

Abstract
At nerve terminals, synaptic vesicle components are retrieved from the cell surface and recycled for local reuse soon after exocytosis. The kinetics of this coupling is critical for the proper functioning of synapses during repetitive action potential firing, because deficiencies in this process lead to abnormal depletion of the releasable vesicle pool. Although the molecular basis of this coupling is poorly understood, numerous biochemical data point to a role for synaptotagmin I (SytI), an essential synaptic vesicle protein required for fast calcium-dependent exocytosis. Here, using synapto-pHluorin in an approach that allows the dissection of endocytosis and exocytosis into separate components during periods of stimulation, we examined exocytic–endocytic coupling in synapses from SytI knockout mice and their WT littermates. We show that endocytosis is significantly impaired in the absence of SytI with the relative rates of endocytosis compared with exocytosis reduced ≈3-fold with respect to WT. Thus, in addition to regulating exocytosis, SytI also controls the kinetic efficiency of endocytosis at nerve terminals.
After release of neutrotransmitters, synaptic vesicles are locally endocytosed, refilled, and made available for subsequent rounds of release at nerve terminals (1–4). During repetitive stimulation, depletion of the vesicle pool is prevented when the rate of endocytosis is able to compensate for the rate of exocytosis, and blockade of endocytosis leads to vesicle pool depletion (5). Recently, it was shown that the speed of endocytosis is fast enough to compensate for exocytosis, even during continuous stimulation at physiological frequencies; however, the maximum rate of endocytosis can be exceeded by the rate of exocytosis above a given stimulus frequency (6). Although the kinetic coupling of exocytosis and endocytosis is crucial to maintaining synaptic function, details of the molecular mechanisms regulating this balance remain to be elucidated.
The synaptic vesicle protein synaptotagmin I (SytI) is a known key regulator of evoked, calcium-dependent exocytosis at synapses (7–13). In addition, a body of evidence implicates synaptotagmin as a regulator of endocytosis (14–17). Caenorhabditis elegans lacking a functional SytI gene show a depletion of synaptic vesicles at nerve terminals, consistent with a defect in endocytosis (14). Biochemical studies suggest synaptotagmin may be involved in clathrin-coated pit formation by interaction with AP-2 (15–16). Most recently, it has been shown that SytI is required for endocytosis of synaptic vesicles in Drosophila synapses by photoinactivation of SytI after exocytosis with functional protein (18). Together, these data imply that SytI is a multifunctional protein regulating both exocytosis and endocytosis at nerve terminals.
Here, using synapto-pHluorin (spH) (19) we demonstrate that endocytosis is slowed by 2- to 3-fold in SytI-deficient terminals relative to WT synapses in dissociated primary cortical neuronal cultures. This endocytic defect is evident both in the poststimulus phase of spH fluorescence signal decay and during the stimulus period. We hypothesize that SytI, functioning as a regulator of both exocytosis and endocytosis at nerve terminals, may provide a means of controlling the balance between the two processes.
Materials and Methods
Cell Culture and Transfection. SytI+/– mice (7) purchased from The Jackson Laboratory were mated to obtain WT and SytI-null offspring. Pregnant females from timed matings were killed by asphyxiation with CO2 at embryonic day (E) 18 or E19, and pups were recovered by postmortem caesarian section. The cortex was dissected from each pup, then individually dissociated, plated onto polyornithine-coated coverslips, and cultured as described in refs. 20 and 21. Genomic DNA was isolated from discarded brain tissue with the DNeasy kit (Qiagen, Valencia, CA) and used to genotype each animal by PCR. Primer sequences for PCR detection of WT SytI and the truncated knockout (KO) gene were provided by The Jackson Laboratory. Calcium phosphate-mediated gene transfer was used to transfect 7-day-old WT and KO cultures with a plasmid carrying the gene encoding spH as described in ref. 21. Experiments were performed on cultures 5–10 days posttransfection. All animal use and experiments were approved by the Institutional Animal Care and Use Committee of the Weill Medical College of Cornell University.
Experimental Conditions. Coverslips were mounted in a perfusion/stimulation chamber equipped with platinum–irridium field-stimulus electrodes on the stage of a custom-built laser-scanning confocal microscope as described in ref. 21. Cultures were perfused with a flow rate of ≈1 ml/min at room temperature (≈24°C) with Tyrode's saline solution containing 119 mM NaCl, 2.5 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 25 mM Hepes (pH 7.4), 30 mM glucose, 10 μM 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), and 50 μM d,l-2-amino-5-phosphonovaleric acid (APV). FM 1-43 (Molecular Probes) was used at 15 μM in Tyrode's solution. Synaptic vesicle pools were labeled by action potential stimulation (1-ms current pulses yielding fields of ≈10 V/cm) for 60 s at 10 Hz, unless otherwise noted, followed by an additional 3 min of dye exposure to ensure maximal labeling of all recycling vesicles. Cultures were rinsed for 10 min in dye-free solution before destaining. The unloading phase consisted of continuously imaging at 6- or 3-s intervals before, during, and after a prolonged train of action potentials. All FM 1-43 measurements and most spH experiments were performed blind by using WT and KO littermate pairs. Bafilomycin (Baf) was used at a concentration between 1 and 5 μM, because the potency varied from batch to batch from the manufacturer (Calbiochem).
Optical Measurements, Microscopy, and Analysis. Laser-scanning fluorescence images were acquired as described in ref. 21 by using a 510-nm-long pass filter. Quantitative measurements of fluorescence intensity at individual boutons were obtained by averaging a 4 × 4 area of pixel intensities. FM 1-43 destaining data were normalized to the total loss of fluorescence during the action potential train, determined by subtracting the average of the final five time points from that of the first five for each individual bouton. FM 1-43 destaining curves and the recovery phase of spH curves were fitted to single exponential decays.
Results and Discussion
SytI-Null Nerve Terminals Exhibit Slowed Poststimulus Endocytosis. To examine the role of SytI in synaptic vesicle recycling, we assayed differences in endocytosis kinetics by using spH-transfected dissociated neurons in culture. SpH, a pH-sensitive form of GFP fused to the luminal domain of vesicle-associated membrane protein (VAMP) 2 (19), provides a sensitive optical probe to track the net balance of exocytosis and endocytosis at nerve terminals (20–22). Normally, resting synaptic vesicles are at an acidic pH (≈5.5) that would quench the spH signal. Action potential firing led to a fluorescence increase at spH-expressing nerve terminals because of fusion of acidic synaptic vesicles with the plasma membrane. After fusion, exposure to the more-alkaline extracellular environment (pH ≈7.4) relieves the proton-dependent quenching of the modified GFP (Fig. 1A). The fluorescence recovery after stimulation results from two sequential steps, endocytosis and a much faster vesicle reacidification (21, 23).
Fig. 1.

SpH experiments reveal slowed poststimulus endocytosis in SytI KO synapses. (A) Average fluorescence signals of spH in WT (black) and SytI-null (red) mouse cortical synapses imaged at 3-s intervals before, during, and after stimulation with 300 action potentials at 10 Hz. Error bars represent SEM (one experiment for each genotype with n = 36 boutons for WT and n = 22 boutons for KO). (B) Average time constants of the recovery of spH signal after a 300-action-potential stimulation at 10 Hz in mouse WT (36.7 ± 2.9; n = 9) and KO (68.5 ± 9.8; n = 9) cortical synapses. All error bars represent SEM.
Comparison of the average time course of poststimulus recovery of spH (τ) revealed an ≈2-fold difference between WT and KO terminals (Fig. 1), indicating that synaptotagmin is important for compensatory endocytosis after the stimulus train. In both WT and KO terminals, the speed of endocytosis depends on the amount of spH added to the surface, with rates increasing as more spH is delivered to the surface with longer action potential trains (data not shown), in agreement with previous data reported for rat hippocampal neurons (21). Although the rate of vesicle reacidification in rat hippocampal neurons is much faster than the time scales examined here (21, 23), we verified that poststimulus spH signals in synaptotagmin KO also reflect clearance from the surface and not acidification times by using acid quenching of surface fluorescence (21) (data not shown). In addition, we verified the genotypic differences in endocytosis kinetics by using pulse–chase analysis with FM 1-43 (3). Both WT and SytI KO boutons readily take up FM 1-43 dye and destain efficiently during action potential stimulation (Fig. 2A). The efficient dye loading and unloading in the SytI KO is potentially surprising because this mutant is often viewed as releasing no or very few neurotransmitters. However, it has recently been shown that the amount of transmitters released from cultured hippocampal terminals is the same in WT and SytI KO mice, and only the timing of release is altered (24). In the pulse–chase experiments, the kinetics of endocytosis was measured by determining the amount of FM 1-43 that is taken up at different times (Δt) after the start of a 300-action-potential stimulus train (Fig. 2B Inset). As the interval of Δt is increased, more endocytosis occurs before dye exposure, and, therefore, less dye is incorporated into the vesicle pool. The total uptake is assessed in later maximal unloading rounds of stimulation (900 action potentials followed by 1,200 action potentials). Comparisons of dye uptake kinetics in WT and SytI KO reveal that during the poststimulus period there is also a significant delay in endocytosis in the KO with respect to WT, with 50% of maximal uptake occurring within ≈25 s of the end of the stimulus for WT and within ≈60 s for SytI KO (Fig. 2B). These data indicate an ≈2-fold reduction in endocytosis kinetics and are consistent with the difference of the average rates of spH fluorescence recovery after stimulation. Furthermore, these results are independent of vesicle reacidification rates and imply that the slowed spH recovery rates in the SytI KO terminals are due to slowed endocytosis and not a defect in vesicle reacidification.
Fig. 2.

Pulse–chase FM 1-43 experiments reveal slowed poststimulus endocytosis in SytI KO synapses. (A) WT and SytI-deficient synapses from littermates loaded with FM 1-43 (Left) and destained (Right) after two rounds of stimulation with 900 and 1,200 action potentials. (B) FM 1-43 pulse–chase experiments. (Inset) Dye-loading protocol. During the loading phase, WT or SytI KO cultures were stimulated for 30 s at 10 Hz and exposed to FM 1-43 for 3 min after a variable delay (Δt) from the onset of the stimulus. As Δt increases, more endocytosis occurs during this delay, escaping labeling by the subsequent dye application, and less dye is loaded into the vesicle pool. For Δt = 0, the 3 min of dye application should capture all vesicles endocytosed both during and after the stimulus. After a 10-min wash, two rounds of stimulation (900 and 1,200 action potentials, separated by 2 min) were applied to evoke maximal release. Dye uptake was assessed by measuring the total fluorescence change from images acquired before and after each round of stimulation. Each measurement of Δt was bracketed by runs where Δt = 0, and the value of the labeled fraction of the vesicle pool was normalized to the average of the bracketing controls. Each point represents Δt measurements from 40 to 128 boutons averaged over three coverslips. Error bars are SEM.
Slowed Destaining and Reduced Vesicle Pool Size but Similar Vesicle Repriming Kinetics in SytI KO Synapses Relative to WT. Because previous ultrastructural analyses in Drosophila and C. elegans revealed alteration in vesicle pools in the absence of SytI (14, 25), we measured the total recycling vesicle pool size by using FM dye uptake experiments. To determine the size of the recycling vesicle pool, we measured the amount of FM 1-43 uptake that can be loaded into individual presynaptic terminals during action potential trains, according to the protocol described in the legend of Fig. 3A. Measurements of the amount of dye uptake for a given stimulus period were carried out in further rounds of maximal stimulation (900 action potentials followed by 1,200 action potentials) and normalized to the dye uptake in WT for 600 action potentials. These experiments indicate that there was an ≈15–20% reduction in the total size of the recycling pool in SytI KO terminals as determined by the relative amount of uptake once steady-state loading is achieved (<600 action potentials for both genotypes) (Fig. 3A). In addition, the kinetics of vesicle turnover appeared to be slower in the SytI KO, as would be expected from the known electrophysiological phenotype (7). To assess vesicle pool turnover kinetics more directly, we examined the kinetics of FM 1-43 destaining during repetitive action potential firing in synapses preloaded with the dye to label the entire recycling pool. Destaining of FM 1-43 during a 10-Hz stimulation revealed ≈30% slower destaining kinetics in SytI KO synapses (Fig. 3 B and C). In addition, in 6 of 13 experiments, we observed a significant delay in the onset of destaining in the KO compared with the WT nerve terminals. In those cases, dye destaining was insignificant during the first 3 s of stimulation, followed by a prompt destaining with a time constant that was ≈30% slower than the time constant in the WT, where destaining always commenced with the onset of stimulation. The slower destaining kinetics are consistent with the known importance of SytI in triggering calcium-mediated release. The defect in destaining kinetics is more modest than one might have predicted based on the reduction in excitatory postsynaptic current size reported in electrophysiological experiments (7–9); however, our studies are necessarily carried out at much higher stimulus frequencies than in those studies. Under these conditions, asynchronous release becomes much more prominent (26). Because SytI has been proposed to operate largely for fast synchronous release and not asynchronous release (7), high-frequency stimulation should partly overcome the defect due to SytI.
Fig. 3.

The kinetics of vesicle pool turnover is reduced in SytI KO terminals. (A) Measurement of the recycling vesicle pool size in WT and SytI KO synapses. (Inset) Loading protocol. Cultures were stimulated for variable times (Δt) at 10 Hz in the presence of FM 1-43. The dye was left on the culture for an additional 3 min at the conclusion of the stimulus train to allow for completion of endocytosis. Dye uptake was measured as the total fluorescence change (ΔFtot) during the unloading phase, as described in Fig. 1. For each Δt, ΔFtot was normalized to the average ΔFtot of two bracketing control runs where Δt = 60 s (600 action potentials, maximal loading). Each time point represents a measurement of the labeled fraction of the recycling pool averaged over 75–204 boutons from three coverslips. (B) Semilog plot of FM 1-43 destaining kinetics in WT and SytI KO boutons. Terminals were loaded as in A with 600 action potentials at 10 Hz. Destaining kinetics were obtained by imaging at 3-s intervals before, during, and after stimulation with 900 action potentials at 10 Hz followed by 1,200 action potentials at 20 Hz after a 2-min rest. Time constants for FM 1-43 release were obtained by fitting the destaining curves with single exponentials. In the example in B, τWT = 15.0 s (n = 53 boutons) and τKO = 23.7 s (n = 39 boutons). In the case of the KO for this and a total of 6 of 13 experiments, the destaining appeared to be delayed with respect to the onset of the stimulus. In those cases, the FM 1-43 release curves were fit to an exponential with a time delay. (C) Average time constants of FM 1-43 destaining in WT (17.5 ± 0.8 s) and SytI KO (22.4 ± 0.8 s) terminals (n = 13 coverslips for each genotype, each coverslip consisted of measurements from 31–58 boutons.) All error bars are SEM. (D) The time course of vesicle repriming. (Inset) Experimental protocol. WT and SytI KO cultures were stimulated at 20 Hz during a 5-s application of FM 1-43 for a period exceeding the dye exposure by an interval Δt. Dye uptake was measured as ΔFtot during an unloading phase as described for A and B. The dye uptake at Δt = 0 represents the total releasable pool of vesicles (F0). Dye uptake measurements, obtained where Δt > 0, correspond to vesicle depletion due to fusion of reprimed vesicles during the additional action potentials after the dye has been rinsed away (FΔt). The data are plotted as (F0–FΔt)/F0, the fraction depleted, as a function of the interval Δt. Each point represents averaged measurements from 28 to 83 boutons from three coverslips, with the exceptions of Δt = 20 s (four coverslips) and Δt = 60 s (two coverslips).
Synaptic vesicle repriming is defined as the amount of time that elapses before newly endocytosed vesicles become available for rerelease at the presynaptic terminal (3). Therefore, vesicle repriming encompasses everything that occurs immediately after, but not including, endocytosis up until rerelease of the vesicle. The protocol for measuring repriming kinetics is illustrated in Fig. 3D Inset. During the loading phase, FM 1-43 is introduced for a fixed period (5 s) coincident with the onset of a train of action potentials (20 Hz) that outlasts the exposure to the dye by a time interval Δt. Sequential runs with different periods Δt were measured at individual boutons to determine the gradual impact of the additional action potentials upon releasing the dye taken up during the dye exposure period. Single bouton measurements of ΔF, the net amount of dye remaining after the loading phase, were carried out by using a maximal stimulus to destain the bouton during a train of 900 action potentials. The unloading measurement was carried out after a 10-min rest after the loading phase. The data are normalized with respect to a run during which the stimulation was terminated precisely at the same time at which the extracellular dye was washed away (Δt = 0, ΔF0). The ratio (ΔF0–ΔFΔt)/ΔF0 gives the fraction of fluorescence depleted by the action potentials fired during the period Δt and was measured for both WT and SytI KO terminals. This plot represents the time course of the gain of rerelease competence of recently endocytosed vesicles, and a perturbation in repriming will shift the kinetics. The resulting time course reveals that there is no significant difference in repriming kinetics, with ≈75% of endocytosed vesicles rereleased within 60 s of continuous stimulation after the dye washout for both WT and KO synaptic vesicle reuptake. Therefore, it seems unlikely that the slowed endocytosis phenotype is attributable to a repriming defect.
SytI Is Required for Compensatory Endocytosis During the Stimulus Train. A critical determinant of synaptic function during repetitive stimulation is the extent to which the rate of endocytosis can compensate for the rate of exocytosis, avoiding vesicle pool depletion. Because the time scale for endocytosis has been shown in a number of systems to depend on the amount of net accumulation of synaptic membrane on the cell surface (21, 27) and synaptotagmin disruption decreases the rates of exocytosis, we surmised that to test the hypothesis that synaptotagmin is important in exocytic–endocytic coupling, we would need to measure the relative rates of exocytosis compared with endocytosis during and after the stimulus. To determine the quantitative relationship between rates of exocytosis and endocytosis during periods of stimulation, we made use of spH transfected into dissociated neurons in culture and of Baf, a membrane-permeant blocker of the V-type ATPase whose action is required for vesicle reacidification and neurotransmitter refilling (20), to trap vesicles in the alkaline and, hence fluorescent state after fusion (Fig. 4A, green trace). Action potential-stimulated fluorescence increases under these conditions provide a measure of the total amount of exocytosis, because Baf does not perturb exocytosis or endocytosis in our system (20, 28, 29); thus,
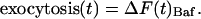 |
[1] |
This measurement tracks only the first round of exocytosis because any vesicles that become endocytosed and undergo a second round of fusion would not provide further increases in fluorescence. However, we have previously shown that the reuse rate for vesicles under continuous stimulation at 10 Hz would result in ≤10% of the vesicles undergoing a second round of exocytosis under these conditions (30). Under control conditions, the net rise in fluorescence represents the balance of exocytosis and endocytosis:
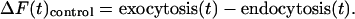 |
[2] |
The fluorescence increases stimulated by action potential firing in Baf were larger than in controls because the effects of endocytosis and reacidification occurring during the stimulus have been eliminated (Fig. 4A). The difference between runs with and without Baf provides a measure of the kinetics of endocytosis during the stimulus period, and the time course of endocytosis can be computed as
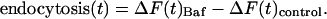 |
[3] |
This approach thus provides a measure of the time course of endocytosis (Fig. 4A, WT red trace) that can be directly compared with the time course of exocytosis (Fig. 4A, WT green trace) at the same nerve terminals. The ratio of the slopes of endocytic and exocytic time courses for the stimulus period provides a measure of the exocytic–endocytic balance.
Fig. 4.

The exocytic–endocytic balance is perturbed in SytI KO synapses. (A) SpH signals in cultured cortical neurons from E19 WT (Upper) and SytI KO (Lower) littermates. (Left) The average of two control runs (black circles) and two Baf runs (green triangles) imaged at 3-s intervals before, during, and after stimulation. (Right) The averaged control was subtracted from the Baf trace to yield a difference curve (red circles) that represents the contribution of endocytosis during stimulation. Rates of endocytosis and exocytosis were obtained by measuring the slope (dashed lines) of the Baf curve and the difference trace during the stimulus period. For WT synapses, exocytosis = 0.35 a.u.·s–1, endocytosis = 0.013 a.u.·s–1, and endocytosis/exocytosis = 0.371. For SytI KO terminals, exocytosis = 0.034 a.u.·s–1, endocytosis = 0.002 a.u.·s–1, and endocytosis/exocytosis = 0.059. For both WT and KO, n = 7 boutons. (B) Average endocytosis/exocytosis ratios from E18–E19 mouse WT (0.33 ± 0.03; n = 4 coverslips) and SytI-deficient (0.10 ± 0.02; n = 6 coverslips) cortical neurons.
We used this approach to compare the balance of exocytosis and endocytosis in synapses formed between neurons derived from either SytI KO mice (7) or their WT littermates (Fig. 4A). This analysis indicated that on average, during the stimulus period, the ratio of the rate of endocytosis compared with exocytosis was reduced from ≈0.35 in WT to ≈0.10 in SytI KO (Fig. 4B). These findings indicate that SytI is important for compensatory synaptic vesicle endocytosis both during and after the stimulus period.
A Role for SytI in Exocytic–Endocytic Coupling? Although a number of genetic, biochemical, and physiological experiments have demonstrated a critical role for SytI in calcium-triggered exocytosis, its role in endocytosis has only recently become more clearly defined. Until recently, slower progress in this area was due in part to the lack of a direct measure of endocytosis rates at synapses deficient in this protein. In C. elegans, mutations in the gene-encoding synaptotagmin resulted in a depletion of synaptic vesicles at nerve terminals, suggestive of a role in endocytosis; however, it has not been possible to provide direct physiological assays of endocytosis in that system. SpH measurements, in combination with Baf, provided the means to examine the kinetic components of exocytosis and endocytosis separately in both mammalian synapses, as shown here and in Drosophila nerve terminals (18). The molecular basis for synaptoagmin's function in endocytosis has not been addressed in physiological experiments; however, results from biochemical experiments have shown that synaptoagmin binds to the clathrin adaptor protein AP-2 (15) and this may in turn regulate recruitment of clathrin-coated pit cargo (16). In addition, because calcium levels can modulate endocytosis rates (20), it is possible that one of synaptotagmin's C2 domains acts as a calcium sensor for endocytosis, in addition to their proposed role for exocytosis. Finally, it was shown that one of the effectors of synaptotagmin is phosphatidylinositol-4,5-bisphosphate (31), a moiety also thought to be important for clathrin-mediated endocytosis (32). Together with our observations that endocytosis is significantly impaired in SytI KO terminals with an ≈3-fold reduction in the kinetics of endocytosis compared with WT terminals, these findings strongly advocate a role for SytI in synaptic vesicle endocytosis. However, the possibility remains that asynchronous exocytosis, the apparent mechanism of release in SytI KO terminals, constrains vesicles to use an alternative path for endocytosis that is inherently slower than endocytosis after synchronous release in WT terminals.
One of the central problems regarding the functioning of synaptic terminals is the mechanism by which endocytosis is closely coupled to exocytosis. It appears that part of the solution is the use of certain synaptic vesicle proteins for dual purposes. In the case of SytI, it appears to act as a key regulator of both exocytosis and endocytosis providing a potential pathway for this coupling.
Acknowledgments
We thank James Rothman (Columbia University) for providing us with the spH construct, members of the Ryan lab for useful discussions, and Wayne Yan for technical assistance. This work was supported by grants from the National Institutes of Health and the Hirsch Trust (to T.A.R.) and a National Institutes of Health postdoctoral training grant (to K.N.-T.).
Author contributions: K.N.-T. and T.A.R. designed research; K.N.-T. performed research; K.N.-T. and T.A.R. analyzed data; and K.N.-T. and T.A.R. wrote the paper.
Abbreviations: Baf, bafilomycin; En, embryonic day n; KO, knockout; SytI, synaptotagmin I; spH, synapto-pHluorin.
See Commentary on page 16401.
References
- 1.Heuser, J. E. & Reese, T. S. (1973) J. Cell Biol. 57, 315–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miller, T. M. & Heuser, J. E. (1984) J. Cell Biol. 98, 685–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ryan, T. A., Reuter, H., Wendland, B., Schweizer, F. E., Tsien, R. W. & Smith, S. J. (1993) Neuron 11, 713–724. [DOI] [PubMed] [Google Scholar]
- 4.Wu, L. G. & Betz, W. J. (1996) Neuron 17, 769–779. [DOI] [PubMed] [Google Scholar]
- 5.Koenig, J. H. & Ikeda, K. (1989) J. Neurosci. 9, 3844–3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fernandez-Alfonzo, T. & Ryan, T. A. (2004) Neuron 41, 939–953. [DOI] [PubMed] [Google Scholar]
- 7.Geppert, M., Goda, Y., Hammer, R. E., Li, C., Rosahl, T. W., Stevens, C. F. & Sudhof, T. C. (1994) Cell 79, 717–727. [DOI] [PubMed] [Google Scholar]
- 8.DiAntonio, A. & Schwarz, T. L. (1994) Neuron 12, 909–920. [DOI] [PubMed] [Google Scholar]
- 9.Littleton, J. T., Stern, M., Perin, M. & Bellen, H. J. (1994) Proc. Natl. Acad. Sci. USA 91, 10888–10892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fernandez-Chacon, R., Konigstorfer, A., Gerber, S. H., Garcia, J., Matos, M. F., Stevens, C. F., Brose, N., Rizo, J., Rosenmund, C. & Sudhof, T. C. (2001) Nature 410, 41–49. [DOI] [PubMed] [Google Scholar]
- 11.Robinson, I. M., Ranjan, R. & Schwarz, T. L. (2002) Nature 418, 336–340. [DOI] [PubMed] [Google Scholar]
- 12.Mackler, J. M., Drummond, J. A., Loewen, C. A., Robinson, I. M. & Reist, N. E. (2002) Nature 418, 340–344. [DOI] [PubMed] [Google Scholar]
- 13.Stevens, C. F. & Sullivan, J. M. (2003) Neuron 39, 299–308. [DOI] [PubMed] [Google Scholar]
- 14.Jorgensen, E. M. Hartwieg, E., Schuske, K., Nonet, M. L., Jin, Y. & Horvitz, H. R. (1995) Nature 378, 196–199. [DOI] [PubMed] [Google Scholar]
- 15.Zhang, J. Z., Davletov, B. A., Sudhof, T. C. & Anderson, R. G. (1994) Cell 78, 751–760. [DOI] [PubMed] [Google Scholar]
- 16.Haucke, V. & De Camilli, P. (1999) Science 285, 1268–1271. [DOI] [PubMed] [Google Scholar]
- 17.Jarousse, N., Wilson, J. D., Arac, D., Rizo, J. & Kelly, R. B. (2003) Traffic 4, 468–478. [DOI] [PubMed] [Google Scholar]
- 18.Poskanzer, K. E., Marek, K. W., Sweeney, S. T. & Davis, G. W. (2003) Nature 426, 559–563. [DOI] [PubMed] [Google Scholar]
- 19.Miesenbock, G., De Angelis, D. A. & Rothman, J. E. (1998) Nature 394, 192–195. [DOI] [PubMed] [Google Scholar]
- 20.Sankaranarayanan, S. & Ryan, T. A. (2001) Nat. Neurosci. 4, 129–136. [DOI] [PubMed] [Google Scholar]
- 21.Sankaranarayanan, S. & Ryan, T. A. (2000) Nat. Cell Biol. 2, 197–204. [DOI] [PubMed] [Google Scholar]
- 22.Li, Z. & Murthy, V. N. (2001) Neuron 31, 593–605. [DOI] [PubMed] [Google Scholar]
- 23.Gandhi, S. P. & Stevens, C. F. (2003) Nature 423, 607–613. [DOI] [PubMed] [Google Scholar]
- 24.Nishiki, T. & Augustine, G. J. (2004) J. Neurosci. 24, 6127–6132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reist, N. E., Buchanan, J., Li, J., DiAntonio, A., Buxton, E. M. & Schwarz, T. L. (1998) J. Neurosci. 18, 7662–7673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hagler, D. J., Jr., & Goda, Y. (2001) J. Neurophysiol. 85, 2324–2334. [DOI] [PubMed] [Google Scholar]
- 27.Sun, J. Y., Wu, X. S. & Wu, L. G. (2002) Nature 417, 555–559. [DOI] [PubMed] [Google Scholar]
- 28.Zhou, Q., Petersen, C. C. H. & Nicoll, R. A. (2000) J. Physiol. 525, 195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cousin, M. A. & Nicholls, D. G. (1997) J. Neurochem. 69, 1927–1935. [DOI] [PubMed] [Google Scholar]
- 30.Ryan, T. A. & Smith, S. J. (1995) Neuron 14, 983–989. [DOI] [PubMed] [Google Scholar]
- 31.Tucker, W. C., Edwardson, J. M., Bai, J., Kim, H. J., Martin, T. F. & Chapman, E. R. (2003) J. Cell Biol. 162, 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krauss, M., Kinuta, M., Wenk, M. R., De Camilli, P., Takei, K. & Haucke, V. (2003) J. Cell Biol. 162, 113–124. [DOI] [PMC free article] [PubMed] [Google Scholar]