked into the fibrin network. Thrombin activatable fibrinolysis inhibitor (TAFI) cleaves C-terminal lysine residues from partially degraded fibrin, decreasing the number of available plasminogen binding sites [26]. The main factors involved in clot formation and lysis are summarised in Fig. 1. Altered level and/or activity of these antifibrinolytic factors can modulate plasma clot lysis, and thereby thrombosis risk. Several studies have shown an association between hypofibrinolysis and increased thrombosis risk [27–30].
Fig. 1.
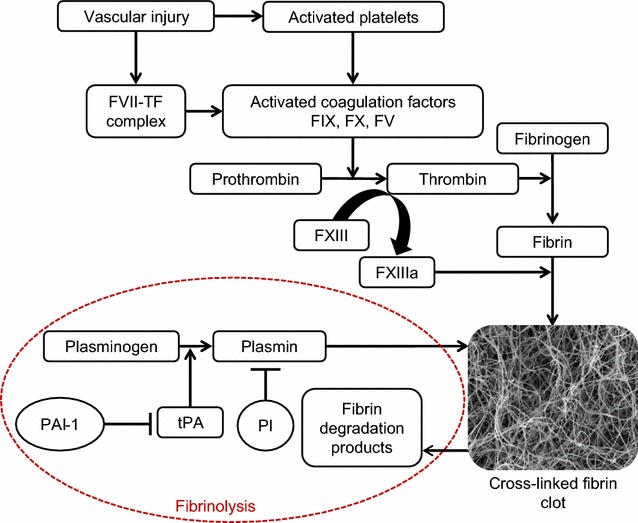
Fibrin clot formation and lysis. Following vascular injury, tissue factor (TF) is released followed by a complex interaction between the cellular and protein arms of coagulation, resulting in activation of various coagulation factors, including FV, FVII, FIX and FX, culminating in the formation of thrombin, which converts soluble fibrinogen into a network of insoluble fibrin fibres. Thrombin also activates factor XIII (FXIII) to active FXIII (FXIIIa), which crosslinks the fibrin fibres and also incorporates antifibrinolytic proteins into the clot. Plasmin, derived from plasminogen by the action of tissue plasminogen activator (tPA), is the enzyme responsible for fibrin clot lysis, resulting in the generation of fibrin degradation products. The main inhibitors of fibrinolysis are plasmin inhibitor (PI) and plasminogen activator inhibitor (PAI-1)
Factors involved in increased vascular risk in diabetes
Individuals with diabetes suffer from premature atherosclerosis, a key contributing factor to increased cardiovascular risk in this population. This is treated in the acute stage with revascularisation followed by preventative therapy in the long-term, including control of blood pressure, lipid and glucose levels. To control thrombosis risk following an acute vascular event, dual antiplatelet therapy is usually used [31, 32], whereas treatment targeting the fibrin network is not considered except in cases of cardiac arrhythmias, valvular heart disease or history of venous thromboembolism. Factors leading to increased vascular events in diabetes and current treatment modalities are summarised in Fig. 2.
Fig. 2.
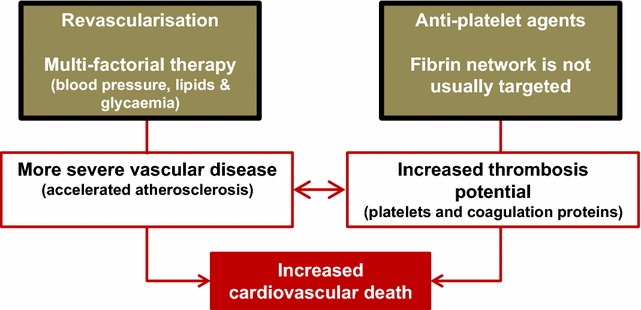
Mechanisms for increased risk of vascular occlusive events in diabetes. Individuals with diabetes have premature atherosclerosis, which increases the risk of a coronary event. Individuals with coronary artery occlusion are treated with revascularisation in the acute stage followed by multifactorial therapy to halt the atherosclerotic process including control of blood pressure, blood glucose and lipid levels. Diabetes is also associated with an enhanced thrombotic milieu, secondary to increased activation of both platelets and coagulation factors. This is treated with anti-platelet therapy and the protein arm of coagulation is not usually targeted except in those with arrhythmias, valvular heart disease or a history of venous occlusion
The risk of aggressive anti-thrombotic therapy is increased bleeding which may negate any beneficial effect of such therapy. A prime example is the TRITON study that has shown that the combination of prasugrel and aspirin is superior to clopidogrel and aspirin at reducing vascular ischaemia following myocardial infarction but there was an increased risk of bleeding with the former combination making widespread clinical use problematic [33]. Interestingly, patients with diabetes showed a benefit with prasugrel/aspirin therapy without increased risk of bleeding suggesting altered response to anti-thrombotic therapy in this population [32, 34].
Current anti-thrombotic strategies: pitfalls
Reducing the thrombotic environment in arterial occlusive disease relies mainly on antiplatelet therapy, using the cyclooxygenase inhibitor aspirin with or without P2Y12 antagonists clopidogrel, prasugrel or ticagrelor. While all anti-platelet agents are likely to alter clot structure through indirect effects on platelet-dependent thrombin formation, aspirin is unique amongst anti-platelet agents as it directly alters clot structure and enhances fibrinolysis, related, at least in part, to acetylation of fibrinogen. This makes aspirin an agent that independently targets both the cellular and protein phase of coagulation [35–37]. It remains unclear, however, whether the pro-fibrinolytic effects of aspirin are important clinically and this remains an area for future research. It should be noted that other agents used in diabetes such as hypoglycaemia therapies, anti-hypertensive agents and statins may affect the thrombotic milieu, which is reviewed elsewhere and is not the focus of the current review [38].
Antithrombotic therapy in diabetes can be divided into primary prevention, which includes individuals without a previous history of vascular ischaemia, and secondary prevention in those who have had a vascular occlusive event. Until relatively recently, aspirin has been used for primary vascular protection in diabetes but a number of studies suggest a lack of significant success with such an approach [39]. Therefore, guidelines generally advocate aspirin for primary vascular protection in subgroups of diabetes patients at a higher vascular risk, without clearly identifying these subgroups [8]. Until data becomes available from large primary prevention studies in diabetes, such as ASCEND (NCT00135226), routine aspirin therapy for primary vascular protection in diabetes should be avoided and limited to individuals with multiple risk factors [40].
In individuals sustaining a coronary event, current guidelines advocate the use of dual antiplatelet therapy (DAT) to reduce thrombosis risk for a period of 1 year, followed by lifelong antiplatelet monotherapy [8]. Interestingly, there is generally no differentiation between individuals with and without diabetes when it comes to DAT, despite the documented increased thrombotic environment in those with impaired glucose metabolism. Limited evidence suggests that prasugrel is more effective than clopidogrel at reducing further atherothrombosis in those with diabetes, however ticagrelor appears to have the best risk/benefit profile in those with and without diabetes, and therefore many centres use this agent for all patients regardless of glycaemic status [6, 41].
One year after the vascular event, P2Y12 inhibitor therapy is usually stopped and patients continue on aspirin treatment. However, this approach is questionable in diabetes as the efficacy of aspirin appears to be compromised, secondary to a number of factors including increased platelet protein glycation and higher platelet turnover in diabetes [42]. Improving glycaemia may address the first point but the latter can only be overcome by more frequent aspirin dosing. Indeed, a number of studies have shown a benefit in twice daily aspirin administration on platelet function profile, although outcome studies employing this dosing regimen are lacking [43–45].
One striking observation of clinical studies aiming to reduce atherothrombosis risk is the concentration on antiplatelet therapy, generally ignoring the contribution of the protein arm of coagulation to the vascular ischaemic event. Early work has shown that warfarin, an agent that inhibits the production of various coagulation proteins, can indeed improve outcome after arterial occlusion. However this agent is associated with a narrow safe therapeutic window and the combination with antiplatelet agents greatly increases bleeding risk [46]. More modern agents that affect coagulation proteins, such as FX inhibitor, show a greater promise although their use is still limited by the high bleeding risk when used with DAT [47].
FXII inhibitors offer a potential alternative antithrombotic strategy in diabetes that do not carry the bleeding risk associated with the aforementioned agents. FXII initiates the intrinsic coagulation pathway, however, FXII deficiency in mice is not associated with increased bleeding and offers protection against thrombosis. FXII inhibition may therefore be useful in instances involving activation of the intrinsic pathway (such as stent thrombosis) but may not be appropriate if the underlying pathology is plaque rupture and future research in this area is certainly needed [48].
It should be remembered that acute fibrinolytic therapy has been successfully employed as a treatment for acute vascular occlusion and therefore strategies that facilitate clot lysis on a long-term basis have the potential to control the risk of atherothrombosis, particularly in conditions characterised by impaired fibrinolysis.
Although the current review concentrates on macrovascular complications in diabetes, studies suggest that hypofibrinolysis is also associated with microvascular complications [49–51]. Therefore, addressing the hypofibrinolytic environment in diabetes may help not only by ameliorating macrovascular complications but also by reducing microvascular disease.
Factors involved in increased thrombosis risk in diabetes and current anti-thrombotic strategies are summarised in Table 1.
Table 1.
Current therapeutic interventions to reduce thrombosis risk in diabetes with their main limitations
| Risk factor | Cause | Current therapeutic intervention | Pitfalls of current therapeutic strategy |
|---|---|---|---|
| Premature atherosclerosis | Metabolic abnormalities Inflammatory changes resulting in endothelial dysfunction |
Treated in the acute stage with revascularisation Control of blood pressure, lipid and glucose levels in the long term |
Mainly preventative Limited ability to reverse vascular pathological changes |
| Prothrombotic environment | Enhanced platelet activity Altered levels of coagulation factors |
Dual antiplatelet therapy (DAT) for 1 year following an acute coronary event Lifelong platelet monotherapy after 1 year |
Aspirin resistance in individuals with diabetes Bleeding risk No targeting of the fibrin network |
| Hypofibrinolysis | Formation of more compact clots Impaired fibrinolytic system |
Not usually targeted Agents that modulate the coagulation cascade, which are likely to affect fibrinolysis, are used in the presence of cardiac arrhythmias, valvular heart disease or venous thrombosis |
Adding an agent that targets the fibrin network, particularly on a background of DAT, increases the risk of bleeding complications |
Mechanisms of hypofibrinolysis
Hypofibrinolysis is a key abnormality in individuals with diabetes and therefore modulation of this pathological process may offer therapeutic benefits. The main factors influencing fibrinolysis in diabetes include altered fibrin network structure, increased incorporation of antifibrinolytic proteins into the clot and compromised activity of the fibrinolytic system.
Clot structure in diabetes and factors contributing to altered clot phenotype
Compact fibrin networks with densely packed thin fibres are associated with increased risk of cardiovascular events [52–57], which may be due to reduced permeation of fibrinolytic enzymes into clots with these structures [58]. Although single thick fibrin fibres are cleaved at a slower rate than thin fibres, clots made from an increased number of more densely packed thin fibres are slower to lyse [59]. Additionally, thinner fibrin fibres support a slower rate of tPA-mediated plasmin generation than thick fibres, and a slower rate of fibrin digestion by plasmin [60, 61]. It should be noted that increased incorporation of antifibrinolytic proteins into clots with compact structure further contributes to their resistance to breakdown and therefore the relationship between structure and lysis is more complex than initially envisaged [62].
Individuals with type 1 and type 2 diabetes have prothrombotic compact fibrin networks which correlate with glycaemic control measured as HbA1c [63–66]. To complicate matters, glycaemia-independent factors, such as gender, can also determine fibrin clot structure with female patients exhibiting denser fibrin clots and prolonged lysis time [67]. This may be one of the mechanisms contributing to the loss of cardiovascular protection in women who develop diabetes. Prolonged duration of T2DM (>5 years) is further associated with hypofibrinolysis and a prothrombotic clot phenotype, even if glycaemic control is adequate, adding yet more complexity to studying fibrin network characteristics in this population [68]. These examples emphasise the heterogeneity in thrombosis potential in diabetes and highlight some of the challenges faced in developing safe and effective anti-thrombotic agents in this condition.
Mechanisms for altered clot structure in diabetes
A number of mechanisms are responsible for altered clot structure in diabetes including quantitative and qualitative changes in coagulation factors as well as altered thrombin generation.
Plasma fibrinogen levels
Previous studies have observed a relationship between elevated plasma levels of fibrinogen and the risk of CVD [69–72]. Evidence suggests that high plasma fibrinogen levels influence clot structure, by modulating network density and rigidity through increasing fibre number and branch points. The increased risk of MI associated with elevated plasma fibrinogen levels may be attributed, in part, to the formation of stiffer clots in these individuals [73]. In vivo work has shown that elevated fibrinogen levels reduce time to vascular occlusion, and increase clot fibrin content, network density and resistance to fibrinolysis in a murine model [74], linking changes in fibrin network to increased risk of thrombosis.
Plasma levels of fibrinogen in diabetes subjects has been a focus of much research, however, results are not always in agreement. The majority of, but not all, studies found elevated plasma fibrinogen levels in individuals with T2DM, and some reported higher levels in individuals with T1DM (summarised in [38]). The Rotterdam study found no significant difference in fibrinogen levels between individuals with T2DM and those with normal glucose metabolism after adjusting for age. However, plasma fibrinogen was significantly elevated in insulin-treated T2DM patients, which may be due to poorer metabolic control in these individuals or longer disease duration [75].
Taken together, elevated fibrinogen levels appear to contribute to the compact fibrin networks observed in diabetes, at least in some patients.
Fibrinogen glycation
Protein glycation is a post translational modification, the extent of which is determined by ambient glucose levels and the duration of protein exposure. Glucose is able to bind non-enzymatically to fibrinogen by condensation of the carbonyl groups with amino groups on the fibrinogen molecule. Elevated glucose levels increase plasma protein glycation, including that of fibrinogen [76–78], with a number of lysine residues becoming glycated [79]. This may contribute to more compact clots in diabetes, which in turn increases resistance to fibrinolysis [80, 81]. Moreover, as lysine is involved in the binding of tPA and plasminogen to fibrin, it is possible that fibrinogen glycation reduces binding of fibrinolytic proteins further impairing clot lysis [81]. In support of this hypothesis, Dunn et al. observed a reduction in plasmin generation in individuals with diabetes, attributed to reduced tPA and plasminogen binding to fibrin [65].
Fibrinogen glycation in subjects with diabetes is modulated by improving glycaemic control [76, 78, 82]. Pieters et al. observed that improving glycaemia had no effect on the structure of fibrin clots made from T2DM plasma although an effect on clots made from purified fibrinogen was observed [80, 83]. The discrepancy between plasma and purified systems can be explained by the heterogeneity observed in plasma of patients with T2DM and the relatively small number of samples analysed.
It should be noted, however, that glycation is not the only post-translational modification of fibrinogen in diabetes [63]. Diabetes is also associated with elevated levels of reactive oxygen species [84], which are able to covalently modify protein structure, including that of fibrinogen [85]. Elevated levels of oxidative stress markers have been documented in T2DM, levels of which were inversely correlated with clot permeability and positively correlated with clot lysis time [86]. Therefore, both glycation and oxidation influence fibrin network properties in diabetes.
Thrombin generation
Fibrin fibre diameter decreases with increasing thrombin levels. Low thrombin concentrations (<1 nM, <0.1 U/ml) produce clots composed of loosely woven, thick fibres, whilst clots formed at high thrombin tend to be made up of thin, tightly packed fibrin strands which are relatively resistant to lysis [87, 88]. The concentration of thrombin observed during a coagulation reaction ranges from <1 to >500 nM [89], although low levels of thrombin (~2 nM) are sufficient for fibrin polymerisation [90].
Elevated thrombin generation has been reported in subjects with T2DM [68, 91–93]. In acute coronary syndrome, hyperglycaemia is associated with enhanced thrombin generation at the site of vascular injury and unfavourably altered clot features in patients with and without a history of diabetes [94]. Indeed, controlling glucose levels results in reduced thrombin generation [95]. On the other hand, over treatment of hyperglycaemia and precipitation of hypoglycaemia is associated with enhanced thrombin formation and the formation of more dense fibrin clots with resistance to lysis [96]. This indicates that both hyper and hypoglycaemia are prothrombotic and caution should be exercised to avoid low blood glucose with hypoglycaemic therapy.
The mechanisms for enhanced thrombin generation in individuals with diabetes are not entirely clear. A study involving T1DM and T2DM patients showed that high levels of coagulation factors II,V,VII,VIII and X, coupled with low levels of anticoagulation factor protein C in diabetes are likely to be factors contributing to enhanced thrombin generation [91]. In another study, a modest elevation in thrombin generation in individuals with T2DM as well as those with impaired glucose tolerance was observed. In these individuals, central adiposity and related low grade inflammation, rather than glucose levels per se, were the likely explanations for enhanced thrombin generation [97].
Interaction between inflammatory and thrombotic proteins
It is well established that low grade inflammation predisposes to atherothrombosis and a number of pathways have been implicated describing interactions between inflammatory and thrombotic molecules, reviewed elsewhere [98–100]. For example, cytokines, important mediators of inflammation, can directly affect thrombosis risk by creating a hypercoagulable milieu and enhancing platelet reactivity [101]. Another example is complement C3, a key regulator of inflammatory responses, a molecule that is incorporated into the clot and modulates fibrinolytic potential (discussed below).
Increased incorporation of anti-fibrinolytic proteins
A potentially important but overlooked diabetes-related mechanism for hypofibrinolysis is increased incorporation of antifibrinolytic proteins into the clots, specifically complement C3 and PI.
Complement C3
Until recently, complement C3 was regarded as an inflammatory protein, but evidence suggests key interactions between the complement and coagulation systems [102]. Two studies, using a proteomics approach, demonstrated the presence of C3 and its metabolites in plasma clots [103, 104]. C3 protein can be covalently crosslinked to fibrin by FXIIIa, as well as weakly associating with the clot via non-covalent interactions [105, 106].
C3 binding/cross-linking into the fibrin network increases resistance of the clot to lysis [104]. Further work from our laboratory has demonstrated that incorporation of C3 into the fibrin clots of T1DM subjects has a greater effect on prolongation of clot lysis compared with clots from healthy controls [107]. The association between elevated C3 plasma levels and prolonged clot lysis has also been observed in T2DM [108, 109]. In the study by Hess et al. of 875 patients with T2DM, a regression analysis involving PAI-1, fibrinogen, C3 and CRP plasma levels, demonstrated that C3 was an independent predictor of fibrinolysis potential in contrast to CRP which failed to show an independent association with clot lysis.
The mechanisms for C3-induced compromise in clot lysis are not entirely clear but one may involve C3 interference with tPA and/or plasminogen binding sites on the fibrin network. Alternatively, C3 may affect the availability of plasmin to act on fibrin fibres, as C3 is a known substrate for plasmin [110]. Finally, the presence of elevated levels of C3 in the fibrin clot may simply increase mechanical resistance of the clot to lysis [107]. From the therapeutic point of view, these findings suggest that targeted disruption of C3-fibrinogen interaction may offer a diabetes-specific anti-thrombotic treatment strategy.
Plasmin inhibitor (PI)
Plasmin inhibitor (α2-antiplasmin, α2-plasmin inhibitor) belongs to the serpin superfamily of proteins. PI is a key protein in blood haemostasis, as evidenced by the disorders caused by homozygous deficiency or by protein over-production. Congenital deficiency of PI results in a severe bleeding disorder [111, 112], whilst elevated levels of PI are associated with an increased risk of first MI [113]. PI is the main biological inhibitor of plasmin, free PI is able to bind plasmin and form an irreversible stable complex, however, the fibrin-bound form of PI seems to be the main regulator of clot lysis [114]. Cross-linking of PI to the fibrin clot by FXIIIa has been shown to be a key determinant of resistance to lysis [25].
Agren et al. found increased incorporation of PI into the fibrin network in individuals with T1DM, although these individuals exhibited a paradoxical reduction in clot lysis time, which the authors attributed to reduced PAI-1/fibrinogen levels [115]. An earlier study by Dunn et al. found increased cross-linking of PI into the fibrin networks in T2DM, which increased resistance to lysis compared with controls [65]. This provides yet another diabetes-specific therapeutic target by manipulating PI incorporation into fibrin networks.
Compromised efficiency of the fibrinolytic system
A number of abnormalities in the fibrinolytic system have been reported in diabetes including elevated PAI-1 and TAFI levels, and deranged plasminogen function.
TAFI in diabetes
There is some evidence to suggest that antifibrinolytic protein TAFI is upregulated in diabetes, which could contribute to the hypofibrinolytic state. Elevated plasma levels of TAFI are associated with increased risk of cardiovascular diseases [116–120]. In diabetes, TAFI levels appear to correlate with HbA1c, indicating an association between TAFI levels and poor glycaemic control [121]. Plasma concentration and activity of TAFI are significantly increased in T2DM patients compared with healthy controls, particularly in obese patients [121]. TAFI levels have also been linked in T2DM to the presence of microvascular complications manifesting as microalbuminuria [122, 123]. However, Chudý et al. found no significant difference in TAFI levels when normoalbuminuric T2DM patients were compared with controls, and Yener and colleagues have demonstrated that TAFI levels do not increase in normotensive T2DM subjects without diabetic complications [123, 124]. To further complicate matters, others found TAFI levels to be decreased in nonobese T2DM individuals [125]. Taken together, these data reflect inconsistencies in the relationship between TAFI and diabetes, which is perhaps related to the heterogeneity observed in these patients. Therefore, studies including large numbers of diabetes patients are required, which would allow appropriate subgroup analysis, to fully understand the role of TAFI in diabetes.
PAI-1 in diabetes
PAI-1 levels are associated with increased risk of CVD [126] with elevated protein levels found in young (<45 years) survivors of myocardial infarction [127] and those with recurrent MI [128]. For many years, PAI-1, produced by endothelial cells and adipose tissue, has been regarded as the main inhibitor of fibrinolysis in diabetes. However, there is a difference between T1DM and T2DM as PAI-1 levels appear to be only elevated in the latter and correlate with glycaemic parameters as well as markers of insulin resistance [115, 129–133]. Hormonal (hyperinsulinemia) and metabolic (hyperglycemia and hypertriglyceridemia) derangements, typically found in T2DM patients, seem to have a role in elevated levels of PAI-1 levels in this population [130]. Stegenga et al. demonstrated that in healthy subjects, hyperinsulinemia inhibits fibrinolysis, primarily by enhancing PAI-1 secretion, whilst hyperglycaemia stimulates coagulation [134]. This group suggests that in T2DM subjects, the presence of both hyperinsulinemia and hyperglycaemia has a procoagulant effect with the simultaneous inhibition of fibrinolysis.
There is also an association of elevated PAI-1 levels and abdominal obesity as adipose tissue expresses PAI-1 and represents an important source of plasma PAI-1 in obese subjects [131–133]. This explains that the combination of T2DM and obesity contribute to an even greater elevation of PAI-1 than obesity or diabetes alone [135].
Plasminogen in diabetes
The hyperglycaemic environment in diabetes is responsible for glycation of plasminogen, which compromises protein function. Plasminogen purified from plasma of individuals with T1DM has a decreased rate of conversion to plasmin, and the plasmin generated has altered proteolytic activity, resulting in impaired fibrinolysis. Moreover, improving glycaemic control partially restores plasminogen conversion to plasmin and the activity of the enzyme, further emphasising the importance of maintaining good glycaemic control to reduce thrombosis risk. Interestingly, only a modest improvement in glycaemic control is enough to result in significant improvement in plasmin activity [136]. This suggests small improvements in glycaemic control translate clinically into significant reduction in thrombosis risk.
Therefore, hyperglycaemia enhances thrombosis potential by increasing plasma levels of pro-coagulant and antifibrinolytic factors as well as modulating protein activity by introducing post-translational modifications in coagulation proteins. It should be noted, however, that not every single factor that modulates fibrinolysis has been comprehensively studied in diabetes, particularly in relation to the various subgroups of patients. Therefore, in addition to developing agents that target the large number of anti-fibrinolytic proteins, future studies are required to understand the most relevant anti-fibrinolytic strategy in the different subgroups of diabetes patients. This may indeed involve targeting multiple proteins/pathways, particularly in the higher risk groups, to maximise clinical benefit.
A summary of the mechanisms involved in hypofibrinolysis in diabetes is provided in Fig. 3.
Fig. 3.
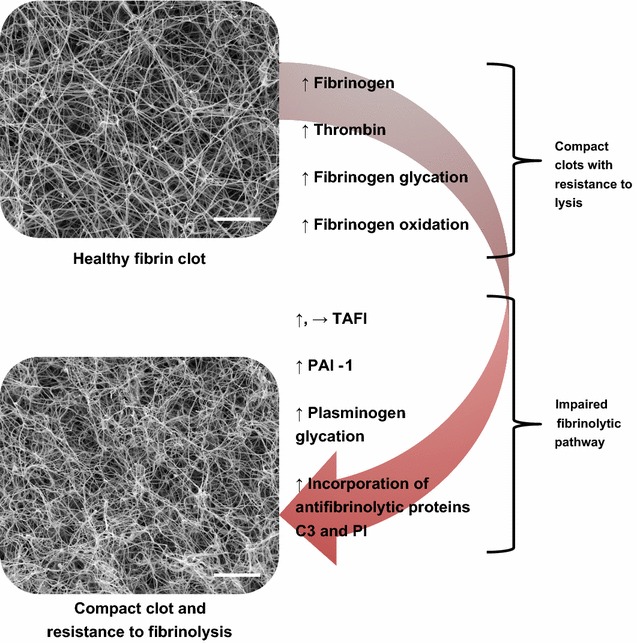
Mechanisms involved in hypofibrinolysis in diabetes. The main factors influencing hypofibrinolysis in diabetes include altered fibrin network structure and an impaired fibrinolytic system. Factors contributing to altered clot structure include elevated levels of thrombin, and both quantitative and qualitative alterations in fibrinogen, including glycation and oxidation of fibrinogen molecules. Elevated levels of plasminogen activator inhibitor (PAI-1), glycation of plasminogen and increased incorporation of antifibrinolytic proteins plasmin inhibitor (PI) and complement C3 into clots in individuals with diabetes all contribute to impaired fibrinolysis. Levels of thrombin activatable fibrinolysis inhibitor (TAFI) can be raised in diabetes but studies are conflicting with some showing no change. Scale bar 5 μm. ↑, increase; →, no change
Current approaches to reduce hypofibrinolysis in diabetes
The potential role of various hypoglycaemic agents in thrombosis risk is beyond the scope of this review and will therefore concentrate on the effect of glycaemia per se on thrombosis potential.
Role of glycaemia
From the evidence presented above, it is clear that hyperglycaemia results in a prothrombotic and hypofibrinolytic environment. Moreover, relatively modest improvement in glycaemia appears to have a significant effect on fibrin network structure and/or resistance to lysis. It should be noted, however, that overtreatment of hyperglycaemia, and precipitation of hypoglycaemia, can also be prothrombotic [137]. Studies have shown that hypoglycaemia results in elevated fibrinogen and PAI-1 levels [138]. This is consistent with our findings of impaired fibrinolysis following hypoglycaemic clamps in diabetes individuals, with this enhanced prothrombotic milieu lasting for up to one week after the hypoglycaemic event [139]. The observation that both hyper and hypoglycaemia are prothrombotic adds another dimension to the management of this risk factor, particularly as glucose levels can fluctuate significantly in diabetes patients secondary to daily activities, diet and hypoglycaemic therapies.
Taken together, the above findings may offer mechanistic explanations for the disappointing clinical outcome trials investigating the role for tight glycaemic control in reduction of vascular ischaemic events in diabetes [140]. It is plausible that mild improvement in glucose is all that is needed to control the prothrombotic environment in diabetes. Trying to achieve too tight control runs the risk of repeated hypoglycaemia, predisposing to an enhanced thrombotic environment, thus negating any beneficial effect for reducing blood glucose levels.
From the practical point of view, using agents that are less likely to cause hypoglycaemia may have the advantage of reducing the thrombotic environment in diabetes and protecting against vascular ischaemic events. We have limited evidence to suggest that agents that do not cause hypoglycaemia, such as metformin, pioglitazone, empagliflozin and liraglutide are associated with favourable cardiovascular profile [141–144]. In contrast, agents that may result in hypoglycaemia such as sulphonylurea and insulin have been linked to increased cardiovascular risk [145]. However, there are complexities encountered in dissecting out the effect of each agent, given that most high risk individuals are on combination therapy, and therefore further research in this area is needed before concrete conclusions can be made.
Potential diabetes–specific therapeutic targets to reduce hypofibrinolysis
Given that diabetes is associated with increased plasma levels of PAI-1 and TAFI, and increased incorporation of PI and C3 into the clot, targeting these proteins may alleviate the hypofibrinolytic environment, consequently decreasing atherothrombotic risk.
TAFI as a drug target
TAFI circulates in an inactive zymogen form and is activated by thrombin, plasmin, or the thrombin-thrombomodulin complex. Activated TAFI cleaves C-terminal lysine residues from partially degraded fibrin, which are critical for the binding of plasminogen and as a result, plasmin generation is reduced [146].
Inhibition of TAFI was considered as a therapeutic strategy in thrombotic disorders but only a limited number of drug candidates have made it to clinical trials, which were then discontinued (reviewed elsewhere [147, 148]). More recent work has investigated the TAFI-inhibitory ability of TAFI-derived peptides on the protein’s activation and activity. Peptides with the ability to prevent TAFI activation, and inhibit TAFIa activity directly were identified [149]. An alternative anti-TAFI approach by Buelens et al. created a panel of inhibitory nanobodies effective against the various modes of TAFI activation and activity. Nanobodies are single domain antibodies from the sera of members of the Camilidae family which have advantageous properties such as low immunogenicity and high affinity, solubility and stability [150]. Two nanobodies showed a potent profibrinolytic effect in an in vitro clot lysis assay and their interaction with TAFI was later characterised using X-ray crystallography. One nanobody was shown to bind close to the TAFI activation site, and the other close to a possible thrombomodulin binding site. These findings explained the interference of the two nanobodies with TAFI activation, and thrombin-thrombomodulin-mediated activation, respectively [151]. Although these studies are promising and shed light on the mechanistic properties of TAFI-inhibitory compounds, in vivo data are lacking.
PAI-1 as a drug target
PAI-1 is present in an active and a latent state in vivo with the active form having high affinity for vitronectin, which binds PAI-1 in the blood [152]. PAI-1 has a relatively short half-life of ~ 1 h in physiological conditions [153, 154] which some may argue would limit its use as a therapeutic target. The counter-argument, however, is that the short half-life makes it a suitable target for acute vascular thrombosis. Several PAI-1 inhibitors have been tested in vivo and in vitro and displayed inhibitory activity, but to date no PAI-1 inhibitor is clinically available. Prevention of cellular PAI-1 biosynthesis is another strategy which has been partially explored.
Small molecule inhibitors of PAI-1 synthesis
Small molecule inhibitors of PAI-1 synthesis include synthetic and natural products such as niacin, fibrates and butadiene derivatives (reviewed in [114]). However, these compounds are not PAI-1 specific, having other metabolic effects and therefore dissecting out their role on clinical outcome is difficult. Also, in vitro studies do not always translate into in vivo benefits and an example of the disparity can be seen with fibrate use; a recent meta-analysis has reported that fibrates do not cause a decrease in PAI-1 levels or activity, despite their in vitro effects [155]. Therefore, direct PAI-1 inhibitors may have an advantage by targeting plasma PAI-1.
Direct PAI-1 inhibitors
Antibodies against PAI-1
There have been numerous attempts at inhibiting PAI-1 with antibodies, reviewed elsewhere [148, 156] but none have been taken forward to the clinical arena. Recent work in mouse models of thrombotic stroke have shown that simultaneous inhibition of PAI-1 and TAFI with a bispecific antibody results in a significant enhancement of fibrinolysis in mice, without increased bleeding [157]. To dissect out the contribution of PAI-1 inhibition and TAFI inhibition during ischaemic stroke, Denorme et al. used monoclonal antibodies to identify the effect of inhibiting each of these antifibrinolytic factors. In a mouse model of transient middle cerebral artery occlusion, inhibition of TAFI or PAI-1 significantly decreased fibrin(ogen) deposition in the ischaemic brain, thereby improving reperfusion, but the combined inhibition had an additive beneficial effect [158]. PAI-1 inhibition is also possible with nanobodies, which was demonstrated by Zhou et al. who used nanobodies against PAI-1 to inhibit profibrinolytic activity in an in vitro clot lysis assay [159]. Clearly there are some promising benefits in the inhibition of PAI-1 with antibodies, which merit further in vitro and in vivo analysis.
Other PAI-1 inhibitors
A number of groups have employed different methods for identification of PAI-1 inhibitors, including high throughput and in silico screening, reviewed extensively elsewhere [114, 160]. Using this methodology, Elokdah and colleagues identified Tiplaxtinin as a potent and selective PAI-1 inhibitor [161], which became one of the most studied PAI-1 inhibitors to date. Tiplaxtinin binds specifically to the active conformation of PAI-1, its activity, however, is blocked by vitronectin, suggesting their binding sites on PAI-1 are overlapping [162]. In a canine model of occlusive thrombus formation, administration of Tiplaxtinin caused spontaneous coronary reperfusion, indicating active fibrinolysis. When Tiplaxtinin was administered to rats following induction of an occlusive arterial thrombosis, thrombus weight was reduced and blood flow facilitated with increased occlusion time in the affected vessel. In both these models, there were no side effects reported such as changes in heart rate, blood pressure or increased bleeding [161].
Other small molecule inhibitors have been identified, such as ZK4044 which binds directly to PAI-1, preventing its interaction with target proteases [163], WAY-140312 which binds and inactivates PAI-1 [164], and S35225 a benzothiophene derivative which, in contrast to WAY-140312 and Tiplaxtinin is able to inhibit PAI-1 in the presence of vitronectin [165]. Despite promising candidates, there has been no progression of PAI-1 inhibitors to clinical trials in man [166]. One explanation for this lack of progress could be the unique structural properties of PAI-1. Crystallographic data of PAI-1 in complex with vitronectin is still incomplete and thus hampers rational design of small molecules able to bind and inactivate PAI-1 in its vitronectin-bound state. Additionally, the active form of PAI-1 is yet to be crystallised due to its instability, and the mechanism by which active PAI-1 transitions to its latent form is not fully understood. Although numerous PAI-1 inhibitory compounds have been investigated, for many, their mechanism of action remains elusive, providing yet another obstacle in the clinical application of PAI-1 inhibitors.
Plasmin inhibitor as a drug target
The reported increased incorporation of PI into fibrin clots in diabetes renders this protein a potential diabetes-specific therapeutic target. Numerous attempts at targeting PI have been made, including the use of antibodies and mutant forms of the protein.
Antibodies
Kumada et al. reported that repeated injection of polyclonal anti-PI F(ab’)2 fragments reduced circulating PI levels and led to an acceleration of thrombolysis by enhancing fibrinolytic activity [167]. A more targeted approach by Sakata et al. saw the creation of a monoclonal antibody that was able to interfere with the formation of PI-plasmin complexes by recognising an epitope either in or close to the reactive site of PI. This antibody, termed JTPI-1, was able to increase the effectiveness of tPA-mediated clot lysis in plasma [168]. Similarly, an antibody capable of inhibiting both PI in plasma and clot-bound PI caused spontaneous clot lysis and also enhanced tPA-mediated clot lysis in vitro [169]. An extension of these studies in vivo by Reed et al. used an antibody targeted to clot-bound PI, which was found to enhance lysis of a human clot in a rabbit jugular vein thrombosis model [170].
Plasmin inhibitor mutants
Arginine residue 364 is the main reactive site on PI, which forms a covalent bond with the active site serine in plasmin, resulting in an inactive protease-inhibitor complex [171, 172]. Lee et al. generated PI with a chemically modified reactive site arginine [173] and an Arg-Ala mutant [174]. Both of these modified PI molecules lost their plasmin-inhibitory activity, but still competed with native PI for crosslinking into the fibrin clot by FXIIIa, thereby enhancing fibrinolysis.
N and C terminal peptides of plasmin inhibitor
The N-terminal region of PI is crucial for cross-linking to fibrinogen [175, 176], whereas the C-terminal of the protein mediates interaction with plasmin [177]. Kimura et al. created a 12 residue synthetic N-terminal peptide of PI that was able to reduce incorporation of native PI into fibrin networks by FXIIIa in vitro. Cross-linking of the synthetic peptide to fibrin accelerated spontaneous as well as tPA-induced fibrinolysis. The quantity of native PI cross-linked to fibrin was proportionally reduced by the presence of the synthetic N-terminal peptide, indicating PI specificity of the synthetic peptide [178].
Others attempted to use the C-terminus of PI as a means of enhancing fibrinolysis. A 26 amino acid peptide composed of the carboxy terminal region of PI was found to enhance in vitro fibrin clot lysis by increasing conversion of plasminogen to plasmin approximately fivefold [179]. Similarly, Udvardy et al. observed that a peptide containing the 26 amino acid C-terminal of PI, coupled with RGD (Arg-Gly-Asp) peptide was capable of simultaneously promoting fibrinolysis and inhibiting platelet aggregation. Fibrinogen RGD sequence mediates its interaction with platelets, RGD peptides are able to disrupt fibrinogen-platelet interactions and have platelet inhibitory effects, as observed in this study [180].
Inhibition of plasmin inhibitor cleavage
Plasmin inhibitor is present in two forms in human plasma, a 464-residue protein with methionine at the N-terminus (Met-PI) and a truncated 452-residue protein with asparagine at its N-terminus (Asn-PI). Human plasma contains around 30% Met-PI and 70% Asn-PI form. Asn-PI, formed from Met-PI by the action of antiplasmin cleaving enzyme (APCE), is more rapidly cross-linked to fibrin by FXIIIa [181–184]. APCE represents a therapeutic target, since inhibition of this enzyme could reduce circulating levels of Asn-PI and therefore decrease incorporation of PI into the fibrin clot. Indeed, Lee et al. observed an enhancement of plasma clot lysis when APCE was inhibited in vitro [185] but in vivo studies are lacking.
Despite promising preliminary data, none of these approaches of PI-related therapeutic targets have been clinically adopted, indicating that translating in vitro findings, and even animal work, into application in man is difficult and more complex than initially envisaged.
Complement C3 as a drug target
Therapeutic intervention in the complement system has been recognised as a potential strategy for the treatment of a series of inflammatory and autoimmune diseases [186, 187] whilst the role of C3 in fibrinolysis has yet to be widely recognised as an antithrombotic therapeutic target. Early work has shown that one approach in modulating the antifibrinolytic action of C3 has involved using a phage display system of small conformational peptides termed Adhirons. One Adhiron was identified which abolished C3-induced prolongation of fibrin clot lysis by interfering with the C3-fibrinogen interaction. Importantly, this Adhiron, being specific for fibrinogen potentially avoids targeting of C3 systemically, narrowing the therapeutic window and lessening the risk of off-target effects [188]. It remains to be seen whether this approach proves to be suitable for future clinical application.
Potential future strategies to enhance the fibrinolytic process, and reduce thrombosis risk in diabetes are summarised in Table 2.
Table 2.
Current and potential future strategies to enhance fibrinolysis and reduce thrombosis risk in diabetes
| Modulatory mechanism | Current/potential therapeutic strategies |
|---|---|
| Improve glycaemic control | Agents which control blood glucose while avoiding hypoglycaemia |
| Decrease levels of active TAFI | Inhibit activation of TAFI [147–151] Direct TAFIa inhibitors [147–151] |
| Decrease levels of active PAI-1 | Inhibition of PAI-1 synthesis [114] Direct PAI-1 inhibition [114, 148, 156–165] |
| Decrease incorporation of plasmin inhibitor (PI) into the clot | Antibodies [167–170] PI mutants [173, 174] N- and C-terminal peptides of PI [178–180] Inhibition of antiplasmin cleaving enzyme [185] |
| Decrease incorporation of C3 into the clot | Targeted disruption of the fibrinogen-C3 interaction [188] |
Conclusions/summary
Despite advances in treatment, vascular mortality in patients with diabetes remains unacceptably high, which is partly related to the enhanced thrombotic environment that is not fully controlled with current anti-platelet therapies. Rather than developing more effective anti-platelet agents, which runs the risk of bleeding complications, an alternative approach is to target the hypofibrinolytic state in diabetes, a key abnormality in this condition. Therefore, understanding the mechanisms for increased fibrin-related thrombosis risk in diabetes is key to develop effective and safe novel antithrombotic therapies.
Diabetes is associated with both quantitative and qualitative changes in clotting factors resulting in denser fibrin networks that are more difficult to lyse. In addition to changes in clot structure, diabetes directly impairs the function of the fibrinolytic system and shows increased incorporation of anti-fibrinolytic proteins into the clot. More specifically, recent studies have shown increased incorporation of PI and complement C3 into diabetes clots, which compromise fibrin clot lysis. The increased incorporation of anti-fibrinolytic proteins into fibrin networks represents a novel diabetes-specific mechanistic pathway that may be a target for a new generation of anti-thrombotic agents.
In summary, targeting alternative and diabetes-specific thrombotic pathways may be the best approach to reduce the residual thrombosis risk in diabetes and improve vascular outcome in this population.
Authors’ contributions
KK designed the review, undertook the literature search and wrote the manuscript, RA designed the review and critically reviewed the manuscript, DT and KS critically reviewed the manuscript. All authors read and approved the final manuscript.
Acknowledgements
Research work in Ajjan's laboratory is currently funded by the NIHR, BHF, Diabetes UK, Abbott Diabetes Care and AVACTA.
Competing interests
The authors declare that they have no competing interests.
Funding
The British Heart Foundation (BHF) is currently funding the studies of KK.
Contributor Information
Katherine Kearney, Email: umkke@leeds.ac.uk.
Darren Tomlinson, Email: D.c.Tomlinson@leeds.ac.uk.
Kerrie Smith, Email: k.a.smith@leeds.ac.uk.
Ramzi Ajjan, Phone: 0113 343 7475, Email: R.Ajjan@leeds.ac.uk.
References
- 1.Booth GL, Kapral MK, Fung K, Tu JV. Relation between age and cardiovascular disease in men and women with diabetes compared with non-diabetic people: a population-based retrospective cohort study. Lancet. 2006;368(9529):29–36. doi: 10.1016/S0140-6736(06)68967-8. [DOI] [PubMed] [Google Scholar]
- 2.Di Angelantonio E, Kaptoge S, Wormser D, Willeit P, Butterworth AS, Bansal N, O’Keeffe LM, Gao P, Wood AM, Burgess S, et al. Association of cardiometabolic multimorbidity with mortality. JAMA. 2015;314(1):52–60. doi: 10.1001/jama.2015.7008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cubbon RM, Wheatcroft SB, Grant PJ, Gale CP, Barth JH, Sapsford RJ, Ajjan R, Kearney MT, Hall AS, et al. Evaluation of M et al. temporal trends in mortality of patients with diabetes mellitus suffering acute myocardial infarction: a comparison of over 3000 patients between 1995 and 2003. Eur Heart J. 2007;28(5):540–545. doi: 10.1093/eurheartj/ehl510. [DOI] [PubMed] [Google Scholar]
- 4.Kahn MB, Cubbon RM, Mercer B, Wheatcroft AC, Gherardi G, Aziz A, Baliga V, Blaxill JM, McLenachan JM, Blackman DJ, et al. Association of diabetes with increased all-cause mortality following primary percutaneous coronary intervention for ST-segment elevation myocardial infarction in the contemporary era. Diabetes Vasc Dis Res. 2012;9(1):3–9. doi: 10.1177/1479164111427752. [DOI] [PubMed] [Google Scholar]
- 5.van Straten AH, Soliman Hamad MA, van Zundert AA, Martens EJ, Schonberger JP, ter Woorst JF, de Wolf AM. Diabetes and survival after coronary artery bypass grafting: comparison with an age- and sex-matched population. Eur J Cardiothorac Surg. 2010;37(5):1068–1074. doi: 10.1016/j.ejcts.2009.11.042. [DOI] [PubMed] [Google Scholar]
- 6.James S, Angiolillo DJ, Cornel JH, Erlinge D, Husted S, Kontny F, Maya J, Nicolau JC, Spinar J, Storey RF, et al. Ticagrelor vs. clopidogrel in patients with acute coronary syndromes and diabetes: a substudy from the PLATelet inhibition and patient Outcomes (PLATO) trial. Eur Heart J. 2010;31(24):3006–3016. doi: 10.1093/eurheartj/ehq325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kohli P, Wallentin L, Reyes E, Horrow J, Husted S, Angiolillo DJ, Ardissino D, Maurer G, Morais J, Nicolau JC, et al. Reduction in first and recurrent cardiovascular events with ticagrelor compared with clopidogrel in the PLATO Study. Circulation. 2013;127(6):673–680. doi: 10.1161/CIRCULATIONAHA.112.124248. [DOI] [PubMed] [Google Scholar]
- 8.Ryden L, Grant PJ, Anker SD, Berne C, Cosentino F, Danchin N, Deaton C, Escaned J, Hammes HP, Huikuri H, et al. ESC Guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD: the Task Force on diabetes, pre-diabetes, and cardiovascular diseases of the European Society of Cardiology (ESC) and developed in collaboration with the European Association for the Study of Diabetes (EASD) Eur Heart J. 2013;34(39):3035–3087. doi: 10.1093/eurheartj/eht108. [DOI] [PubMed] [Google Scholar]
- 9.Cleland SJ, Fisher BM, Colhoun HM, Sattar N, Petrie JR. Insulin resistance in type 1 diabetes: what is ‘double diabetes’ and what are the risks? Diabetologia. 2013;56(7):1462–1470. doi: 10.1007/s00125-013-2904-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rollini F, Franchi F, Muniz-Lozano A, Angiolillo DJ. Platelet function profiles in patients with diabetes mellitus. J Cardiovasc Transl Res. 2013;6(3):329–345. doi: 10.1007/s12265-013-9449-0. [DOI] [PubMed] [Google Scholar]
- 11.Hess K. The vulnerable blood: coagulation and clot structure in diabetes mellitus. Hamostaseologie. 2015;35(1):25–33. doi: 10.5482/HAMO-14-09-0039. [DOI] [PubMed] [Google Scholar]
- 12.Soma P, Swanepoel AC, du Plooy JN, Mqoco T, Pretorius E. Flow cytometric analysis of platelets type 2 diabetes mellitus reveals ‘angry’ platelets. Cardiovasc Diabetol. 2016;15:52. doi: 10.1186/s12933-016-0373-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pretorius E, Bester J, Vermeulen N, Alummoottil S, Soma P, Buys AV, Kell DB. Poorly controlled type 2 diabetes is accompanied by significant morphological and ultrastructural changes in both erythrocytes and in thrombin-generated fibrin: implications for diagnostics. Cardiovasc Diabetol. 2015;14:30. doi: 10.1186/s12933-015-0192-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pretorius E, Oberholzer HM, van der Spuy WJ, Swanepoel AC, Soma P. Qualitative scanning electron microscopy analysis of fibrin networks and platelet abnormalities in diabetes. Blood Coagul Fibrinolysis. 2011;22(6):463–467. doi: 10.1097/MBC.0b013e3283468a0d. [DOI] [PubMed] [Google Scholar]
- 15.Soma P, Pretorius E. Interplay between ultrastructural findings and atherothrombotic complications in type 2 diabetes mellitus. Cardiovasc Diabetol. 2015;14:96. doi: 10.1186/s12933-015-0261-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Versteeg HH, Heemskerk JW, Levi M, Reitsma PH. New fundamentals in hemostasis. Physiol Rev. 2013;93(1):327–358. doi: 10.1152/physrev.00016.2011. [DOI] [PubMed] [Google Scholar]
- 17.Hethershaw EL, Cilia La Corte AL, Duval C, Ali M, Grant PJ, Ariens RA, Philippou H. The effect of blood coagulation factor XIII on fibrin clot structure and fibrinolysis. J Thromb Haemost. 2014;12(2):197–205. doi: 10.1111/jth.12455. [DOI] [PubMed] [Google Scholar]
- 18.Sakata Y, Aoki N. Cross-linking of alpha 2-plasmin inhibitor to fibrin by fibrin-stabilizing factor. J Clin Invest. 1980;65(2):290–297. doi: 10.1172/JCI109671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Valnickova Z, Enghild JJ. Human procarboxypeptidase U, or thrombin-activable fibrinolysis inhibitor, is a substrate for transglutaminases. Evidence for transglutaminase-catalyzed cross-linking to fibrin. J Biol Chem. 1998;273(42):27220–27224. doi: 10.1074/jbc.273.42.27220. [DOI] [PubMed] [Google Scholar]
- 20.Ritchie H, Lawrie LC, Crombie PW, Mosesson MW, Booth NA. Cross-linking of plasminogen activator inhibitor 2 and alpha 2-antiplasmin to fibrin(ogen) J Biol Chem. 2000;275(32):24915–24920. doi: 10.1074/jbc.M002901200. [DOI] [PubMed] [Google Scholar]
- 21.Chapin JC, Hajjar KA. Fibrinolysis and the control of blood coagulation. Blood Rev. 2015;29(1):17–24. doi: 10.1016/j.blre.2014.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weisel J, Litvinov R. The biochemical and physical process of fibrinolysis and effects of clot structure and stability on the lysis rate. Cardiovasc Hematol Agents Med Chem. 2008;6(3):161–180. doi: 10.2174/187152508784871963. [DOI] [PubMed] [Google Scholar]
- 23.Lawrence DA, Ginsburg D, Day DE, Berkenpas MB, Verhamme IM, Kvassman JO, Shore JD. Serpin-protease complexes are trapped as stable acyl-enzyme intermediates. J Biol Chem. 1995;270(43):25309–25312. doi: 10.1074/jbc.270.43.25309. [DOI] [PubMed] [Google Scholar]
- 24.Sakata Y, Aoki N. Significance of cross-linking of alpha 2-plasmin inhibitor to fibrin in inhibition of fibrinolysis and in hemostasis. J Clin Invest. 1982;69(3):536–542. doi: 10.1172/JCI110479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fraser SR, Booth NA, Mutch NJ. The antifibrinolytic function of factor XIII is exclusively expressed through alpha(2)-antiplasmin cross-linking. Blood. 2011;117(23):6371–6374. doi: 10.1182/blood-2011-02-333203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sakharov DV, Plow EF, Rijken DC. On the mechanism of the antifibrinolytic activity of plasma carboxypeptidase B. J Biol Chem. 1997;272(22):14477–14482. doi: 10.1074/jbc.272.22.14477. [DOI] [PubMed] [Google Scholar]
- 27.Meade TW, Ruddock V, Stirling Y, Chakrabarti R, Miller GJ. Fibrinolytic activity, clotting factors, and long-term incidence of ischaemic heart disease in the Northwick Park Heart Study. Lancet. 1993;342(8879):1076–1079. doi: 10.1016/0140-6736(93)92062-X. [DOI] [PubMed] [Google Scholar]
- 28.Lisman T, de Groot PG, Meijers JC, Rosendaal FR. Reduced plasma fibrinolytic potential is a risk factor for venous thrombosis. Blood. 2005;105(3):1102–1105. doi: 10.1182/blood-2004-08-3253. [DOI] [PubMed] [Google Scholar]
- 29.Meltzer ME, Doggen CJ, de Groot PG, Rosendaal FR, Lisman T. Reduced plasma fibrinolytic capacity as a potential risk factor for a first myocardial infarction in young men. Br J Haematol. 2009;145(1):121–127. doi: 10.1111/j.1365-2141.2008.07569.x. [DOI] [PubMed] [Google Scholar]
- 30.Guimaraes AH, de Bruijne EL, Lisman T, Dippel DW, Deckers JW, Poldermans D, Rijken DC, Leebeek FW. Hypofibrinolysis is a risk factor for arterial thrombosis at young age. Br J Haematol. 2009;145(1):115–120. doi: 10.1111/j.1365-2141.2008.07568.x. [DOI] [PubMed] [Google Scholar]
- 31.Katz P, Leiter LA, Mellbin L, Ryden L. The clinical burden of type 2 diabetes in patients with acute coronary syndromes: prognosis and implications for short- and long-term management. Diabetes Vasc Dis Res. 2014;11(6):395–409. doi: 10.1177/1479164114546854. [DOI] [PubMed] [Google Scholar]
- 32.Clemmensen P, Dridi NP, Holmvang L. Dual antiplatelet therapy with prasugrel or ticagrelor versus clopidogrel in interventional cardiology. Cardiovasc Drugs Ther. 2013;27(3):239–245. doi: 10.1007/s10557-013-6444-2. [DOI] [PubMed] [Google Scholar]
- 33.Wiviott SD, Braunwald E, McCabe CH, Montalescot G, Ruzyllo W, Gottlieb S, Neumann FJ, Ardissino D, De Servi S, Murphy SA, et al. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2007;357(20):2001–2015. doi: 10.1056/NEJMoa0706482. [DOI] [PubMed] [Google Scholar]
- 34.Wiviott SD, Braunwald E, Angiolillo DJ, Meisel S, Dalby AJ, Verheugt FW, Goodman SG, Corbalan R, Purdy DA, Murphy SA, et al. Greater clinical benefit of more intensive oral antiplatelet therapy with prasugrel in patients with diabetes mellitus in the trial to assess improvement in therapeutic outcomes by optimizing platelet inhibition with prasugrel-Thrombolysis in Myocardial Infarction 38. Circulation. 2008;118(16):1626–1636. doi: 10.1161/CIRCULATIONAHA.108.791061. [DOI] [PubMed] [Google Scholar]
- 35.Ajjan RA, Standeven KF, Khanbhai M, Phoenix F, Gersh KC, Weisel JW, Kearney MT, Ariens RA, Grant PJ. Effects of aspirin on clot structure and fibrinolysis using a novel in vitro cellular system. Arterioscler Thromb Vasc Biol. 2009;29(5):712–717. doi: 10.1161/ATVBAHA.109.183707. [DOI] [PubMed] [Google Scholar]
- 36.Bailey MA, Aggarwal R, Bridge KI, Griffin KJ, Iqbal F, Phoenix F, Purdell-Lewis J, Thomas T, Johnson AB, Ariens RA, et al. Aspirin therapy is associated with less compact fibrin networks and enhanced fibrinolysis in patients with abdominal aortic aneurysm. J Thromb Haemost. 2015;13(5):795–801. doi: 10.1111/jth.12872. [DOI] [PubMed] [Google Scholar]
- 37.Tehrani S, Antovic A, Mobarrez F, Mageed K, Lins PE, Adamson U, Wallen HN, Jorneskog G. High-dose aspirin is required to influence plasma fibrin network structure in patients with type 1 diabetes. Diabetes Care. 2012;35(2):404–408. doi: 10.2337/dc11-1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alzahrani SH, Ajjan RA. Coagulation and fibrinolysis in diabetes. Diab Vasc Dis Res. 2010;7(4):260–273. doi: 10.1177/1479164110383723. [DOI] [PubMed] [Google Scholar]
- 39.Kunutsor SK, Seidu S, Khunti K. Aspirin for primary prevention of cardiovascular and all-cause mortality events in diabetes: updated meta-analysis of randomized controlled trials. Diabet Med. 2016;23:579–593. doi: 10.1111/dme.13133. [DOI] [PubMed] [Google Scholar]
- 40.Fox CS, Golden SH, Anderson C, Bray GA, Burke LE, de Boer IH, Deedwania P, Eckel RH, Ershow AG, Fradkin J, et al. Update on prevention of cardiovascular disease in adults with type 2 diabetes mellitus in light of recent evidence: a scientific statement from the American Heart Association and the American Diabetes Association. Diabetes Care. 2015;38(9):1777–1803. doi: 10.2337/dci15-0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wallentin L, Becker RC, Budaj A, Cannon CP, Emanuelsson H, Held C, Horrow J, Husted S, James S, Katus H, et al. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2009;361(11):1045–1057. doi: 10.1056/NEJMoa0904327. [DOI] [PubMed] [Google Scholar]
- 42.Ferreiro JL, Angiolillo DJ. Diabetes and antiplatelet therapy in acute coronary syndrome. Circulation. 2011;123(7):798–813. doi: 10.1161/CIRCULATIONAHA.109.913376. [DOI] [PubMed] [Google Scholar]
- 43.Capodanno D, Patel A, Dharmashankar K, Ferreiro JL, Ueno M, Kodali M, Tomasello SD, Capranzano P, Seecheran N, Darlington A, et al. Pharmacodynamic effects of different aspirin dosing regimens in type 2 diabetes mellitus patients with coronary artery disease. Circ Cardiovasc Interv. 2011;4(2):180–187. doi: 10.1161/CIRCINTERVENTIONS.110.960187. [DOI] [PubMed] [Google Scholar]
- 44.Dillinger JG, Drissa A, Sideris G, dit Sollier CB, Voicu S, Silberman SM, Logeart D, Drouet L, Henry P. Biological efficacy of twice daily aspirin in type 2 diabetic patients with coronary artery disease. Am Heart J. 2012;164(4):600. doi: 10.1016/j.ahj.2012.06.008. [DOI] [PubMed] [Google Scholar]
- 45.Rocca B, Santilli F, Pitocco D, Mucci L, Petrucci G, Vitacolonna E, Lattanzio S, Mattoscio D, Zaccardi F, Liani R, et al. The recovery of platelet cyclooxygenase activity explains interindividual variability in responsiveness to low-dose aspirin in patients with and without diabetes. J Thromb Haemost. 2012;10(7):1220–1230. doi: 10.1111/j.1538-7836.2012.04723.x. [DOI] [PubMed] [Google Scholar]
- 46.Schulman S, Spencer FA. Antithrombotic drugs in coronary artery disease: risk benefit ratio and bleeding. J Thromb Haemost. 2010;8(4):641–650. doi: 10.1111/j.1538-7836.2010.03737.x. [DOI] [PubMed] [Google Scholar]
- 47.Mega JL, Braunwald E, Wiviott SD, Bassand JP, Bhatt DL, Bode C, Burton P, Cohen M, Cook-Bruns N, Fox KA, et al. Rivaroxaban in patients with a recent acute coronary syndrome. N Engl J Med. 2012;366(1):9–19. doi: 10.1056/NEJMoa1112277. [DOI] [PubMed] [Google Scholar]
- 48.Baeriswyl V, Calzavarini S, Chen S, Zorzi A, Bologna L, Angelillo-Scherrer A, Heinis C. A synthetic factor XIIa inhibitor blocks selectively intrinsic coagulation initiation. ACS Chem Biol. 2015;10(8):1861–1870. doi: 10.1021/acschembio.5b00103. [DOI] [PubMed] [Google Scholar]
- 49.Tehrani S, Jorneskog G, Agren A, Lins PE, Wallen H, Antovic A. Fibrin clot properties and haemostatic function in men and women with type 1 diabetes. Thromb Haemost. 2015;113(2):312–318. doi: 10.1160/TH14-05-0404. [DOI] [PubMed] [Google Scholar]
- 50.Sherif EM, Elbarbary NS, Abd Al Aziz MM, Mohamed SF. Plasma thrombin-activatable fibrinolysis inhibitor levels in children and adolescents with type 1 diabetes mellitus: possible relation to diabetic microvascular complications. Blood Coagul Fibrinolysis. 2014;25(5):451–457. doi: 10.1097/MBC.0000000000000080. [DOI] [PubMed] [Google Scholar]
- 51.Zheng N, Shi X, Chen X, Lv W. Associations between inflammatory markers, hemostatic markers, and microvascular complications in 182 Chinese patients with type 2 diabetes mellitus. Lab Med. 2015;46(3):214–220. doi: 10.1309/LMF8R2KSTOW3FLKD. [DOI] [PubMed] [Google Scholar]
- 52.Collet JP, Allali Y, Lesty C, Tanguy ML, Silvain J, Ankri A, Blanchet B, Dumaine R, Gianetti J, Payot L, et al. Altered fibrin architecture is associated with hypofibrinolysis and premature coronary atherothrombosis. Arterioscler Thromb Vasc Biol. 2006;26(11):2567–2573. doi: 10.1161/01.ATV.0000241589.52950.4c. [DOI] [PubMed] [Google Scholar]
- 53.Undas A, Plicner D, Stepien E, Drwila R, Sadowski J. Altered fibrin clot structure in patients with advanced coronary artery disease: a role of C-reactive protein, lipoprotein(a) and homocysteine. J Thromb Haemost. 2007;5(9):1988–1990. doi: 10.1111/j.1538-7836.2007.02637.x. [DOI] [PubMed] [Google Scholar]
- 54.Fatah K, Silveira A, Tornvall P, Karpe F, Blomback M, Hamsten A. Proneness to formation of tight and rigid fibrin gel structures in men with myocardial infarction at a young age. Thromb Haemost. 1996;76(4):535–540. [PubMed] [Google Scholar]
- 55.Leander K, Blomback M, Wallen H, He S. Impaired fibrinolytic capacity and increased fibrin formation associate with myocardial infarction. Thromb Haemost. 2012;107(6):1092–1099. doi: 10.1160/TH11-11-0760. [DOI] [PubMed] [Google Scholar]
- 56.Undas A, Kolarz M, Kopec G, Tracz W. Altered fibrin clot properties in patients on long-term haemodialysis: relation to cardiovascular mortality. Nephrol Dial Transplant. 2008;23(6):2010–2015. doi: 10.1093/ndt/gfm884. [DOI] [PubMed] [Google Scholar]
- 57.Mills JD, Ariens RA, Mansfield MW, Grant PJ. Altered fibrin clot structure in the healthy relatives of patients with premature coronary artery disease. Circulation. 2002;106(15):1938–1942. doi: 10.1161/01.CIR.0000033221.73082.06. [DOI] [PubMed] [Google Scholar]
- 58.Lord ST. Molecular mechanisms affecting fibrin structure and stability. Arterioscler Thromb Vasc Biol. 2011;31(3):494–499. doi: 10.1161/ATVBAHA.110.213389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Collet JP, Park D, Lesty C, Soria J, Soria C, Montalescot G, Weisel JW. Influence of fibrin network conformation and fibrin fiber diameter on fibrinolysis speed: dynamic and structural approaches by confocal microscopy. Arterioscler Thromb Vasc Biol. 2000;20(5):1354–1361. doi: 10.1161/01.ATV.20.5.1354. [DOI] [PubMed] [Google Scholar]
- 60.Gabriel DA, Muga K, Boothroyd EM. The effect of fibrin structure on fibrinolysis. J Biol Chem. 1992;267(34):24259–24263. [PubMed] [Google Scholar]
- 61.Carr ME, Jr, Alving BM. Effect of fibrin structure on plasmin-mediated dissolution of plasma clots. Blood Coagul Fibrinolysis. 1995;6(6):567–573. doi: 10.1097/00001721-199509000-00011. [DOI] [PubMed] [Google Scholar]
- 62.Ajjan R, Lim BC, Standeven KF, Harrand R, Dolling S, Phoenix F, Greaves R, Abou-Saleh RH, Connell S, Smith DA, et al. Common variation in the C-terminal region of the fibrinogen beta-chain: effects on fibrin structure, fibrinolysis and clot rigidity. Blood. 2008;111(2):643–650. doi: 10.1182/blood-2007-05-091231. [DOI] [PubMed] [Google Scholar]
- 63.Dunn EJ, Ariens RA, Grant PJ. The influence of type 2 diabetes on fibrin structure and function. Diabetologia. 2005;48(6):1198–1206. doi: 10.1007/s00125-005-1742-2. [DOI] [PubMed] [Google Scholar]
- 64.Jorneskog G, Egberg N, Fagrell B, Fatah K, Hessel B, Johnsson H, Brismar K, Blomback M. Altered properties of the fibrin gel structure in patients with IDDM. Diabetologia. 1996;39(12):1519–1523. doi: 10.1007/s001250050607. [DOI] [PubMed] [Google Scholar]
- 65.Dunn EJ, Philippou H, Ariens RA, Grant PJ. Molecular mechanisms involved in the resistance of fibrin to clot lysis by plasmin in subjects with type 2 diabetes mellitus. Diabetologia. 2006;49(5):1071–1080. doi: 10.1007/s00125-006-0197-4. [DOI] [PubMed] [Google Scholar]
- 66.Nair CH, Azhar A, Wilson JD, Dhall DP. Studies on fibrin network structure in human plasma. Part II-clinical application: diabetes and antidiabetic drugs. Thromb Res. 1991;64(4):477–485. doi: 10.1016/0049-3848(91)90347-Y. [DOI] [PubMed] [Google Scholar]
- 67.Alzahrani SH, Hess K, Price JF, Strachan M, Baxter PD, Cubbon R, Phoenix F, Gamlen T, Ariens RA, Grant PJ, et al. Gender-specific alterations in fibrin structure function in type 2 diabetes: associations with cardiometabolic and vascular markers. J Clin Endocrinol Metab. 2012;97(12):E2282–E2287. doi: 10.1210/jc.2012-2128. [DOI] [PubMed] [Google Scholar]
- 68.Konieczynska M, Fil K, Bazanek M, Undas A. Prolonged duration of type 2 diabetes is associated with increased thrombin generation, prothrombotic fibrin clot phenotype and impaired fibrinolysis. Thromb Haemost. 2014;111(4):685–693. doi: 10.1160/TH13-07-0566. [DOI] [PubMed] [Google Scholar]
- 69.Danesh J, Lewington S, Thompson SG, Lowe GD, Collins R, Kostis JB, Wilson AC, Folsom AR, Wu K, Benderly M, et al. Plasma fibrinogen level and the risk of major cardiovascular diseases and nonvascular mortality: an individual participant meta-analysis. JAMA. 2005;294(14):1799–1809. doi: 10.1001/jama.294.14.1799. [DOI] [PubMed] [Google Scholar]
- 70.Kannel WB, Wolf PA, Castelli WP, D’Agostino RB. Fibrinogen and risk of cardiovascular disease. The Framingham Study. JAMA. 1987;258(9):1183–1186. doi: 10.1001/jama.1987.03400090067035. [DOI] [PubMed] [Google Scholar]
- 71.van Holten TC, Waanders LF, de Groot PG, Vissers J, Hoefer IE, Pasterkamp G, Prins MW, Roest M. Circulating biomarkers for predicting cardiovascular disease risk; a systematic review and comprehensive overview of meta-analyses. PLoS ONE. 2013;8(4):e62080. doi: 10.1371/journal.pone.0062080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ernst E, Resch KL. Fibrinogen as a cardiovascular risk factor: a meta-analysis and review of the literature. Ann Intern Med. 1993;118(12):956–963. doi: 10.7326/0003-4819-118-12-199306150-00008. [DOI] [PubMed] [Google Scholar]
- 73.Weisel JW. Structure of fibrin: impact on clot stability. J Thromb Haemost. 2007;5(Suppl 1):116–124. doi: 10.1111/j.1538-7836.2007.02504.x. [DOI] [PubMed] [Google Scholar]
- 74.Machlus KR, Cardenas JC, Church FC, Wolberg AS. Causal relationship between hyperfibrinogenemia, thrombosis, and resistance to thrombolysis in mice. Blood. 2011;117(18):4953–4963. doi: 10.1182/blood-2010-11-316885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Missov RM, Stolk RP, van der Bom JG, Hofman A, Bots ML, Pols HA, Grobbee DE. Plasma fibrinogen in NIDDM: the Rotterdam study. Diabetes Care. 1996;19(2):157–159. doi: 10.2337/diacare.19.2.157. [DOI] [PubMed] [Google Scholar]
- 76.Pieters M, van Zyl DG, Rheeder P, Jerling JC, du Loots T, van der Westhuizen FH, Gottsche LT, Weisel JW. Glycation of fibrinogen in uncontrolled diabetic patients and the effects of glycaemic control on fibrinogen glycation. Thromb Res. 2007;120(3):439–446. doi: 10.1016/j.thromres.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 77.Lutjens A, te Velde AA, vd Veen EA, vd Meer J. Glycosylation of human fibrinogen in vivo. Diabetologia. 1985;28(2):87–89. doi: 10.1007/BF00279921. [DOI] [PubMed] [Google Scholar]
- 78.Hammer MR, John PN, Flynn MD, Bellingham AJ, Leslie RD. Glycated fibrinogen: a new index of short-term diabetic control. Ann Clin Biochem. 1989;26(Pt 1):58–62. doi: 10.1177/000456328902600108. [DOI] [PubMed] [Google Scholar]
- 79.Svensson J, Bergman AC, Adamson U, Blomback M, Wallen H, Jorneskog G. Acetylation and glycation of fibrinogen in vitro occur at specific lysine residues in a concentration dependent manner: a mass spectrometric and isotope labeling study. Biochem Biophys Res Commun. 2012;421(2):335–342. doi: 10.1016/j.bbrc.2012.03.154. [DOI] [PubMed] [Google Scholar]
- 80.Pieters M, Covic N, van der Westhuizen FH, Nagaswami C, Baras Y, Toit Loots D, Jerling JC, Elgar D, Edmondson KS, van Zyl DG, et al. Glycaemic control improves fibrin network characteristics in type 2 diabetes—a purified fibrinogen model. Thromb Haemost. 2008;99(4):691–700. doi: 10.1160/TH07-11-0699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Henschen-Edman AH. Fibrinogen non-inherited heterogeneity and its relationship to function in health and disease. Ann N Y Acad Sci. 2001;936:580–593. doi: 10.1111/j.1749-6632.2001.tb03546.x. [DOI] [PubMed] [Google Scholar]
- 82.Ardawi MS, Nasrat HN, Mira SA, Fatani HH. Comparison of glycosylated fibrinogen, albumin, and haemoglobin as indices of blood glucose control in diabetic patients. Diabet Med. 1990;7(9):819–824. doi: 10.1111/j.1464-5491.1990.tb01499.x. [DOI] [PubMed] [Google Scholar]
- 83.Pieters M, Covic N, du Loots T, van der Westhuizen FH, van Zyl DG, Rheeder P, Jerling JC, Weisel JW. The effect of glycaemic control on fibrin network structure of type 2 diabetic subjects. Thromb Haemost. 2006;96(5):623–629. [PubMed] [Google Scholar]
- 84.Lipinski B. Pathophysiology of oxidative stress in diabetes mellitus. J Diabetes Complic. 2001;15(4):203–210. doi: 10.1016/S1056-8727(01)00143-X. [DOI] [PubMed] [Google Scholar]
- 85.Shacter E, Williams JA, Levine RL. Oxidative modification of fibrinogen inhibits thrombin-catalyzed clot formation. Free Radic Biol Med. 1995;18(4):815–821. doi: 10.1016/0891-5849(95)93872-4. [DOI] [PubMed] [Google Scholar]
- 86.Lados-Krupa A, Konieczynska M, Chmiel A, Undas A. Increased oxidation as an additional mechanism underlying reduced clot permeability and impaired fibrinolysis in type 2 diabetes. J Diabetes Res. 2015;2015:456189. doi: 10.1155/2015/456189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Blomback B, Carlsson K, Hessel B, Liljeborg A, Procyk R, Aslund N. Native fibrin gel networks observed by 3D microscopy, permeation and turbidity. Biochim Biophys Acta. 1989;997(1–2):96–110. doi: 10.1016/0167-4838(89)90140-4. [DOI] [PubMed] [Google Scholar]
- 88.Wolberg AS, Monroe DM, Roberts HR, Hoffman M. Elevated prothrombin results in clots with an altered fiber structure: a possible mechanism of the increased thrombotic risk. Blood. 2003;101(8):3008–3013. doi: 10.1182/blood-2002-08-2527. [DOI] [PubMed] [Google Scholar]
- 89.Wolberg AS. Thrombin generation and fibrin clot structure. Blood Rev. 2007;21(3):131–142. doi: 10.1016/j.blre.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 90.Brummel KE, Paradis SG, Butenas S, Mann KG. Thrombin functions during tissue factor-induced blood coagulation. Blood. 2002;100(1):148–152. doi: 10.1182/blood.V100.1.148. [DOI] [PubMed] [Google Scholar]
- 91.Kim HK, Kim JE, Park SH, Kim YI, Nam-Goong IS, Kim ES. High coagulation factor levels and low protein C levels contribute to enhanced thrombin generation in patients with diabetes who do not have macrovascular complications. J Diabetes Complic. 2014;28(3):365–369. doi: 10.1016/j.jdiacomp.2014.01.006. [DOI] [PubMed] [Google Scholar]
- 92.Boden G, Vaidyula VR, Homko C, Cheung P, Rao AK. Circulating tissue factor procoagulant activity and thrombin generation in patients with type 2 diabetes: effects of insulin and glucose. J Clin Endocrinol Metab. 2007;92(11):4352–4358. doi: 10.1210/jc.2007-0933. [DOI] [PubMed] [Google Scholar]
- 93.Tripodi A, Branchi A, Chantarangkul V, Clerici M, Merati G, Artoni A, Mannucci PM. Hypercoagulability in patients with type 2 diabetes mellitus detected by a thrombin generation assay. J Thromb Thrombolysis. 2011;31(2):165–172. doi: 10.1007/s11239-010-0506-0. [DOI] [PubMed] [Google Scholar]
- 94.Undas A, Wiek I, Stepien E, Zmudka K, Tracz W. Hyperglycemia is associated with enhanced thrombin formation, platelet activation, and fibrin clot resistance to lysis in patients with acute coronary syndrome. Diabetes Care. 2008;31(8):1590–1595. doi: 10.2337/dc08-0282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ceriello A, Esposito K, Ihnat M, Zhang J, Giugliano D. Simultaneous control of hyperglycemia and oxidative stress normalizes enhanced thrombin generation in type 1 diabetes. J Thromb Haemost. 2009;7(7):1228–1230. doi: 10.1111/j.1538-7836.2009.03445.x. [DOI] [PubMed] [Google Scholar]
- 96.Gajos G, Konieczynska M, Zalewski J, Undas A. Low fasting glucose is associated with enhanced thrombin generation and unfavorable fibrin clot properties in type 2 diabetic patients with high cardiovascular risk. Cardiovasc Diabetol. 2015;14:44. doi: 10.1186/s12933-015-0207-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Beijers HJ, Ferreira I, Spronk HM, Bravenboer B, Dekker JM, Nijpels G, ten Cate H, Stehouwer CD. Impaired glucose metabolism and type 2 diabetes are associated with hypercoagulability: potential role of central adiposity and low-grade inflammation–The Hoorn Study. Thromb Res. 2012;129(5):557–562. doi: 10.1016/j.thromres.2011.07.033. [DOI] [PubMed] [Google Scholar]
- 98.Marini MG, Sonnino C, Previtero M, Biasucci LM. Targeting inflammation: impact on atherothrombosis. J Cardiovasc Transl Res. 2014;7(1):9–18. doi: 10.1007/s12265-013-9523-7. [DOI] [PubMed] [Google Scholar]
- 99.Hansson GK, Libby P, Tabas I. Inflammation and plaque vulnerability. J Intern Med. 2015;278(5):483–493. doi: 10.1111/joim.12406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Badimon L, Suades R, Fuentes E, Palomo I, Padro T. Role of platelet-derived microvesicles as crosstalk mediators in atherothrombosis and future pharmacology targets: a link between inflammation, atherosclerosis, and thrombosis. Front Pharmacol. 2016;7:293. doi: 10.3389/fphar.2016.00293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bester J, Pretorius E. Effects of IL-1beta, IL-6 and IL-8 on erythrocytes, platelets and clot viscoelasticity. Sci Rep. 2016;6:32188. doi: 10.1038/srep32188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Foley JH, Conway EM. Cross talk pathways between coagulation and inflammation. Circ Res. 2016;118(9):1392–1408. doi: 10.1161/CIRCRESAHA.116.306853. [DOI] [PubMed] [Google Scholar]
- 103.Distelmaier K, Adlbrecht C, Jakowitsch J, Winkler S, Dunkler D, Gerner C, Wagner O, Lang IM, Kubicek M. Local complement activation triggers neutrophil recruitment to the site of thrombus formation in acute myocardial infarction. Thromb Haemost. 2009;102(3):564–572. doi: 10.1160/TH09-02-0103. [DOI] [PubMed] [Google Scholar]
- 104.Howes JM, Richardson VR, Smith KA, Schroeder V, Somani R, Shore A, Hess K, Ajjan R, Pease RJ, Keen JN, et al. Complement C3 is a novel plasma clot component with anti-fibrinolytic properties. Diab Vasc Dis Res. 2012;9(3):216–225. doi: 10.1177/1479164111432788. [DOI] [PubMed] [Google Scholar]
- 105.Nikolajsen CL, Scavenius C, Enghild JJ. Human complement C3 is a substrate for transglutaminases. A functional link between non-protease-based members of the coagulation and complement cascades. Biochemistry. 2012;51(23):4735–4742. doi: 10.1021/bi3004022. [DOI] [PubMed] [Google Scholar]
- 106.Richardson VR, Schroeder V, Grant PJ, Standeven KF, Carter AM. Complement C3 is a substrate for activated factor XIII that is cross-linked to fibrin during clot formation. Br J Haematol. 2013;160(1):116–119. doi: 10.1111/bjh.12096. [DOI] [PubMed] [Google Scholar]
- 107.Hess K, Alzahrani SH, Mathai M, Schroeder V, Carter AM, Howell G, Koko T, Strachan MW, Price JF, Smith KA, et al. A novel mechanism for hypofibrinolysis in diabetes: the role of complement C3. Diabetologia. 2012;55(4):1103–1113. doi: 10.1007/s00125-011-2301-7. [DOI] [PubMed] [Google Scholar]
- 108.Hess K, Alzahrani SH, Price JF, Strachan MW, Oxley N, King R, Gamlen T, Schroeder V, Baxter PD, Ajjan RA. Hypofibrinolysis in type 2 diabetes: the role of the inflammatory pathway and complement C3. Diabetologia. 2014;57(8):1737–1741. doi: 10.1007/s00125-014-3267-z. [DOI] [PubMed] [Google Scholar]
- 109.Neergaard-Petersen S, Hvas AM, Kristensen SD, Grove EL, Larsen SB, Phoenix F, Kurdee Z, Grant PJ, Ajjan RA. The influence of type 2 diabetes on fibrin clot properties in patients with coronary artery disease. Thromb Haemost. 2014;112(6):1142–1150. doi: 10.1160/TH14-05-0468. [DOI] [PubMed] [Google Scholar]
- 110.Amara U, Flierl MA, Rittirsch D, Klos A, Chen H, Acker B, Bruckner UB, Nilsson B, Gebhard F, Lambris JD, et al. Molecular intercommunication between the complement and coagulation systems. J Immunol. 2010;185(9):5628–5636. doi: 10.4049/jimmunol.0903678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Favier R, Aoki N, de Moerloose P. Congenital alpha(2)-plasmin inhibitor deficiencies: a review. Br J Haematol. 2001;114(1):4–10. doi: 10.1046/j.1365-2141.2001.02845.x. [DOI] [PubMed] [Google Scholar]
- 112.Carpenter SL, Mathew P. Alpha2-antiplasmin and its deficiency: fibrinolysis out of balance. Haemophilia. 2008;14(6):1250–1254. doi: 10.1111/j.1365-2516.2008.01766.x. [DOI] [PubMed] [Google Scholar]
- 113.Meltzer ME, Doggen CJ, de Groot PG, Rosendaal FR, Lisman T. Plasma levels of fibrinolytic proteins and the risk of myocardial infarction in men. Blood. 2010;116(4):529–536. doi: 10.1182/blood-2010-01-263103. [DOI] [PubMed] [Google Scholar]
- 114.Al-Horani RA. Serpin regulation of fibrinolytic system: implications for therapeutic applications in cardiovascular diseases. Cardiovasc Hematol Agents Med Chem. 2014;12(2):91–125. doi: 10.2174/1871525712666141106095927. [DOI] [PubMed] [Google Scholar]
- 115.Agren A, Jorneskog G, Elgue G, Henriksson P, Wallen H, Wiman B. Increased incorporation of antiplasmin into the fibrin network in patients with type 1 diabetes. Diabetes Care. 2014;37(7):2007–2014. doi: 10.2337/dc13-1776. [DOI] [PubMed] [Google Scholar]
- 116.Meltzer ME, Lisman T, de Groot PG, Meijers JC, le Cessie S, Doggen CJ, Rosendaal FR. Venous thrombosis risk associated with plasma hypofibrinolysis is explained by elevated plasma levels of TAFI and PAI-1. Blood. 2010;116(1):113–121. doi: 10.1182/blood-2010-02-267740. [DOI] [PubMed] [Google Scholar]
- 117.Brouwers GJ, Leebeek FW, Tanck MW, Wouter Jukema J, Kluft C, de Maat MP. Association between thrombin-activatable fibrinolysis inhibitor (TAFI) and clinical outcome in patients with unstable angina pectoris. Thromb Haemost. 2003;90(1):92–100. [PubMed] [Google Scholar]
- 118.Montaner J, Ribo M, Monasterio J, Molina CA, Alvarez-Sabin J. Thrombin-activable fibrinolysis inhibitor levels in the acute phase of ischemic stroke. Stroke. 2003;34(4):1038–1040. doi: 10.1161/01.STR.0000063139.06585.45. [DOI] [PubMed] [Google Scholar]
- 119.van Tilburg NH, Rosendaal FR, Bertina RM. Thrombin activatable fibrinolysis inhibitor and the risk for deep vein thrombosis. Blood. 2000;95(9):2855–2859. [PubMed] [Google Scholar]
- 120.Leenaerts D, Bosmans JM, van der Veken P, Sim Y, Lambeir AM, Hendriks D. Plasma levels of carboxypeptidase U (CPU, CPB2 or TAFIa) are elevated in patients with acute myocardial infarction. J Thromb Haemost. 2015;13(12):2227–2232. doi: 10.1111/jth.13135. [DOI] [PubMed] [Google Scholar]
- 121.Hori Y, Gabazza EC, Yano Y, Katsuki A, Suzuki K, Adachi Y, Sumida Y. Insulin resistance is associated with increased circulating level of thrombin-activatable fibrinolysis inhibitor in type 2 diabetic patients. J Clin Endocrinol Metab. 2002;87(2):660–665. doi: 10.1210/jcem.87.2.8214. [DOI] [PubMed] [Google Scholar]
- 122.Yano Y, Kitagawa N, Gabazza EC, Morioka K, Urakawa H, Tanaka T, Katsuki A, Araki-Sasaki R, Hori Y, Nakatani K, et al. Increased plasma thrombin-activatable fibrinolysis inhibitor levels in normotensive type 2 diabetic patients with microalbuminuria. J Clin Endocrinol Metab. 2003;88(2):736–741. doi: 10.1210/jc.2002-020691. [DOI] [PubMed] [Google Scholar]
- 123.Chudy P, Kotulicova D, Stasko J, Kubisz P. The relationship among TAFI, t-PA, PAI-1 and F1+ 2 in type 2 diabetic patients with normoalbuminuria and microalbuminuria. Blood Coagul Fibrinolysis. 2011;22(6):493–498. doi: 10.1097/MBC.0b013e328346f8ca. [DOI] [PubMed] [Google Scholar]
- 124.Yener S, Comlekci A, Akinci B, Demir T, Yuksel F, Ozcan MA, Bayraktar F, Yesil S. Soluble CD40 ligand, plasminogen activator inhibitor-1 and thrombin-activatable fibrinolysis inhibitor-1-antigen in normotensive type 2 diabetic subjects without diabetic complications. Effects of metformin and rosiglitazone. Med Princ Pract. 2009;18(4):266–271. doi: 10.1159/000215722. [DOI] [PubMed] [Google Scholar]
- 125.Rigla M, Wagner AM, Borrell M, Mateo J, Foncuberta J, de Leiva A, Ordonez-Llanos J, Perez A. Postprandial thrombin activatable fibrinolysis inhibitor and markers of endothelial dysfunction in type 2 diabetic patients. Metabolism. 2006;55(11):1437–1442. doi: 10.1016/j.metabol.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 126.Leander K, Wiman B, Hallqvist J, Sten-Linder M, de Faire U. PAI-1 level and the PAI-1 4G/5G polymorphism in relation to risk of non-fatal myocardial infarction: results from the Stockholm Heart Epidemiology Program (SHEEP) Thromb Haemost. 2003;89(6):1064–1071. [PubMed] [Google Scholar]
- 127.Hamsten A, Wiman B, de Faire U, Blomback M. Increased plasma levels of a rapid inhibitor of tissue plasminogen activator in young survivors of myocardial infarction. N Engl J Med. 1985;313(25):1557–1563. doi: 10.1056/NEJM198512193132501. [DOI] [PubMed] [Google Scholar]
- 128.Hamsten A, de Faire U, Walldius G, Dahlen G, Szamosi A, Landou C, Blomback M, Wiman B. Plasminogen activator inhibitor in plasma: risk factor for recurrent myocardial infarction. Lancet. 1987;2(8549):3–9. doi: 10.1016/S0140-6736(87)93050-9. [DOI] [PubMed] [Google Scholar]
- 129.Juhan-Vague I, Roul C, Alessi MC, Ardissone JP, Heim M, Vague P. Increased plasminogen activator inhibitor activity in non insulin dependent diabetic patients-relationship with plasma insulin. Thromb Haemost. 1989;61(3):370–373. [PubMed] [Google Scholar]
- 130.Schneider DJ, Sobel BE. PAI-1 and diabetes: a journey from the bench to the bedside. Diabetes Care. 2012;35(10):1961–1967. doi: 10.2337/dc12-0638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Shimomura I, Funahashi T, Takahashi M, Maeda K, Kotani K, Nakamura T, Yamashita S, Miura M, Fukuda Y, Takemura K, et al. Enhanced expression of PAI-1 in visceral fat: possible contributor to vascular disease in obesity. Nat Med. 1996;2(7):800–803. doi: 10.1038/nm0796-800. [DOI] [PubMed] [Google Scholar]
- 132.Alessi MC, Peiretti F, Morange P, Henry M, Nalbone G, Juhan-Vague I. Production of plasminogen activator inhibitor 1 by human adipose tissue: possible link between visceral fat accumulation and vascular disease. Diabetes. 1997;46(5):860–867. doi: 10.2337/diab.46.5.860. [DOI] [PubMed] [Google Scholar]
- 133.Lundgren CH, Brown SL, Nordt TK, Sobel BE, Fujii S. Elaboration of type-1 plasminogen activator inhibitor from adipocytes. A potential pathogenetic link between obesity and cardiovascular disease. Circulation. 1996;93(1):106–110. doi: 10.1161/01.CIR.93.1.106. [DOI] [PubMed] [Google Scholar]
- 134.Stegenga ME, van der Crabben SN, Levi M, de Vos AF, Tanck MW, Sauerwein HP, van der Poll T. Hyperglycemia stimulates coagulation, whereas hyperinsulinemia impairs fibrinolysis in healthy humans. Diabetes. 2006;55(6):1807–1812. doi: 10.2337/db05-1543. [DOI] [PubMed] [Google Scholar]
- 135.Belalcazar LM, Ballantyne CM, Lang W, Haffner SM, Rushing J, Schwenke DC, Pi-Sunyer FX, Tracy RP. Metabolic factors, adipose tissue, and plasminogen activator inhibitor-1 levels in type 2 diabetes: findings from the look AHEAD study. Arterioscler Thromb Vasc Biol. 2011;31(7):1689–1695. doi: 10.1161/ATVBAHA.111.224386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ajjan RA, Gamlen T, Standeven KF, Mughal S, Hess K, Smith KA, Dunn EJ, Anwar MM, Rabbani N, Thornalley PJ, et al. Diabetes is associated with posttranslational modifications in plasminogen resulting in reduced plasmin generation and enzyme-specific activity. Blood. 2013;122(1):134–142. doi: 10.1182/blood-2013-04-494641. [DOI] [PubMed] [Google Scholar]
- 137.Gerstein HC, Miller ME, Byington RP, Goff DC, Jr, Bigger JT, Buse JB, Cushman WC, Genuth S, Ismail-Beigi F, Grimm RH, Jr, et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med. 2008;358(24):2545–2559. doi: 10.1056/NEJMoa0802743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Gogitidze Joy N, Hedrington MS, Briscoe VJ, Tate DB, Ertl AC, Davis SN. Effects of acute hypoglycemia on inflammatory and pro-atherothrombotic biomarkers in individuals with type 1 diabetes and healthy individuals. Diabetes Care. 2010;33(7):1529–1535. doi: 10.2337/dc09-0354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Chow E, Iqbal A, Bernjak A, Ajjan R, Heller SR. Effect of hypoglycaemia on thrombosis and inflammation in patients with type 2 diabetes. Lancet. 2014;383:S35. doi: 10.1016/S0140-6736(14)60298-1. [DOI] [Google Scholar]
- 140.Hill D, Fisher M. The effect of intensive glycaemic control on cardiovascular outcomes. Diabetes Obes Metab. 2010;12(8):641–647. doi: 10.1111/j.1463-1326.2010.01199.x. [DOI] [PubMed] [Google Scholar]
- 141.Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med. 2008;359(15):1577–1589. doi: 10.1056/NEJMoa0806470. [DOI] [PubMed] [Google Scholar]
- 142.Dormandy JA, Charbonnel B, Eckland DJ, Erdmann E, Massi-Benedetti M, Moules IK, Skene AM, Tan MH, Lefebvre PJ, Murray GD, et al. Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial In macroVascular Events): a randomised controlled trial. Lancet. 2005;366(9493):1279–1289. doi: 10.1016/S0140-6736(05)67528-9. [DOI] [PubMed] [Google Scholar]
- 143.Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, Mattheus M, Devins T, Johansen OE, Woerle HJ, et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015;373(22):2117–2128. doi: 10.1056/NEJMoa1504720. [DOI] [PubMed] [Google Scholar]
- 144.Marso SP, Daniels GH, Brown-Frandsen K, Kristensen P, Mann JF, Nauck MA, Nissen SE, Pocock S, Poulter NR, Ravn LS, et al. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2016;375(4):311–322. doi: 10.1056/NEJMoa1603827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Phung OJ, Schwartzman E, Allen RW, Engel SS, Rajpathak SN. Sulphonylureas and risk of cardiovascular disease: systematic review and meta-analysis. Diabet Med. 2013;30(10):1160–1171. doi: 10.1111/dme.12232. [DOI] [PubMed] [Google Scholar]
- 146.Longstaff C, Kolev K. Basic mechanisms and regulation of fibrinolysis. J Thromb Haemost. 2015;13(Suppl 1):S98–S105. doi: 10.1111/jth.12935. [DOI] [PubMed] [Google Scholar]
- 147.Vercauteren E, Gils A, Declerck PJ. Thrombin activatable fibrinolysis inhibitor: a putative target to enhance fibrinolysis. Semin Thromb Hemost. 2013;39(4):365–372. doi: 10.1055/s-0033-1334488. [DOI] [PubMed] [Google Scholar]
- 148.Wyseure T, Declerck PJ. Novel or expanding current targets in fibrinolysis. Drug Discov Today. 2014;19(9):1476–1482. doi: 10.1016/j.drudis.2014.05.025. [DOI] [PubMed] [Google Scholar]
- 149.Plug T, Marquart JA, Marx PF, Meijers JC. Selective modulation of thrombin-activatable fibrinolysis inhibitor (TAFI) activation by thrombin or the thrombin-thrombomodulin complex using TAFI-derived peptides. J Thromb Haemost. 2015;13(11):2093–2101. doi: 10.1111/jth.13133. [DOI] [PubMed] [Google Scholar]
- 150.Buelens K, Hassanzadeh-Ghassabeh G, Muyldermans S, Gils A, Declerck PJ. Generation and characterization of inhibitory nanobodies towards thrombin activatable fibrinolysis inhibitor. J Thromb Haemost. 2010;8(6):1302–1312. doi: 10.1111/j.1538-7836.2010.03816.x. [DOI] [PubMed] [Google Scholar]
- 151.Zhou X, Weeks SD, Ameloot P, Callewaert N, Strelkov SV, Declerck PJ. Elucidation of the molecular mechanisms of two nanobodies that inhibit thrombin-activatable fibrinolysis inhibitor activation and activated thrombin-activatable fibrinolysis inhibitor activity. J Thromb Haemost. 2016;14(8):1629–1638. doi: 10.1111/jth.13381. [DOI] [PubMed] [Google Scholar]
- 152.Sigurdardottir O, Wiman B. Identification of a PAI-1 binding site in vitronectin. Biochim Biophys Acta. 1994;1208(1):104–110. doi: 10.1016/0167-4838(94)90166-X. [DOI] [PubMed] [Google Scholar]
- 153.Levin EG, Santell L. Conversion of the active to latent plasminogen activator inhibitor from human endothelial cells. Blood. 1987;70(4):1090–1098. [PubMed] [Google Scholar]
- 154.Lawrence DA, Olson ST, Palaniappan S, Ginsburg D. Engineering plasminogen activator inhibitor 1 mutants with increased functional stability. Biochemistry. 1994;33(12):3643–3648. doi: 10.1021/bi00178a022. [DOI] [PubMed] [Google Scholar]
- 155.Sahebkar A, Simental-Mendia LE, Watts GF, Golledge J. Impact of fibrate therapy on plasma plasminogen activator inhibitor-1: a systematic review and meta-analysis of randomized controlled trials. Atherosclerosis. 2015;240(1):284–296. doi: 10.1016/j.atherosclerosis.2015.03.016. [DOI] [PubMed] [Google Scholar]
- 156.Van De Craen B, Declerck PJ, Gils A. The Biochemistry, Physiology and Pathological roles of PAI-1 and the requirements for PAI-1 inhibition in vivo. Thromb Res. 2012;130(4):576–585. doi: 10.1016/j.thromres.2012.06.023. [DOI] [PubMed] [Google Scholar]
- 157.Wyseure T, Rubio M, Denorme F, Martinez de Lizarrondo S, Peeters M, Gils A, De Meyer SF, Vivien D, Declerck PJ. Innovative thrombolytic strategy using a heterodimer diabody against TAFI and PAI-1 in mouse models of thrombosis and stroke. Blood. 2015;125(8):1325–1332. doi: 10.1182/blood-2014-07-588319. [DOI] [PubMed] [Google Scholar]
- 158.Denorme F, Wyseure T, Peeters M, Vandeputte N, Gils A, Deckmyn H, Vanhoorelbeke K, Declerck PJ, De Meyer SF. Inhibition of thrombin-activatable fibrinolysis inhibitor and plasminogen activator inhibitor-1 reduces ischemic brain damage in mice. Stroke. 2016;47(9):2419–2422. doi: 10.1161/STROKEAHA.116.014091. [DOI] [PubMed] [Google Scholar]
- 159.Zhou X, Hendrickx ML, Hassanzadeh-Ghassabeh G, Muyldermans S, Declerck PJ. Generation and in vitro characterisation of inhibitory nanobodies towards plasminogen activator inhibitor 1. Thromb Haemost. 2016;116(6):1032–1040. doi: 10.1160/TH16-04-0306. [DOI] [PubMed] [Google Scholar]
- 160.Rouch A, Vanucci-Bacque C, Bedos-Belval F, Baltas M. Small molecules inhibitors of plasminogen activator inhibitor-1—an overview. Eur J Med Chem. 2015;92:619–636. doi: 10.1016/j.ejmech.2015.01.010. [DOI] [PubMed] [Google Scholar]
- 161.Elokdah H, Abou-Gharbia M, Hennan JK, McFarlane G, Mugford CP, Krishnamurthy G, Crandall DL. Tiplaxtinin, a novel, orally efficacious inhibitor of plasminogen activator inhibitor-1: design, synthesis, and preclinical characterization. J Med Chem. 2004;47(14):3491–3494. doi: 10.1021/jm049766q. [DOI] [PubMed] [Google Scholar]
- 162.Gorlatova NV, Cale JM, Elokdah H, Li D, Fan K, Warnock M, Crandall DL, Lawrence DA. Mechanism of inactivation of plasminogen activator inhibitor-1 by a small molecule inhibitor. J Biol Chem. 2007;282(12):9288–9296. doi: 10.1074/jbc.M611642200. [DOI] [PubMed] [Google Scholar]
- 163.Liang A, Wu F, Tran K, Jones SW, Deng G, Ye B, Zhao Z, Snider RM, Dole WP, Morser J, et al. Characterization of a small molecule PAI-1 inhibitor, ZK4044. Thromb Res. 2005;115(4):341–350. doi: 10.1016/j.thromres.2004.09.021. [DOI] [PubMed] [Google Scholar]
- 164.Crandall DL, Elokdah H, Di L, Hennan JK, Gorlatova NV, Lawrence DA. Characterization and comparative evaluation of a structurally unique PAI-1 inhibitor exhibiting oral in vivo efficacy. J Thromb Haemost. 2004;2(8):1422–1428. doi: 10.1111/j.1538-7836.2004.00829.x. [DOI] [PubMed] [Google Scholar]
- 165.Rupin A, Gaertner R, Mennecier P, Richard I, Benoist A, De Nanteuil G, Verbeuren TJ. S35225 is a direct inhibitor of plasminogen activator inhibitor type-1 activity in the blood. Thromb Res. 2008;122(2):265–270. doi: 10.1016/j.thromres.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 166.Fortenberry YM. Plasminogen activator inhibitor-1 inhibitors: a patent review (2006-present) Expert Opin Ther Pat. 2013;23(7):801–815. doi: 10.1517/13543776.2013.782393. [DOI] [PubMed] [Google Scholar]
- 167.Kumada T, Abiko Y. Physiological role of alpha 2-plasmin inhibitor in rats. Thromb Res. 1984;36(2):153–163. doi: 10.1016/0049-3848(84)90337-2. [DOI] [PubMed] [Google Scholar]
- 168.Sakata Y, Eguchi Y, Mimuro J, Matsuda M, Sumi Y. Clot lysis induced by a monoclonal antibody against alpha 2-plasmin inhibitor. Blood. 1989;74(8):2692–2697. [PubMed] [Google Scholar]
- 169.Reed GL, 3rd, Matsueda GR, Haber E. Synergistic fibrinolysis: combined effects of plasminogen activators and an antibody that inhibits alpha 2-antiplasmin. Proc Natl Acad Sci USA. 1990;87(3):1114–1118. doi: 10.1073/pnas.87.3.1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Reed GL, 3rd, Matsueda GR, Haber E. Inhibition of clot-bound alpha 2-antiplasmin enhances in vivo thrombolysis. Circulation. 1990;82(1):164–168. doi: 10.1161/01.CIR.82.1.164. [DOI] [PubMed] [Google Scholar]
- 171.Wiman B, Collen D. On the mechanism of the reaction between human alpha 2-antiplasmin and plasmin. J Biol Chem. 1979;254(18):9291–9297. [PubMed] [Google Scholar]
- 172.Shieh BH, Travis J. The reactive site of human alpha 2-antiplasmin. J Biol Chem. 1987;262(13):6055–6059. [PubMed] [Google Scholar]
- 173.Lee KN, Lee SC, Jackson KW, Tae WC, Schwartzott DG, McKee PA. Effect of phenylglyoxal-modified alpha2-antiplasmin on urokinase-induced fibrinolysis. Thromb Haemost. 1998;80(4):637–644. [PubMed] [Google Scholar]
- 174.Lee KN, Tae WC, Jackson KW, Kwon SH, McKee PA. Characterization of wild-type and mutant alpha2-antiplasmins: fibrinolysis enhancement by reactive site mutant. Blood. 1999;94(1):164–171. [PubMed] [Google Scholar]
- 175.Kimura S, Aoki N. Cross-linking site in fibrinogen for alpha 2-plasmin inhibitor. J Biol Chem. 1986;261(33):15591–15595. [PubMed] [Google Scholar]
- 176.Tamaki T, Aoki N. Cross-linking of alpha 2-plasmin inhibitor to fibrin catalyzed by activated fibrin-stabilizing factor. J Biol Chem. 1982;257(24):14767–14772. [PubMed] [Google Scholar]
- 177.Sasaki T, Morita T, Iwanaga S. Identification of the plasminogen-binding site of human alpha 2-plasmin inhibitor. J Biochem. 1986;99(6):1699–1705. doi: 10.1093/oxfordjournals.jbchem.a135645. [DOI] [PubMed] [Google Scholar]
- 178.Kimura S, Tamaki T, Aoki N. Acceleration of fibrinolysis by the N-terminal peptide of alpha 2-plasmin inhibitor. Blood. 1985;66(1):157–160. [PubMed] [Google Scholar]
- 179.Lee KN, Jackson KW, McKee PA. Effect of a synthetic carboxy-terminal peptide of alpha(2)-antiplasmin on urokinase-induced fibrinolysis. Thromb Res. 2002;105(3):263–270. doi: 10.1016/S0049-3848(02)00030-0. [DOI] [PubMed] [Google Scholar]
- 180.Udvardy M, Schwartzott D, Jackson K, McKee PA. Hybrid peptide containing RGDF (Arg-Gly-Asp-Phe) coupled with the carboxy terminal part of alpha 2-antiplasmin capable of inhibiting platelet aggregation and promoting fibrinolysis. Blood Coagul Fibrinolysis. 1995;6(1):11–16. doi: 10.1097/00001721-199502000-00002. [DOI] [PubMed] [Google Scholar]
- 181.Sumi Y, Ichikawa Y, Nakamura Y, Miura O, Aoki N. Expression and characterization of pro alpha 2-plasmin inhibitor. J Biochem. 1989;106(4):703–707. doi: 10.1093/oxfordjournals.jbchem.a122920. [DOI] [PubMed] [Google Scholar]
- 182.Koyama T, Koike Y, Toyota S, Miyagi F, Suzuki N, Aoki N. Different NH2-terminal form with 12 additional residues of alpha 2-plasmin inhibitor from human plasma and culture media of Hep G2 cells. Biochem Biophys Res Commun. 1994;200(1):417–422. doi: 10.1006/bbrc.1994.1465. [DOI] [PubMed] [Google Scholar]
- 183.Bangert K, Johnsen AH, Christensen U, Thorsen S. Different N-terminal forms of alpha 2-plasmin inhibitor in human plasma. Biochem J. 1993;291(pt 2):623–625. doi: 10.1042/bj2910623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Lee KN, Jackson KW, Christiansen VJ, Chung KH, McKee PA. A novel plasma proteinase potentiates alpha2-antiplasmin inhibition of fibrin digestion. Blood. 2004;103(10):3783–3788. doi: 10.1182/blood-2003-12-4240. [DOI] [PubMed] [Google Scholar]
- 185.Lee KN, Jackson KW, Christiansen VJ, Dolence EK, McKee PA. Enhancement of fibrinolysis by inhibiting enzymatic cleavage of precursor alpha2-antiplasmin. J Thromb Haemost. 2011;9(5):987–996. doi: 10.1111/j.1538-7836.2011.04195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Ricklin D, Lambris JD. Complement-targeted therapeutics. Nat Biotechnol. 2007;25(11):1265–1275. doi: 10.1038/nbt1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Rother RP, Rollins SA, Mojcik CF, Brodsky RA, Bell L. Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nat Biotechnol. 2007;25(11):1256–1264. doi: 10.1038/nbt1344. [DOI] [PubMed] [Google Scholar]
- 188.King R, Tiede C, Simmons K, Fishwick C, Tomlinson D, Ajjan R. Inhibition of complement C3 and fibrinogen interaction: a potential novel therapeutic target to reduce cardiovascular disease in diabetes. Lancet. 2015;385(Suppl 1):S57. doi: 10.1016/S0140-6736(15)60372-5. [DOI] [PubMed] [Google Scholar]