

Abstract
The prevalence of diabetes mellitus (DM) is increasing secondary to increased consumption of food and decreased physical activity worldwide. Hyperglycaemia, insulin resistance and hypertrophy of pancreatic beta cells occur in the early phase of diabetes. However, with the progression of diabetes, dysfunction and loss of beta cells occur in both types 1 and 2 DM. Programmed cell death also named apoptosis is found to be associated with diabetes, and apoptosis of beta cells might be the main mechanism of relative insulin deficiency in DM. Autophagic cell death and apoptosis are not entirely distinct programmed cell death mechanisms and share many of the regulator proteins. These processes can occur in both physiologic and pathologic conditions including DM. Besides these two important pathways, endoplasmic reticulum (ER) also acts as a cell sensor to monitor and maintain cellular homeostasis. ER stress has been found to be associated with autophagy and apoptosis. This review was aimed to describe the interactions between apoptosis, autophagy and ER stress pathways in DM.
Keywords: Apoptosis, autophagy, cell survival and death, diabetes mellitus, endoplasmic reticulum stress
Introduction
Despite the availability of diagnostic approaches and therapeutic progress, the prevalence of diabetes mellitus (DM) is increasing globally. It has been demonstrated that increased plasma glucose levels secondary to insulin resistance might be the major contributing factors in the pathogenesis of diabetes1. To overcome hyperglycaemia and insulin resistance, pancreatic β-cell hypertrophy occurs in the early phase of diabetes. However, with the progression of diabetes, dysfunction and loss of beta cells occur in response to increased metabolic load. Though DM is traditionally introduced as a non-immune disorder, novel evidence showed that inflammatory mechanisms, apoptosis, autophagy and endoplasmic reticulum (ER) stress could be the main pathways in the pathogenesis and progression of this chronic metabolic disease2.
Apoptosis was found to be closely related with caspase system3. Accumulating evidence suggests that loss of pancreatic beta cells is closely associated with increased apoptosis of beta cells, especially secondary to increased glucotoxicity, lipotoxicity, chronic ongoing inflammation and oxidative stress in both types 1 (T1) and 2 (T2) DM. Autophagy is a process of cellular proteins and macromolecules which are delivered by autophagosomes to lysosome for degradation4. The autophagy has been shown to protect pancreatic beta cells against metabolic load5,6. In this context, impairment of autophagy could cause diabetes in susceptible individuals. Besides these two important pathways, ER is also acting as a cell sensor to monitor and maintain cellular homeostasis. ER stress was found to be associated with autophagy and apoptosis and might also contribute in the pathogenesis of diabetes2.
In the first part of this review, the machinery of apoptosis, autophagy and ER stress in normal physiology and in the pathogenesis and progression of DM is discussed. The second part describes the interactions between apoptosis, autophagy and ER stress contributing to the pathogenesis of DM.
Apoptosis
The major determinants of apoptosis include cell volume reduction, chromatin condensation and cleavage of DNA. As a result of these changes, apoptotic and pyknotic cells are generated. These changes are mainly mediated by cysteine proteases namely caspases. Caspases induce apoptosis by two different pathways including death receptor pathway and mitochondrial pathway. In the mitochondrial pathway, the release of cytochrome c from mitochondria is controlled by the balance between anti- and pro-apoptotic Bcl-2 (B-cell lymphoma 2) proteins7. Apoptosis protease-activating factor-1 (APAF-1) and cytochrome c form a complex procaspace-9. Thereafter, APAF-1 and procaspase-9 generate another complex named ‘apoptosome’. Within this complex, caspase-9 becomes activated8 (Fig. 1). In the death receptor pathway, Fas-Fas ligand (FasL) interaction can activate Fas-associated death domain (FADD) and FADD may induce pro-caspase 8. These two molecules form a death-inducing signalling complex (DISC)8. In the following step, both caspases-8 and -9 could induce caspase-3 production9.
Fig. 1.
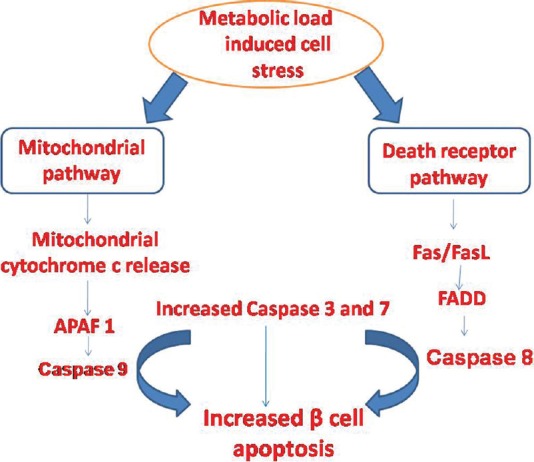
Mechanisms of apoptosis. In the mitochondrial pathway, pro- and antiapoptotic Bcl-2 proteins control cytochrome c release from mitochondria. Cytochrome c binds to APAF-1, which forms a complex with procaspase-9. Thereafter, caspase-9 becomes activated. In the death receptor pathway, the binding of a ligand to its death receptor recruits an adaptor protein that in turn activates procaspase-8. FasL binds to Fas that activates FADD. FADD activates caspase-8. Caspases-8 and -9 in turn activate caspase-3. Caspase-3 plays a crucial role in the promotion of apoptotic cell death. APAF-1, apoptosis protease-activating factor 1; FADD, Fas-associated death domain; FasL, Fas ligand.
Bcl-2 family of proteins consists of three major domains. These domains have pro- and anti-apoptotic properties. As their names describe, first subgroup has anti-apoptotic properties including Bcl-2, Bcl-xL, Bcl2A1, Bcl-w and Mcl-1. The second subgroup acts as pro-apoptic and consists of Bax, Bak and Bok proteins. The last group includes Bim (Bcl 2-interacting mediator of cell death), Bid (Bcl2-interacting domain death agonist), Bad (Bcl2-interacting associated death promoter), Bik (Bcl2-interacting killer), Hrk (Harakiri), Puma (p53-upregulated modulator of apoptosis), Bmf (Bcl2-modifying factor), and Noxa (meaning damage in Latin)10.
The mechanisms of apoptosis in diabetes
Apoptosis of islet beta cells occurs in immune-mediated T1DM11. β-cell apoptosis was found to be the final step in the pathogenesis of T1DM12. Grunnet et al13 suggested the mitochondrial pathway to be the prominent one in the pathogenesis of cytokine-induced β-cell apoptosis when compared with death cell receptor-mediated apoptosis. Others suggested that soluble or membrane bound death starting molecules might be responsible for the main pathway of β apoptosis in T1DM. Interleukin (IL) 1-β, tumour necrosis factor (TNF)-α, interferon-γ (IFN-γ) and Fas ligand (FasL) are the mostly encountered molecules that are responsible in the death cell receptor-mediated β-cell apoptosis12,14. Further, IFN-γ-induced signal transducer and activator of transcription 1 (STAT1) modulation via a gene named ubiquitin-specific proteases 18 has been found to be closely associated with the apoptosis of beta cells15. In this pathway of apoptosis when IFN-γ is bound to cell membrane, tyrosine phosphorylation activity of STAT-1 and apoptosis occur. Thus, STAT1 knockout mice were found to be resistant to develop hyperglycaemia and diabetes in streptozotocin-induced T1DM and non-obese diabetic model of mice12. However, this pathway is not the only one that plays a role in the pathogenesis of β-cell apoptosis-associated T1DM. It has been demonstrated that combined effects of IFN-γ with IL1-β via Hrk and PUMA (which are the members of BH3 domain only protein) activation might be the alternative pathways of β-cell apoptosis in T1DM16,17. Gurzov et al16 showed that Hrk inhibited Bcl-xL which was an antiapoptotic protein and therefore, blocked apoptosis.
In contrast to T1DM, the information regarding the exact role of β-cell apoptosis in the pathogenesis of T2DM is scant. In the early phase of T2DM, insulin resistance rather than insulin deficiency occurs. Pancreatic beta cell hyperplasia and hyperinsulinaemia develop secondary to insulin resistance. In the following years, relative insulin deficiency secondary to increased insulin demand will establish and eventually overt diabetes will develop. Experimental as well as clinical studies demonstrated that β-cell apoptosis and dedifferentiation might be the responsible mechanisms of relative insulin deficiency in T2DM12,18,19.
The question is what are the main triggers that activate pancreatic β-cell apoptosis in the T2DM? Besides the inflammatory cytokines, a couple of molecules including long-chain free fatty acids (FFAs) and their end products including ceramide, triacylglycerol and diacylglycerol, islet amyloid protein and advanced glycation end products are identified. Among these, FFAs via c-Jun N-terminal kinase (JNK) was found to play an important role in the pathogenesis of β-cell apoptosis also named lipoapoptosis20,21. Besides FFAs, their metabolites such as ceramide, triacylglycerol and diacylglycerol might contribute to lipoapoptosis in T2DM22.
Autophagy
Autophagy is a cellular degradation process which involves the fusion of autophagosomes and lysosomes23. This tightly regulated lysosomal pathway is crucial for homeostasis, development and survival of cell24. With the discovery of autophagy-related genes (ATG) autophagy as a programmed cell death became popular in the 1990s25. In a single-cell organism, autophagy can be activated if no food is available to digest. However, in humans, this process is more complicated to understand because autophagy takes place in both cell survival and death. Autophagy may have a central role in the integrated pathways including apoptosis and mammalian target of rapamycin (mTOR) signalling which are also cornerstones of cell homeostasis6,26. In addition, eukaryotic cells must adapt to various deleterious effects of external stimuli including ultraviolet light, microbial pathogens, starvation and fluctuations of conditions such as temperature, ion concentration, pH, cytokines and hormones26. Eukaryotic cells can fight with sublethal stress and undergo rapid changes to protect themselves against deleterious attacks successfully. In this stress-induced adaptation, autophagy plays an important role by eliminating damaged or harmful components through mechanism including lysosome-associated digestion27. Hence, exaggerated or exiguous autophagy can be deleterious to cells.
Autophagy is a physiological cascade that ends with a bulk degradation of long-lived unuseful proteins, defective organelles and soluble molecules by integration of lysosomes and double-membraned autophagosomes24. This process is tightly regulated and highly conserved and can be active at low levels in all cells23. Potent stimulators of autophagy include starvation, energy depletion and hypoxaemia. Autophagy is essential for recycling of essential amino acids28. A specific gene family called autophagy-related genes are related with initiation, formation and maturation of autophagosomes. Autophagosomes subsequently fuse with lysosomes for hydrolysis or degradation of enwrapped materials through the process of autophagy29 (Fig. 2).
Fig. 2.
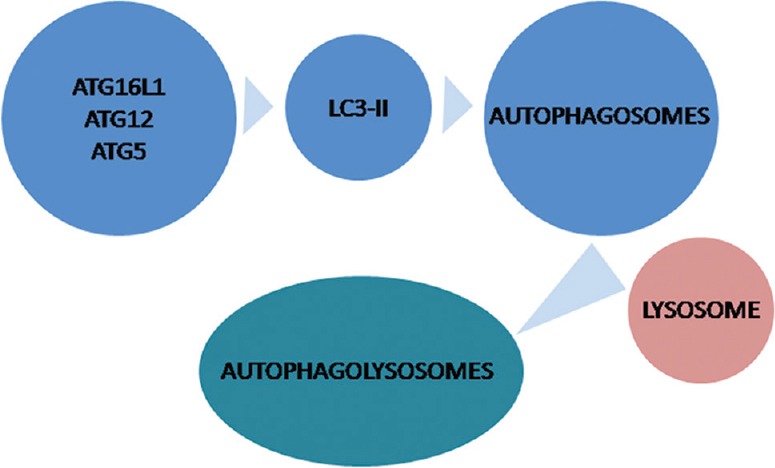
Molecular machinery of autophagy. Proteins encoded by autophagy-related genes (ATGs), ATG5, ATG12, ATG16 and LC3-II are responsible for initiation, formation, and maturation of autophagosomes, which subsequently fuse with lysosomes for hydrolysis or degradation of enwrapped materials through the process of autophagy. LC3, light chain 3.
Autophagy and apoptosis axis in physiologic conditions
This process can occur in both physiologic and pathologic conditions26. Autophagic cell death and apoptosis are not entirely distinct programmed cell death mechanisms which can merge in the pathogenic mechanisms and share many of the regulator proteins. Hence, these can act in synergy against a particular death stimulus.
S6 protein which is a substrate of p70S6K can inactivate Bcl-2-associated death promoter (BAD) by preventing phosphorylation of Ser136 and blocking cell survival30.
Autophagy and apoptosis as close friends
In this scenario, autophagy and apoptosis work together to reach the dead end of cells secondary to any deleterious insult. One of these can lead to cell death and the other helps or one of these can be activated upon the failure of the other. In the pathogenesis of T1DM, both autophagy and apoptosis were found to be activated5,12.
Wei et al31 demonstrated that JNK-1 mediated phosphorylation of Bcl-2 had a dual role in autophagy and apoptosis. They examined the relationship between Beclin-1, Bcl-2, Bax and caspase-3 activation during starvation. In this study, they showed that autophagy was activated after four hours of starvation and ultimately apoptosis was initiated after 16 h of nutrient deprivation. Wei et al31 highlighted that autophagy came first, and after a certain point of starvation when autophagy was not able to keep cells alive, apoptosis was started in such a physiological condition.
Apoptosis and autophagy as enemies
In this scenario, autophagy antagonizes apoptosis in many physiological circumstances. For instance, autophagy is activated to remove damaged organelles and mis/unfolded proteins in ER stress and thus inhibits further activation of apoptosis32,33.
The mechanisms of autophagy in diabetes
In metabolic diseases, autophagic machinery has been found to be protective in deleterious stresses of pancreatic beta cells5. Nutrient paucity, anti-ageing, control of cell growth, pathological stress factors including hypoxia, depletion of energy, mTOR inhibitors (e.g., sirolimus) might induce autophagy. In contrast, hyperglycaemic conditions might impair autophagic machinary6. Atg 7 knockout mice model has been generated to show the role of autophagy in diabetes. According to the results of these studies, hyperglycaemia and glucose intolerance occur especially secondary to impairment of insulin production which is found to be closely associated with diminished β-cell mass in the pancreas of Atg 7 knockout mice34,35. In a mouse model of db/db and Zucker diabetic mice, accumulation of autophagosomes has been shown in the pancreatic beta cells34. Fujitani et al5 showed similar results that reduced insulin secretion was closely related with pancreatic beta cell degeneration and impaired glucose in autophagy-deficient mice. The results of this study have demonstrated that degradation and reuse of the cellular organelles and proteins are essential for the function of pancreas in normal physiologic conditions. However, if autophagy persists, detrimental effects to β-cells occur. The question is which organelles and mechanisms organize the two face effect of autophagy and apoptosis in the cell? In this regard, the relationship between ER and autophagy might unfold the mystery.
Endoplasmic reticulum stress
ER is one of the most important organelles of the cell that have functions involved in the modification of the proteins primarily secreted through the cellular membranes, lipid production and calcium storage in physiologic conditions36. Folding process of proteins is essential because if the proteins are modified improperly, accumulation of these mutant proteins may induce ER stress. The ER stress can be described as a situation including abnormal protein folding, nutrient deprivation or excess, oxidative stress and chronic low-grade inflammation that affects the cell stability37. In response to increased ER stress, an appropriate adaptive change called the unfolded protein response (UPR) occurs. In eukaryotes, this adaptive response is mediated by three ER-membrane bound enzymes. These proteins are (i) protein kinase R (PKR)-like eukaryotic inhibition factor 2a kinase (PERK), (ii) activating transcription factor 6 (ATF-6), and (iii) inositol-requiring enzyme 1α (IRE-1α)38.
What is the appropriate response to ER stress?
In normal physiologic conditions, the ER-membrane bound kinases interact with an ER chaperone called Bip (also named as GRP78). In the bound form, these kinases are inactive but ready to sense a stress condition. If an unwanted condition occurs, for instance, accumulation of misfolded proteins in the ER, the bond between UPR enzymes and Bip is broken and eventually the downstream pathways of PERK, ATF-6 and IRE-1α are activated39.
PERK rapidly attenuates protein translocation; however, ATF-6 and IRE1α upregulate ER chaperone genes that further activate proper folding and increase ER-associated protein degradation (ERAD)40. In physiologic situations, these three pathways can control misfolded/unfolded protein accumulation, and if sustained ER stress occurs, PERK, ATF-6 and IRE-1α initiate apoptosis41.
Pathways of UPR
Downstream of PERK pathway: PERK organizes eukaryotic translation initiation factor-2 alpha (eIF-2α) by phosphorylation. This activation triggers the synthesis of two important ATFs (Fig. 3). One of these is CHOP (C/EBP homologous protein, also named as GADD153) and the other one is ATF-442,43. CHOP can further activate apoptotic Bim or deactivate the expression of antiapoptotic Bcl-244,45. CHOP is also associated with oxidative stress-induced apoptosis by inducing ER oxidase-1α (ERO1α) that can hyperoxidize the lumina of the ER and eventually cause cell death46.
Fig. 3.

Downstream pathways of inositol requiring enzyme 1α (IRE1α) and PKR-like eukaryotic inhibition factor 2a kinase (PERK) in endoplasmic reticulum stress. UPR- unfolded protein response; ASK1, apoptosis signal-regulating kinase 1; CHOP, C/EBP homologous protein; XBP-1, X box binding protein 1; ERO1α, endoplasmic reticulum oxidase 1alpha; eIF-2α, eukaryotic translation initiation factor-2α; JNK, janus kinase; NF-κB, nuclear factor kappa β; GADD34, growth arrest and DNA damage-inducible protein-34; Bcl2, B-cell lymphoma 2; BIM, Bcl 2-interacting mediator of cell death.
ATF-4 is found to be associated with increased CHOP, GADD34 (growth arrest and DNA damage-inducible protein-34) and TRB3 (Tribless Homolog 3)47,48,49. Among these, GADD34 blocks the effects of eIF-2α and TRB3 is a negative regulator of nuclear factor kappa β (NFkβ) and can also aggravate apoptosis via TNF and TRAIL molecules. In addition, TRB-3 can inhibit serine-threonine kinase AKT1 and, therefore, can negatively regulate cell survival50.
Downstream of IRE-1α pathway: IRE-1 is a serine/threonine kinase which also has endonuclease activity. ER stress activates IRE-1α by dissociation of Bip and as a consequence unbound form of IRE-1α binds to mis/unfolded proteins (Fig. 3). This complex triggers endoribonucleatic cleavage and encodes a transcription factor named X box binding protein 1 (XBP-1)51. XBP-1 urges the transcription of genes associated with UPR including PERK, ERAD, ATF-4 and genes regarding autophagy, inflammation and apoptosis such as JNK (Janus kinase), NF-κB and apoptosis signal-regulating kinase 1 (ASK1), respectively40,52,53. Hetz et al54 investigated signalling of UPR in double knockout mice in terms of proapoptotic Bax and Bak. They showed that ER-stressed knockout cells had deficient signalling of IRE-1α. However, Bax and Bak form a complex with a cytosolic domain of IRE-1α. This complex is found to be essential for the activation of IRE-1α. In conclusion, Bax and Bak were found to be essential in IRE-1α activation and this highlighted a physical link between apoptosis and the response to ER stress.
Downstream of ATF-6 pathway: ATF-6 is the third sensor of UPR that binds to ER membrane in its inactive form. Once activated, it is delivered to golgi apparatus and cleaved by two proteases (site 1 and 2). Cleaved ATF-6 is transferred to the nucleus to stimulate genes that are associated with UPR55.
Among these three pathways of UPR, the data regarding IRE-1α and PERK pathways are of paramount importance, and studies regarding ATF-6 pathway are only a few. Further studies exhibiting the importance of ATF-6 pathway are needed especially for the pathogenesis of DM.
ER stress in diabetes mellitus
As already known, pancreatic β-cell destruction is the main issue regarding the initiation and progression of diabetes. ER stress has been found to be closely related to β-cell dysfunction especially secondary to glucotoxicity, lipotoxicity, chronic inflammation and eventually increased oxidative stress. All of these perturbations might induce improper protein folding and thereby accumulation of these proteins could cause β-cell ER stress. The main consequence of this stress in β-cell is worsening of the quality of proinsulin folding that leads to the onset and advancement of both T1 and T2DM56.
Roles of ER stress response regarding apoptosis and autophagy in diabetes
The relation between ER stress and apoptosis in diabetes is illustrated in Fig. 4. If prolonged ER stress cannot be diminished via UPR pathways, this will exceed ER functional capacity and thereafter homeostasis of the ER cannot be re-established. Eventually, this situation will induce apoptosis-related cell death2. The PERK pathway can activate CHOP, caspase-12 and JNK signalling pathways57, and IRE-1α can trigger proapoptotic Bcl-2, Bak, Bax and induce the recruitment of tumour necrosis factor receptor-associated factor 2 and ASK1 to the cytoplasmic side of the ER membrane to activate apoptosis58. In contrast, persistent ER stress can reduce ATF6 and IRE-1α activity and induce PERK pathway of UPR. Hetz et al54 demonstrated that diminution of IRE-1α RNA ribonuclease might inhibit its protective activity via activating JNK and XBP1 or proapoptotic mediators. Hence, IRE-1α-related degradation of mRNA in ER might be an important determinant of cell survival or death59. In this context, IRE-1α pathway of UPR might be the main moderator of cell fate in ER stress.
Fig. 4.

Relationship between endoplasmic reticulum (ER) stress, autophagy and apoptosis. In diabetes mellitus, various effectors including hyperglycaemia, increased free fatty acids (FFAs), islet amyloid polypeptide (IAPP), chronic low-grade ongoing inflammation and oxidative stress can induce protein misfolding, mammalian target of rapamycin 1 (mTORC1) and decreased lysosomal degradation process. Thereafter, protein (for instance, proinsulin) misfolding induces ER stress and eventually β-cell death occurs via apoptosis. Activation of mTORC1 inhibits autophagy. mTORC1 inhibitors such as rapamycin might stimulate autophagy and prevent ER-stress activated β-cell apoptosis. Eventually, there is a decrease in proinsulin and insulin biosynthesis. PERK, PKR-like eukaryotic inhibition factor 2a kinase; inositol requiring enzyme 1alpha (IRE1α); ATF6, activating transcription factor 6.
Impaired glucose metabolism and insulin resistance were found to be closely related with UPR and ER stress60. This association is partially related to the increased apoptosis that involves the three UPR pathways, especially in diabetes57. Apoptotic degeneration of pancreatic β-cells may cause insulin deprivation leading to T1DM. Experimental data showed that PERK deficient beta cells were very sensitive to ER stress-induced apoptosis. Harding et al61 demonstrated that perk-/- mice had very high levels of plasma glucose, especially secondary to increased beta cell apoptosis early in the life. Back et al62 determined the importance of the absence of eIF2α phosphorylation in pancreatic β-cells. They showed that silencing eIF2α caused severe diabetes secondary to various deleterious cellular effects such as increased dysregulated pro-insulin translation, impaired intracellular trafficking of ER proteins, increased oxidative stress, diminished stress response and apoptosis.
Besides the experimental data, in clinical practice, Wolcott–Rallison syndrome is a disease associated with autosomal recessive mutations of PERK. The phenotypic features of this syndrome include infantile diabetes secondary to pancreatic β-cell apoptosis, skeletal abnormalities, chronic kidney disease and proteinuria in the childhood due to podocytopathy in the childhood63,64.
Relationship between ER stress and autophagy in diabetes
The relation between ER stress and autophagy in diabetes is illustrated in Fig. 4. Various factors including gluco- and lipotoxicity increased levels of islet amyloid polypeptide (IAPP) and chronic low-grade inflammation alter insulin modification in β-cell ER and eventually misfolded protein accumulation might induce ER stress2. In addition, misfolded proinsulin might per se induce autophagic pathways.
Under miscellaneous situations including ER stress, autophagy plays an important role in the elimination of mis/unfolded proteins26. Hence, autophagy could be an adaptive mechanism against increased ER stress to eliminate misfolded proteins. For instance, nascent proinsulin could be eliminated through ERAD and subsequent proteasomal degradation procedures. However, ER stress-induced autophagy might be an alternative degradation process of these mis/unfolded proteins whether ATF-6 and IRE-1α were appropriately working or not65. Furthermore, to identify the exact role of misfolded proinsulin on autophagy in diabetes, a mouse model named Akita can be used. These mice have a mutation in one proinsulin allele that results in protein misfolding. In this model of diabetes, mutant proinsulin accumulates in the β-cell ER and induces ER stress and subsequently decreased levels of insulin are seen66. In clinical practice, mutant INS gene-induced diabetes of youth syndrome is a rare form of congenital diabetes that shows a similar mutation seen in Akita mice67.
In the pathogenesis of DM, gluco- and lipotoxicity, IAPP, chronic low-grade ongoing inflammation induce proinsulin misfolding, mTORC1 and decrease lysosomal degradation process. As seen in Akita mice model, proinsulin misfolding induces ER stress and eventually β-cell death occurs via apoptosis. At this point, ER stress can activate autophagy. This might rescue β-cell from death. Activation of mTORC1 inhibits autophagy. As predicted, mTORC1 inhibitors such as rapamycin and Torin1 might stimulate autophagy and prevent ER stress-induced β cell destruction and apoptosis.
In sequestosome1 (SQSTM1/p62) deficient mice, the role of autophagy has been demonstrated68. In this context, Geetha et al68 showed that SQSTM1/p62deficient mice had severe hyperglycaemia and consequently diabetes. Bachar-Wikstrom et al69 showed that SQSTM1/p62 might be diminished secondary to increased autophagy via mTORC1 inhibition while these changes could augment autophagosomes and autolysosomes. They also showed that the baseline expression of LC3-II was very low and mTORC1 inhibitors could not stimulate LC3-II expression in beta cells. According to the results of this study, LC3-II accumulation was prevented by increased autophagosome formation which was induced with mTORC1 inhibitors. In addition, the appearance of autolysosomes was 2-fold higher than autophagosomes in rapamycin-treated mouse model of Akita β-cells, indicating that rapamycin had a dominant effect on lysosome-autophagosome fusion, rather than the generation of autophagosomes. In accordance with these results, treatment of diabetic Akita mice with rapamycin improved serum glucose levels and increased insulin secretion and decreased β-cell apoptosis. Hence, activation of autophagy might be beneficial in the novel treatment of DM6.
Conclusion
Cell survival and death are complicated processes that involve apoptosis and autophagy pathways. ER may be an intersection of the two pathways. However, there are many points to be highlighted in this puzzle. How does misfolded proinsulin exit from ER and get degraded? Is there any role of mitochondria regarding the association between ER stress and mitophagy? These are some of the important questions that should be addressed. It is anticipated that ongoing research may throw light on these issues. Eventually, the developments and the gained knowledge will improve our therapeutic approaches in both diabetes and diabetes-related disorders.
Footnotes
Conflicts of Interest: None.
References
- 1.Calcutt NA, Cooper ME, Kern TS, Schmidt AM. Therapies for hyperglycaemia-induced diabetic complications: from animal models to clinical trials. Nat Rev Drug Discov. 2009;8:417–29. doi: 10.1038/nrd2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Su J, Zhou L, Kong X, Yang X, Xiang X, Zhang Y, et al. Endoplasmic reticulum is at the crossroads of autophagy, inflammation, and apoptosis signaling pathways and participates in the pathogenesis of diabetes mellitus. J Diabetes Res 2013. 2013:193461. doi: 10.1155/2013/193461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Galluzzi L, Maiuri MC, Vitale I, Zischka H, Castedo M, Zitvogel L, et al. Cell death modalities: classification and pathophysiological implications. Cell Death Differ. 2007;14:1237–43. doi: 10.1038/sj.cdd.4402148. [DOI] [PubMed] [Google Scholar]
- 4.Baehrecke EH. Autophagy: dual roles in life and death? Nat Rev Mol Cell Biol. 2005;6:505–10. doi: 10.1038/nrm1666. [DOI] [PubMed] [Google Scholar]
- 5.Fujitani Y, Kawamori R, Watada H. The role of autophagy in pancreatic beta-cell and diabetes. Autophagy. 2009;5:280–2. doi: 10.4161/auto.5.2.7656. [DOI] [PubMed] [Google Scholar]
- 6.Tanaka Y, Kume S, Kitada M, Kanasaki K, Uzu T, Maegawa H, et al. Autophagy as a therapeutic target in diabetic nephropathy. Exp Diabetes Res 2012. 2012:628978. doi: 10.1155/2012/628978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Adams JM, Cory S. The Bcl-2 protein family: arbiters of cell survival. Science. 1998;281:1322–6. doi: 10.1126/science.281.5381.1322. [DOI] [PubMed] [Google Scholar]
- 8.Shi Y. Mechanisms of caspase activation and inhibition during apoptosis. Mol Cell. 2002;9:459–70. doi: 10.1016/s1097-2765(02)00482-3. [DOI] [PubMed] [Google Scholar]
- 9.Edelstein CL. Mammalian target of rapamycin and caspase inhibitors in polycystic kidney disease. Clin J Am Soc Nephrol. 2008;3:1219–26. doi: 10.2215/CJN.05611207. [DOI] [PubMed] [Google Scholar]
- 10.Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer. 2002;2:647–56. doi: 10.1038/nrc883. [DOI] [PubMed] [Google Scholar]
- 11.Tanaka S, Aida K, Nishida Y, Kobayashi T. Pathophysiological mechanisms involving aggressive islet cell destruction in fulminant type 1 diabetes. Endocr J. 2013;60:837–45. doi: 10.1507/endocrj.ej13-0222. [DOI] [PubMed] [Google Scholar]
- 12.Quan W, Jo EK, Lee MS. Role of pancreatic ß-cell death and inflammation in diabetes. Diabetes Obes Metab. 2013;15(Suppl 3):141–51. doi: 10.1111/dom.12153. [DOI] [PubMed] [Google Scholar]
- 13.Grunnet LG, Aikin R, Tonnesen MF, Paraskevas S, Blaabjerg L, Størling J, et al. Proinflammatory cytokines activate the intrinsic apoptotic pathway in beta-cells. Diabetes. 2009;58:1807–15. doi: 10.2337/db08-0178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cnop M, Welsh N, Jonas JC, Jörns A, Lenzen S, Eizirik DL. Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes. 2005;54(Suppl 2):S97–107. doi: 10.2337/diabetes.54.suppl_2.s97. [DOI] [PubMed] [Google Scholar]
- 15.Santin I, Moore F, Grieco FA, Marchetti P, Brancolini C, Eizirik DL. USP18 is a key regulator of the interferon-driven gene network modulating pancreatic beta cell inflammation and apoptosis. Cell Death Dis. 2012;3:e419. doi: 10.1038/cddis.2012.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gurzov EN, Germano CM, Cunha DA, Ortis F, Vanderwinden JM, Marchetti P, et al. p53 upregulated modulator of apoptosis (PUMA) activation contributes to pancreatic beta-cell apoptosis induced by proinflammatory cytokines and endoplasmic reticulum stress. J Biol Chem. 2010;285:19910–20. doi: 10.1074/jbc.M110.122374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gurzov EN, Ortis F, Cunha DA, Gosset G, Li M, Cardozo AK, et al. Signaling by IL-1beta+IFN-gamma and ER stress converge on DP5/Hrk activation: a novel mechanism for pancreatic beta-cell apoptosis. Cell Death Differ. 2009;16:1539–50. doi: 10.1038/cdd.2009.99. [DOI] [PubMed] [Google Scholar]
- 18.Yoon KH, Ko SH, Cho JH, Lee JM, Ahn YB, Song KH, et al. Selective beta-cell loss and alpha-cell expansion in patients with type 2 diabetes mellitus in Korea. J Clin Endocrinol Metab. 2003;88:2300–8. doi: 10.1210/jc.2002-020735. [DOI] [PubMed] [Google Scholar]
- 19.Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52:102–10. doi: 10.2337/diabetes.52.1.102. [DOI] [PubMed] [Google Scholar]
- 20.Maedler K, Oberholzer J, Bucher P, Spinas GA, Donath MY. Monounsaturated fatty acids prevent the deleterious effects of palmitate and high glucose on human pancreatic beta-cell turnover and function. Diabetes. 2003;52:726–33. doi: 10.2337/diabetes.52.3.726. [DOI] [PubMed] [Google Scholar]
- 21.Holzer RG, Park EJ, Li N, Tran H, Chen M, Choi C, et al. Saturated fatty acids induce c-Src clustering within membrane subdomains, leading to JNK activation. Cell. 2011;147:173–84. doi: 10.1016/j.cell.2011.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koves TR, Ussher JR, Noland RC, Slentz D, Mosedale M, Ilkayeva O, et al. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 2008;7:45–56. doi: 10.1016/j.cmet.2007.10.013. [DOI] [PubMed] [Google Scholar]
- 23.Yang C, Kaushal V, Shah SV, Kaushal GP. Autophagy is associated with apoptosis in cisplatin injury to renal tubular epithelial cells. Am J Physiol Renal Physiol. 2008;294:F777–87. doi: 10.1152/ajprenal.00590.2007. [DOI] [PubMed] [Google Scholar]
- 24.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–77. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 26.Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40:280–93. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;4 3:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–26. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Levine B, Yuan J. Autophagy in cell death: an innocent convict? J Clin Invest. 2005;115:2679–88. doi: 10.1172/JCI26390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harada H, Andersen JS, Mann M, Terada N, Korsmeyer SJ. p70S6 kinase signals cell survival as well as growth, inactivating the pro-apoptotic molecule BAD. Proc Natl Acad Sci U S A. 2001;98:9666–70. doi: 10.1073/pnas.171301998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wei Y, Sinha S, Levine B. Dual role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy. 2008;4:949–51. doi: 10.4161/auto.6788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bernales S, McDonald KL, Walter P. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol. 2006;4:e423. doi: 10.1371/journal.pbio.0040423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ding WX, Ni HM, Gao W, Hou YF, Melan MA, Chen X, et al. Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival. J Biol Chem. 2007;282:4702–10. doi: 10.1074/jbc.M609267200. [DOI] [PubMed] [Google Scholar]
- 34.Ebato C, Uchida T, Arakawa M, Komatsu M, Ueno T, Komiya K, et al. Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab. 2008;8:325–32. doi: 10.1016/j.cmet.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 35.Jung HS, Chung KW, Won Kim J, Kim J, Komatsu M, Tanaka K, et al. Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metab. 2008;8:318–24. doi: 10.1016/j.cmet.2008.08.013. [DOI] [PubMed] [Google Scholar]
- 36.Laybutt DR, Preston AM, Akerfeldt MC, Kench JG, Busch AK, Biankin AV, et al. Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia. 2007;50:752–63. doi: 10.1007/s00125-006-0590-z. [DOI] [PubMed] [Google Scholar]
- 37.Rao RV, Ellerby HM, Bredesen DE. Coupling endoplasmic reticulum stress to the cell death program. Cell Death Differ. 2004;11:372–80. doi: 10.1038/sj.cdd.4401378. [DOI] [PubMed] [Google Scholar]
- 38.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–29. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 39.Beriault DR, Werstuck GH. The role of glucosamine-induced ER stress in diabetic atherogenesis. Exp Diabetes Res 2012. 2012:187018. doi: 10.1155/2012/187018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim R, Emi M, Tanabe K, Murakami S. Role of the unfolded protein response in cell death. Apoptosis. 2006;11:5–13. doi: 10.1007/s10495-005-3088-0. [DOI] [PubMed] [Google Scholar]
- 41.Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006;7:880–5. doi: 10.1038/sj.embor.7400779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11:381–9. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- 43.Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell. 2000;5:897–904. doi: 10.1016/s1097-2765(00)80330-5. [DOI] [PubMed] [Google Scholar]
- 44.Puthalakath H, O’Reilly LA, Gunn P, Lee L, Kelly PN, Huntington ND, et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell. 2007;129:1337–49. doi: 10.1016/j.cell.2007.04.027. [DOI] [PubMed] [Google Scholar]
- 45.McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol. 2001;21:1249–59. doi: 10.1128/MCB.21.4.1249-1259.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, et al. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004;18:3066–77. doi: 10.1101/gad.1250704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ohoka N, Yoshii S, Hattori T, Onozaki K, Hayashi H. TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J. 2005;24:1243–55. doi: 10.1038/sj.emboj.7600596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ma Y, Hendershot LM. Delineation of a negative feedback regulatory loop that controls protein translation during endoplasmic reticulum stress. J Biol Chem. 2003;278:34864–73. doi: 10.1074/jbc.M301107200. [DOI] [PubMed] [Google Scholar]
- 49.Ma Y, Brewer JW, Diehl JA, Hendershot LM. Two distinct stress signaling pathways converge upon the CHOP promoter during the mammalian unfolded protein response. J Mol Biol. 2002;318:1351–65. doi: 10.1016/s0022-2836(02)00234-6. [DOI] [PubMed] [Google Scholar]
- 50.Du K, Herzig S, Kulkarni RN, Montminy M. TRB3: a tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science. 2003;300:1574–7. doi: 10.1126/science.1079817. [DOI] [PubMed] [Google Scholar]
- 51.Ron D, Hubbard SR. How IRE1 reacts to ER stress. Cell. 2008;132:24–6. doi: 10.1016/j.cell.2007.12.017. [DOI] [PubMed] [Google Scholar]
- 52.Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol. 2006;26:9220–31. doi: 10.1128/MCB.01453-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–6. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 54.Hetz C, Bernasconi P, Fisher J, Lee AH, Bassik MC, Antonsson B, et al. Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1alpha. Science. 2006;312:572–6. doi: 10.1126/science.1123480. [DOI] [PubMed] [Google Scholar]
- 55.Nadanaka S, Okada T, Yoshida H, Mori K. Role of disulfide bridges formed in the luminal domain of ATF6 in sensing endoplasmic reticulum stress. Mol Cell Biol. 2007;27:1027–43. doi: 10.1128/MCB.00408-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bachar-Wikstrom E, Wikstrom JD, Kaiser N, Cerasi E, Leibowitz G. Improvement of ER stress-induced diabetes by stimulating autophagy. Autophagy. 2013;9:626–8. doi: 10.4161/auto.23642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gupta S, Read DE, Deepti A, Cawley K, Gupta A, Oommen D, et al. Perk-dependent repression of miR-106b-25 cluster is required for ER stress-induced apoptosis. Cell Death Dis. 2012;3:e333. doi: 10.1038/cddis.2012.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kaneko M, Niinuma Y, Nomura Y. Activation signal of nuclear factor-kappa B in response to endoplasmic reticulum stress is transduced via IRE1 and tumor necrosis factor receptor-associated factor 2. Biol Pharm Bull. 2003;26:931–5. doi: 10.1248/bpb.26.931. [DOI] [PubMed] [Google Scholar]
- 59.Han D, Lerner AG, Vande Walle L, Upton JP, Xu W, Hagen A, et al. IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell. 2009;138:562–75. doi: 10.1016/j.cell.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–61. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 61.Harding HP, Zeng H, Zhang Y, Jungries R, Chung P, Plesken H, et al. Diabetes mellitus and exocrine pancreatic dysfunction in perk-/- mice reveals a role for translational control in secretory cell survival. Mol Cell. 2001;7:1153–63. doi: 10.1016/s1097-2765(01)00264-7. [DOI] [PubMed] [Google Scholar]
- 62.Back SH, Scheuner D, Han J, Song B, Ribick M, Wang J, et al. Translation attenuation through eIF2alpha phosphorylation prevents oxidative stress and maintains the differentiated state in beta cells. Cell Metab. 2009;10:13–26. doi: 10.1016/j.cmet.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang P, McGrath B, Li S, Frank A, Zambito F, Reinert J, et al. The PERK eukaryotic initiation factor 2 alpha kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Mol Cell Biol. 2002;22:3864–74. doi: 10.1128/MCB.22.11.3864-3874.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Iyer S, Korada M, Rainbow L, Kirk J, Brown RM, Shaw N, et al. Wolcott-Rallison syndrome: a clinical and genetic study of three children, novel mutation in EIF2AK3 and a review of the literature. Acta Paediatr. 2004;93:1195–201. [PubMed] [Google Scholar]
- 65.Yin JJ, Li YB, Wang Y, Liu GD, Wang J, Zhu XO, et al. The role of autophagy in endoplasmic reticulum stress-induced pancreatic ß cell death. Autophagy. 2012;8:158–64. doi: 10.4161/auto.8.2.18807. [DOI] [PubMed] [Google Scholar]
- 66.Nozaki Ji, Kubota H, Yoshida H, Naitoh M, Goji J, Yoshinaga T, et al. The endoplasmic reticulum stress response is stimulated through the continuous activation of transcription factors ATF6 and XBP1 in Ins2+/Akita pancreatic beta cells. Genes Cells. 2004;9:261–70. doi: 10.1111/j.1356-9597.2004.00721.x. [DOI] [PubMed] [Google Scholar]
- 67.Liu M, Lara-Lemus R, Shan SO, Wright J, Haataja L, Barbetti F, et al. Impaired cleavage of preproinsulin signal peptide linked to autosomal-dominant diabetes. Diabetes. 2012;61:828–37. doi: 10.2337/db11-0878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Geetha T, Zheng C, Vishwaprakash N, Broderick TL, Babu JR. Sequestosome 1/p62, a scaffolding protein, is a newly identified partner of IRS-1 protein. J Biol Chem. 2012;287:29672–8. doi: 10.1074/jbc.M111.322404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bachar-Wikstrom E, Wikstrom JD, Ariav Y, Tirosh B, Kaiser N, Cerasi E, et al. Stimulation of autophagy improves endoplasmic reticulum stress-induced diabetes. Diabetes. 2013;62:1227–37. doi: 10.2337/db12-1474. [DOI] [PMC free article] [PubMed] [Google Scholar]