

Abstract
Background: Genetic and environmental risk factors are assumed to contribute to the susceptibility to cervical artery dissection (CeAD). To explore the role of genetic imbalance in the etiology of CeAD, copy number variants (CNVs) were identified in high-density microarrays samples from the multicenter CADISP (Cervical Artery Dissection and Ischemic Stroke Patients) study and from control subjects from the CADISP study and the German PopGen biobank. Microarray data from 833 CeAD patients and 2040 control subjects (565 subjects with ischemic stroke due to causes different from CeAD and 1475 disease-free individuals) were analyzed. Rare genic CNVs were equally frequent in CeAD-patients (16.4%; n=137) and in control subjects (17.0%; n=346) but differed with respect to their genetic content. Compared to control subjects, CNVs from CeAD patients were enriched for genes associated with muscle organ development and cell differentiation, which suggests a possible association with arterial development. CNVs affecting cardiovascular system development were more common in CeAD patients than in control subjects (p=0.003; odds ratio (OR) =2.5; 95% confidence interval (95% CI) =1.4-4.5) and more common in patients with a familial history of CeAD than in those with sporadic CeAD (p=0.036; OR=11.2; 95% CI=1.2-107).
Conclusion: The findings suggest that rare genetic imbalance affecting cardiovascular system development may contribute to the risk of CeAD. Validation of these findings in independent study populations is warranted.
Keywords: Copy number variation, Cervical artery dissection, Rare genetic variation, Cardiovascular system development
INTRODUCTION
Copy number variations (CNVs) are structural genetic variants (deletions or duplications), leading to genetic imbalance. CNVs are widespread in the human genome and play an important role in multiple phenotypes [1-3] including vascular diseases [4, 5]. The contribution of structural genetic variation to the risk of ischemic stroke (IS) has not been explored thoroughly: In a CNV study of 263 patients with IS of different causes [6], no common genomic structural variant was unequivocally linked to IS. A pilot study of 70 patients with cervical artery dissection (CeAD) [7] identified several rare CNVs affecting genes involved in arterial development or in connective tissue disorders. CeAD is a bleeding within the arterial wall of the carotid artery or the vertebral artery. CeAD is a rare cause of IS in old patients, but it causes IS in about 10% of patients <50 years [8, 9]. A recent genome-wide association study identified a common variant in the PHACTR1 gene (encoding the phosphatase and actin regulator 1) that increased the risk for CeAD [10]). In the current study we explore rare CNVs in a large set of SNP-microarray data that genome-wide association study [10]. Our findings suggest that rare structural variation associated with different biological functions including muscle organ development, cell differentiation and cardiovascular system development contribute to the risk of CeAD.
Material and methods
Patients and Controls Subjects
A total of 983 patients with a diagnosis of CeAD based upon predefined criteria were included in the CADISP (Cervical Artery Dissection and Ischemic Stroke Patients) study between 2004 and 2009 [11]. After the exclusion of CeAD patients with (i) confirmed diagnosis of vascular Ehlers-Danlos syndrome (vEDS), (ii) non-European origin, or (iii) insufficient DNA quality, 883 patients were successfully genotyped using an Illumina Human 610-Quad or Human 660W-Quad Bead Chip at the Centre National de Génotypage in Evry, France, and passed genotyping quality control.
As control subjects, 658 patients with ischemic stroke attributable to a cause other than CeAD (non-CEAD-IS patients) and 269 Finnish healthy subjects from the CADISP study [11, 12] were included in the study. After the exclusion of individuals with (i) non-European origin or (ii) insufficient DNA quality, 585 control patients and 237 healthy controls passed genotyping quality control. Ancestry of all CADISP subjects (patients as well as controls) was inferred from places of birth of both parents as well as of self-reported information during the interview upon recruitment. Principal component analysis and stringent Hardy-Weinberg Equilibrium filtering during quality control preceding the genome-wide association study [10] enabled identification of population stratification. As additional controls, we used 1262 high-quality Affymetrix 6.0 datasets from Caucasian subjects in the German PopGen biobank [13].
Baseline Characteristics of CeAD Patients
The following clinical variables were derived from the CADISP-database: age, sex, vascular risk factors (as defined before [14]), site of dissection (internal carotid artery, vertebral artery or both), presence or absence of ischemic stroke, familial history of CeAD, defined as self-reported history of CeAD in first-degree relatives (parents, siblings or children). Since the CeAD study sample comprised two families with two affected relatives each, analysis of the familial history of CeAD was also performed by including only one affected individual from each family.
CNV Discovery and Prioritization
CNVs were identified with the PennCNV software (version May 2010) [15]. CNVs comprising >10 SNPs (Illumina data) or >20 SNPs (Affymetrix data) were analyzed further. Datasets with outlier numbers of CNV calls (i.e., >125 or < 15 CNVs) or with a variance >0.2 of the normalized LRR values taken over all autosomal SNPs were excluded from the analysis [16]. The final study sample comprised microarray data from 833 CeAD patients, 565 control patients, and 215 Finnish and 1260 German healthy subjects.
Published CNV data were used to identify common CNVs in the study subjects. CNVs were considered as rare if less than three copies were found among the 3703 disease-free subjects in two published CNV databases [17, 18]. CNVs overlapping > 50% of their physical length were considered as similar.
CNV Validation and Mapping
Signal intensity and B-allele frequencies of all rare CNVs were visualized (http://noise-free-cnv.sourceforge.net/contact.php) to identify putative false-positive calls and to inspect and map the breakpoints. CNV validation was performed in two-steps as described before [7, 16], including visual inspection of each CNV finding in noise-reduced datasets, followed by molecular analysis of a few selected CNVs. For molecular validation by quantitative PCR [19], we randomly selected 10 rare CNVs from the CeAD patients. For each CNV, PCR was carried out in six replicas with DNA of the CNV carrier and three control subjects in 96-well plates with a Bio-Rad SYBR-Green PCR system following standard procedures. Five additional CNVs from CeAD patients were validated by identification and sequence analysis of the joining fragment [20].
The first and the last SNPs of each CNV were mapped onto the human genome (GRCh38) (http://www.ensembl.org/Homo_sapiens). CNVs were classified as genic if they comprised the deletion of at least one coding exon or a duplication that either encompassed an entire coding region or internal exons [21]. Duplications with both breakpoints within genes of similar chromosomal orientation were also considered as genic, because they might encode a fusion protein [22].
CNV findings located in heterochromatic pericentromic regions, in the short arms of the acrocentric chromosomes (numbers 13,14,15,21 and 22), in the subtelomeric regions of chromosomes 9q and 14q, in the MHC region of chromosome 6p and in the Y-chromosome were a-priori exluded from the current analysis.
Functional Enrichment Analysis / Case Control Studies
All genes identified in all rare CNVs from the CeAD patients sample were analyzed for enrichment of predefined gene sets with the Set Distiller software package (http://www.genecards.org/index.php?path=/GeneDecks). Top enrichments for the category “GeneOntology biological process” were analyzed. P-values for enrichment were Bonferroni-adjusted for multiple testing of the total set of tested biological processes (n=7780) by default.
For each analyzed enriched gene set, all rare CNVs affecting one or more genes belonging to the gene set were identified in both study groups (CeAD patients and control subjects). The prevalence of CNVs affecting a selected gene set was compared between patients and control subjects for 10 top findings. Nominal p-values were multiplied by 10 to adjust for multiple testing.
Among the CeAD-patients, we compared carriers of CNVs affecting enriched pathways with non-carriers with regard to their clinical characteristics and risk factors. Also patients with and without a family history of CeAD were compared.
Statistical Analysis
Categorical variables were tested by a χ2 statistic unless expected cell counts were smaller than five, in which case Fisher's exact test was used. Students’ T-test was used for the analysis of continuous variables with a normal distribution (age, body mass index). The SPSS 19.0 statistics software package was used for statistical analysis. Comparisons were adjusted for age and sex in a logistic regression model.
Ethics
The study protocol was approved by relevant local authorities in all participating centers and complied with all national regulations concerning ethics committee approval and informed consent procedures.
RESULTS
The final study sample comprised 2873 microarray datasets (Fig. 1 : 833 patients and 2040 controls), Genic CNVs with low frequency (<0.001) in two published high-quality datasets were prioritized for further analysis and mapped onto the human genome (genome assembly: GRCh37) to identify coding sequences covered by the CNV. In the CeAD group, 147 rare genic CNVs were identified in 137 carriers (16.4%), affecting a total of 433 annotated protein coding genes. In the control subjects we identified 346 (17.0%) carriers of rare genic CNVs, affecting a total of 742 annotated protein-coding genes. All CNV findings were validated one by one by visual inspection of the datasets with the noise-free-CNV software package. This initial validation step included verification of the mere presence of the CNV, of the positions of its start and end SNPs, and of the copy number. Moreover, visual inspection permitted the identification of split calls or somatic mosaicism and gives an immediate impression of the quality of the microarray sample. Additional validation by independent molecular methods was successfully performed in a small selection of the CNV findings supplementary files, (2 and 3).
Fig. (1).
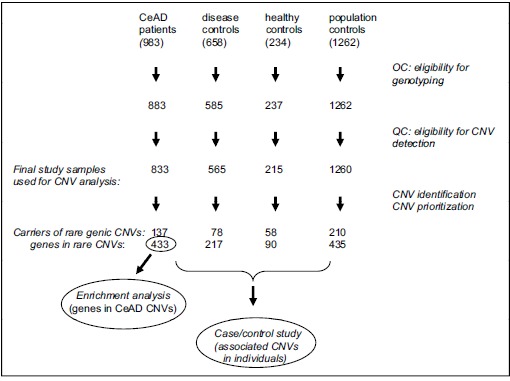
Flow chart of study design.
Genes that were covered by rare CNVs from the CeAD patients were analyzed with the GeneDecks Set Distiller software to search for significant enrichment. Top-10 enrichments for “biological process (GeneOntology)” are presented in (Table 1 ). A case/control study of rare CNVs was then performed to test for disease association. Rare genic CNVs from CeAD patients were significantly more likely to affect genes associated with muscle organ development or with cell differentiation than rare genic CNVs from control subjects. The control sample includes a large population of disease-free German subjects, and two samples that were enrolled by the CADISP study (healthy subjects from Finland and age- and sex-matched IS patients from the main recruiting centers). These three controls were merged into a single large control group (n=2040) and compared with the patients group (n=833). With regard to the CNV-findings associated with the predefined GO groups (Table 1 ), no significant heterogeneity was found across the three control samples (data not shown).
Table 1.
Functional enrichment analysis (FEA) and specificity analysis of rare genic CNVs in CeAD patients. Left hand columns show top findings of GeneDecks enrichment analysis for genes covering rare CNVs of CeAD patients with adjusted p-values. The frequency of rare CNVs at genes belonging to the enriched gene sets was compared between CeAD patients and control subjects (non-CeAD IS disease controls plus population controls). Since the findings suggested CeAD to be associated with cardiovascular system development, this candidate gene set was subsequently tested formally for disease association (see table 2). P-values of FEA were Bonferroni-corrected for multiple testing by default. Crude P-values of Case/Control comparisons were multiplied by 10 to adjust for multiple testing.
| Functional Enrichment Analysis | Case/Control Comparison | |||
|---|---|---|---|---|
| Identified gene sets in GeneOntology – biological processes | adjusted p-value | CeAD (n=833) | Controls (n=2040) | adjusted p-value |
| Small molecule metabolic process | 1.23e-10 | 29 | 41 | 0.234 |
| Muscle organ development | 3.57e-07 | 9 | 3 | 0.013 |
| Multicellular organismal development | 4.91e-07 | 18 | 16 | 0.036 |
| Xenobiotic catabolic process | 9.17e-07 | 1 | 0 | 1.000 |
| Regulation of transcription, DNA-templated | 1.44e-06 | 18 | 41 | 1.000 |
| Cell differentiation | 1.61e-05 | 18 | 13 | 0.010 |
| Signal transduction | 1.93e-05 | 19 | 28 | 1.000 |
| Proteolysis | 3.12e-05 | 8 | 5 | 0.145 |
| Cell adhesion | 3.21e-05 | 13 | 12 | 0.147 |
| Cell surface receptor signaling pathways | 3.59e-05 | 8 | 5 | 0.145 |
| Apoptotic process | 3.86e-05 | 12 | 14 | 0.791 |
Next we tested for genetic association between CeAD and genes involved in cardiovascular system development, a biological process possibly underlying the observed associations with muscle development and cell differentiation. Rare CNVs affecting genes that play a role in cardiovascular system development were identified in 22 (2.6%) CeAD patients and in 22 (1.1%) control subjects (Table 2 : p=0.006; odds ratio = 2.38; 95% confidence interval = 1.30-4.35). Exclusion of Finnish subjects from the analyses didn’t lead to different results.
Table 2.
Carriership of rare CNVs affecting cardiovascular system development in the study samples. p-adj.: p-values (unadjusted) and OR (odds ratio) were calculated for a comparison between patients and all control subjects. 95% CI = 95% confidence interval. *) The CeAD samples included two families with two affected siblings, but only one sibling of each family was included in this analysis.
| CeAD Patients* | Ischemic Stroke Controls | Finnish Healthy Controls | German Healthy Controls |
All
Controls |
p-value | OR | 95% CI | |
|---|---|---|---|---|---|---|---|---|
| n=831 | n=565 | n=215 | n=1260 | n=2040 | ||||
| Carriership of rare CNVs | ||||||||
| CNVs affecting muscle organ development | 8 | 2 | 0 | 1 | 3 | 0.003 | 6.60 | 1.75-24.9 |
| CNVs affecting cell differentiation | 18 | 0 | 1 | 12 | 13 | <0.001 | 3.45 | 1.68-7.08 |
| CNVs affecting cardiovascular system development | 21 | 5 | 2 | 15 | 22 | 0.006 | 2.38 | 1.30-4.35 |
CeAD patients carrying rare CNVs of genes associated with cardiovascular system development did not differ from non-carriers with regard to baseline characteristics, clinical symptoms or risk factors (Table 3 ). However, patients with a family history of CeAD were significantly more likely to carry a rare CNV affecting cardiovascular system development than patients without a family history. The association between familial CeAD and CNVs affecting cardiovascular system development remained significant after exclusion of related CeAD patients (two pairs of affected sibs were enrolled in the CeAD sample). A rare genic duplication in two affected CeAD siblings (Table 4 ) comprised the MKL2 gene (encoding myocardin-like 2) which is associated with muscle organ development as well as with muscle phenotype.
Table 3.
Baseline characteristics and risk factors in CeAD patients with or without rare CNV affecting cardiovascular system development. Adjusted p-values and OR (odds ratio) are from a logistic regression analysis adjusted for age and sex. 95% CI = 95% confidence interval of OR. Values are n (%) or mean ± standard deviation, CeAD = cervical artery dissection, ICAD = internal carotid artery dissection, FH = family history, unrelated = related patients were excluded.
|
Patients with rare CNVs
affecting cardiovascular system development |
Other
patients |
|||||
|---|---|---|---|---|---|---|
| (n=22) | (n=811) | p-value | Adjusted p-value | OR | [95% CI] | |
|---|---|---|---|---|---|---|
| Age | 42.8 ± 10.4 | 44.3 ± 9.9 | 0.494 | |||
| Female sex | 10 (45.5) | 343 (42.3) | 0.828 | |||
| Stroke | 16 (72.7) | 522 (64.4) | 0.503 | 0.425 | 1.47 | [0.57 – 3.81] |
| ICAD | 13 (59.1) | 547 (67.4) | 0.490 | 0.501 | 0.74 | [0.30 – 1.79] |
| Multiple CeAD | 1 (4.5) | 124 (15.3) | 0.230 | 0.184 | 0.25 | [0.03 – 1.91] |
| Hypertension | 5 (22.7 | 214 (26.6) | 0.810 | 0.815 | 0.88 | [0.31 – 2.51] |
| Hypercholesterolemia | 4 (18.2) | 153 (19.3) | 1.000 | 0.955 | 1.03 | [0.33 – 3.23] |
| Migraine | 9 (40.9) | 300 (37.5) | 0.824 | 0.811 | 1.12 | [0.46 – 2.72] |
| BMI | 25.0 ± 6.4 | 24.6 ± 3.9 | 0.743 | 0.469 | 1.04 | [0.94 – 1.15] |
| Previous trauma | 8 (36.8) | 328 (41.2) | 0.827 | 0.592 | 0.78 | [0.32 – 1.91] |
| Previous infection | 7 (31.8) | 150 (18.9) | 0.165 | 0.134 | 2.02 | [0.81 – 5.04] |
| smoking | 1 (50.0) | 418 (52.2) | 1.000 | 0.916 | 0.96 | [0.41 – 2.25] |
| FH of CeAD | 2 (10.5) | 5 (0.7) | 0.011 | 0.001 | 18.5 | [3.3 – 103] |
| FH of CeAD (unrelated) | 1 (5.6) | 4 (0.5) | 0.110 | 0.036 | 11.2 | [1.17 – 107] |
Table 4.
CNVs associated with cardiovascular system development in the patient’ sample. *) Patients B00ACFQ and B00ACKL are siblings.
| Patient ID | CN-state | Locus | Genes |
|---|---|---|---|
| B00ADIK | loss | chr 1: 10.0-12.0 | C1orf127, TARDBP, MASP2, SRM, MTOR, EXOSC10, ANGPTL7, UBIAD1, FBXO44, FBXO6, MAD2L2, DRAXIN, C1orf1 |
| B00ADIC | gain | chr1:12.3-12.8 | AADACL3, AADACL4, DHRS3, PRAMEF1, PRAMEF11, PRAMEF12, C1orf158, HNRNPCL1 |
| B00ADMI | gain | chr1:12.3-12.8 | AADACL3, AADACL4, DHRS3, PRAMEF1, PRAMEF11, PRAMEF12, C1orf158, HNRNPCL1 |
| B00AE14 | gain | chr2:68.9-69.4 | BMP10, GKN1, GKN2, ANTXR1 |
| B00AD9N | gain | chr3:185.5-185.9 | CLCN2, POLR2H, THPO, RP11-433C9.2, EPHB3 |
| B00ACGF | gain | chr5:0.3-0.5 | SDHA, PDCD6, EXOC3, AHRR, C5orf55, CCDC127 |
| B00ACGZ | gain | chr7:16.9-17.4 | AHR |
| B00AD2L | loss | chr7:19.0-19.1 | TWIST1, HDAC9 |
| B00ACEI | gain | chr8:143.8-144.8 | LY6K, THEM6, SLURP1, LYPD2, LYNX1, LY6D, CYP11B2, LY6E, C8orf31, LY6H, GPIHBP1, ZFP41, GLI4, ZNF696, TOP1MT, RHPN1, MAFA |
| B00ACJC | loss | chr8:23.2-23.4 | R3HCC1, ENTPD4, LOXL2 |
| B00ADHH | loss | chr15:82.9-83.5 | ZSCAN2, WDR73, NMB, SEC11A, ZNF92, ALPK3, SLC28A1,PDE8A |
| B00ACFQ* | gain | chr16:14.0-15.6 | MKL2, PARN, BFAR, PLA2G10, NOMO1, NPIP |
| B00ACKL* | gain | chr16:14.0-15.6 | MKL2, PARN, BFAR, PLA2G10, NOMO1, NPIP |
| B00AEME | gain | chr16:15.2-16.2 | PKD1P1, MPV17L, C16orf45, KIAA0430, NDEI, MYH11, FOPNL, ABCC1, ABCC6 |
| B00AEON | gain | chr16:15.2-16.2 | PKD1P1, MPV17L, C16orf45, KIAA0430, NDEI, MYH11, FOPNL, ABCC1, ABCC6 |
| B00ACG3 | loss | chr16:15.4-16.2 | PKD1P1, MPV17L, C16orf45, KIAA0430, NDEI, MYH11, FOPNL, ABCC1, ABCC6 |
| B00ACRB | gain | chr16:15.4-16.2 | PKD1P1, MPV17L, C16orf45, KIAA0430, NDEI, MYH11, FOPNL, ABCC1, ABCC6 |
| B00AE12 | gain | chr16:2.8-3.4 | ZG16B, PRSS22, FLYWCH1,FLYWCH2, KREMEN2, PKMYT1, CLDN9, PAQR4, CLDN6, TNFRSF12A, HCFC1R1, THOC6, CCDC64B, MMP25, ZSCAN10, ZNF205, ZNF213, OR1F1, ZNF263, TIGD7, ZNF75A, OR2C1, MTRNR2L4, ZNF434, ZNF174 |
| B00ADC0 | gain | chr16:84.9-85.3 | MTHFSD, FOXL1, FOXC2, FOXF1 |
| B00ADWH | gain | chr17:0.7-0.9 | TIMM22, NXN, ABR |
| B00AEP8 | gain | chr17:29.0-29.7 | CCL1, CCL2, CCL7, CCL8, CCL11, CCL13 |
| B00AD77 | gain | chr17:75.7-75.8 | GAA, EIF4A3, CARD14, SGSH, SLC26A11 |
The rare CNVs from the CeAD patients that are associated with cardiovascular system development are listed in (Table 4 ). A large duplication in chromosome 16p including MYH11 and ABCC6 was found in three CeAD patients and in a single control subject. A deletion of the same locus was identified in one further CeAD patient.
DISCUSSION
This large genome-wide analysis of rare genetic variants in CeAD patients yielded the following key findings:1) patients with CeAD were more likely to carry rare CNVs associated with muscle development, cell differentiation, or cardiovascular system development than control subjects; 2) patients with a family history of CeAD more often carried a rare CNVs associated with cardiovascular system development than patients with sporadic (non-familial) CeAD and 3) CNVs in CeAD patients were highly heterogeneous, even though three patients carried a recurrent 16p duplication including MYH11 and ABCC6 and one further patient had a deletion of the same region.
The association between CeAD and rare genetic variants affecting cardiovascular system development observed in our study is in agreement with earlier findings of rare COL3A1 and TGFBR2 mutations in some patients [23-26]. Moreover, our pilot study of 70 CeAD patients revealed a rare CNV of the COL3A1/COL5A2 locus and some additional CNVs affecting cardiovascular system development or muscle organ development [7]. In patients with aortic aneurysms and dissections, enrichment of rare copy number variants in similar predefined gene groups has been reported (including enrichment of CNVs in genes associated with smooth muscle cell contraction and in genes associated with cardiovascular diseases) [27].) Interestingly, one patient from our pilot study sample [7] carried the 16p duplication that we identified in three additional patients from the current study. Furthermore, one CeAD patient from the current study had a large deletion of this same region. This 16p duplication is rare in the human population [28] but was previously found to be significantly more common among patients with aortic dissection than in healthy subjects [4]. Our findings suggest that this rare CNV also increases the risk for CeAD. Taken
together, these findings also point towards shared risk factors for dissections of different arterial beds [29].
We are aware of several limitations. The analysis of microarrays data enables the identification of deletions and duplications, but other types of disease-causing mutations (point mutations, indels, translocations, inversions) cannot be detected. Furthermore, smaller CNVs (<50 kb) may have been overlooked, due to the insufficient probe density of the used microarray platforms. The use of different microarray platforms for the analysis of the control groups is a further limitation of our study. Moreover, some of the patient’ characteristics (including history of trauma, and smoking) were self-reported and not validated by medical records. Familial history of CeAD was also self-reported and not confirmed by molecular analysis. The lack of independent patient and control samples to confirm the current findings is another limitation of this study.
A major strength of our study was the large study sample of patients recruited according to uniform criteria with a single, well-characterized stroke etiology, the utilization of large samples of control subjects as well as the careful inspection and validation of each PennCNV finding in noise-reduced datasets in order to reject all artificial CNV findings [7, 16]. Visual inspection of noise-reduced datasets enabled a critical evaluation of PennCNV findings, including their presence, their length and the position of the breakpoints. Moreover, split calls (consecutive calls that in reality represent one single CNV) were identified as CNVs with copy number state = 4, or mosaicism. Last but not least, visual inspection of a dataset gave an immediate impression of the data-quality, including the amplitude of GC-waves and the noise across the BAF-values. For the prioritization of rare CNV findings we compared our data with control subjects from high-quality public databases, to avoid misclassifications [30]. A further strength of our study was the analysis of predefined gene sets associated with biological functions, instead of focusing upon single candidate CNVs. Association of predefined gene sets was identified by different methods: 1) functional enrichment was detected by comparing the frequency of gene sets in the patient sample and in the human genome; 2) genetic association was detected by comparing the frequency of rare CNVs affecting a predefined gene set in patients and control subjects and 3) genetic association was confirmed by comparing the frequency of rare CNVs affecting a predefined gene set in patients with sporadic and familial CeAD.
In summary, our findings suggest that rare genetic imbalance affecting different biological functions contributes to the risk of CeAD. Moreover, the imbalanced genes in this study suggest that developmental defects of the arterial system may predispose to CeAD. To further explore the role of rare genetic variants in the etiology of CeAD, a whole-exome sequencing study of patients with familial CeAD is currently ongoing [31].
ACKNOWLEDGEMENT
The authors thank all patients who participated in this study and all staff and participants at the CADISP centers for their contributions. The authors are grateful to Maja Bucan for critical discussion and to Werner Hacke for excellent working conditions.
SUPPLEMENTARY MATERIAL
Supplementary material is available on the publisher’s web site along with the published article.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
FINANCIAL DISCLOSURE
The CADISP study has been supported by Inserm, Lille 2 University, Institut Pasteur de Lille and Lille University Hospital and received funding from the Contrat de Projet Etat-Region 2007 Région Nord-Pas-de-Calais, Centre National de Genotypage, Emil Aaltonen Foundation, Paavo Ilmari Ahvenainen Foundation, Helsinki University Central Hospital Research Fund, Academy of Finland, Helsinki University Medical Foundation, Päivikki and Sakari Sohlberg Foundation, Aarne Koskelo Foundation, Maire Taponen Foundation, Aarne and Aili Turunen Foundation, Lilly Foundation, Alfred Kordelin Foundation, Finnish Medical Foundation, Biomedicum Helsinki Foundation, Maud Kuistila Foundation, Orion-Farmos Research Foundation, Finnish Brain Foundation Projet Hospitalier de Recherche Clinique Régional, Fondation de France, Génopôle de Lille, Adrinord, Basel Stroke-Funds, Käthe-Zingg-Schwichtenberg-Fonds of the Swiss Academy of Medical Sciences, the Swiss National Science Foundation (33CM30-124119; 33CM30-140340/1) and the Swiss Heart Foundation. Vincent Thijs is supported by FWO Flanders. The PopGen biobank is funded by the State of Schleswig-Holstein and by the German Federal Ministry of Education and Research (BMBF) through a grant to the PopGen 2.0 Network (01EY1103).
REFERENCES
- 1.Mikhail F.M. Copy number variations and human genetic disease. Curr. Opin. Pediatr. 2014;26:646–652. doi: 10.1097/MOP.0000000000000142. [DOI] [PubMed] [Google Scholar]
- 2.Almal S.H., Padh H. Implications of gene copy-number variation in health and diseases. J. Hum. Genet. 2012;57:6–13. doi: 10.1038/jhg.2011.108. [DOI] [PubMed] [Google Scholar]
- 3.Stankiewicz P., Lupski J.R. Structural variation in the human genome and its role in disease. Annu. Rev. Med. 2010;61:437–455. doi: 10.1146/annurev-med-100708-204735. [DOI] [PubMed] [Google Scholar]
- 4.Kuang S.Q., Guo D.C., Prakash S.K., McDonald M.L., Johnson R.J., Wang M., Regalado E.S., Russell L., Cao J.M., Kwartler C., Fraivillig K., Coselli J.S., Safi H.J., Estrera A.L., Leal S.M., LeMaire S.A., Belmont J.W., Milewicz D.M. GenTAC Investigators. Recurrent chromosome 16p13.1 duplications are a risk factor for aortic dissections. PLoS Genet. 2011;7:e1002118. doi: 10.1371/journal.pgen.1002118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Soemedi R., Wilson I.J., Bentham J., Darlay R., Töpf A., Zelenika D., Cosgrove C., Setchfield K., Thornborough C., Granados-Riveron J., Blue G.M., Breckpot J., Hellens S., Zwolinkski S., Glen E., Mamasoula C., Rahman T.J., Hall D., Rauch A., Devriendt K., Gewillig M. O', Sullivan, J.; Winlaw, DS.; Bu'Lock, F.; Brook, JD.; Bhattacharya, S.; Lathrop, M.; Santibanez-Koref, M.; Cordell, H.J.; Goodship, J.A.; Keavney, B.D. Contribution of global rare copy-number variants to the risk of sporadic congenital heart disease. Am. J. Hum. Genet. 2012;91:489–501. doi: 10.1016/j.ajhg.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matarin M., Simon-Sanchez J., Fung H.C., Scholz S., Gibbs J.R., Hernandez D.G., Crews C., Britton A., De Vrieze F.W., Brott T.G., Brown R. D Jr.; Worrall B.B.; Silliman S.; Case L.D.; Hardy J.A.; Rich S.S.; Meschia J.F.; Singleton A.B. Structural genomic variation in ischemic stroke. Neurogenetics. 2008;9:101–108. doi: 10.1007/s10048-008-0119-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grond-Ginsbach C., Chen B., Pjontek R., Wiest T., Burwinkel B. Tchatchou. S.; Krawczak M.; Schreiber S.; Brandt T.; Kloss M.; Arnold M.L.; Hemminki K.; Lichy C.; Lyrer P.A.; Hausser I.; Engelter S.T. Copy number variation in patients with cervical artery dissection. Eur. J. Hum. Genet. 2012;20:1295–1299. doi: 10.1038/ejhg.2012.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blum C.A., Yaghi S. Cervical Artery Dissection. A Review of the Epidemiology, Pathophysiology, Treatment and Outcome. Arch. Neurosci. 2015;2 doi: 10.5812/archneurosci.26670. pii:e26670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Engelter S.T., Traenka C., Von Hessling A., Lyrer P.A. Diagnosis and treatment of cervical artery dissection. Neurol. Clin. 2015;33:421–441. doi: 10.1016/j.ncl.2014.12.002. [DOI] [PubMed] [Google Scholar]
- 10.Debette S., Kamatani Y., Metso T.M., Kloss M., Chauhan G., Engelter S.T., Pezzini A., Thijs V., Markus H.S., Dichgans M., Wolf C., Dittrich R., Touzé E., Southerland A.M., Samson Y., Abboud S., Béjot Y., Caso V., Bersano A., Gschwendtner A., Sessa M., Cole J., Lamy C., Medeiros E., Beretta S., Bonati L.H., Grau A.J., Michel P., Majersik J.J., Sharma P., Kalashnikova L., Nazarova M., Dobrynina L., Bartels E., Guillon B., van den Herik E.G., Fernandez-Cadenas I., Jood K., Nalls M.A., De Leeuw F.E., Jern C., Cheng Y.C., Werner I., Metso A.J., Lichy C., Lyrer P.A., Brandt T., Boncoraglio G.B., Wichmann H.E., Gieger C., Johnson A.D., Böttcher T., Castellano M., Arveiler D., Ikram M.A., Breteler M.M., Padovani A., Meschia J.F., Kuhlenbäumer G., Rolfs A., Worrall B.B., International Stroke Genetics Consortium Ringelstein E.B.; Zelenika D.; Tatlisumak T.; Lathrop M.; Leys D.; Amouyel P.; Dallongeville J; CADISP Group. Common variation in PHACTR1 is associated with susceptibility to cervical artery dissection. Nat. Genet. 2015;47:78–83. doi: 10.1038/ng.3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Debette S., Metso T.M., Pezzini A., Engelter S.T., Leys D., Lyrer P., Metso A.J., Brandt T., Kloss M., Lichy C., Hausser I., Touzé E., Markus H.S., Abboud S., Caso V., Bersano A., Grau A., Altintas A., Amouyel P., Tatlisumak T., Dallongeville J., Grond-Ginsbach C. CADISP-group. CADISP-genetics: an International project searching for genetic risk factors of cervical artery dissections. Int. J. Stroke. 2009;4:224–230. doi: 10.1111/j.1747-4949.2009.00281.x. [DOI] [PubMed] [Google Scholar]
- 12.Engelter S.T., Grond-Ginsbach C., Metso T.M., Metso A.J., Kloss M., Debette S., Leys D., Grau A., Dallongeville J., Bodenant M., Samson Y., Caso V., Pezzini A., Bonati L.H., Thijs V., Gensicke H., Martin J.J., Bersano A., Touzé E., Tatlisumak T., Lyrer P.A., Brandt T., Cervical Artery Dissection and Ischemic Stroke Patients Study Group Cervical Artery Dissection - Trauma and other potential mechanical trigger events. Neurology. 2013;80:1950–1957. doi: 10.1212/WNL.0b013e318293e2eb. [DOI] [PubMed] [Google Scholar]
- 13.Krawczak M., Nikolaus S., von Eberstein H., Croucher P.J., El Mokhtari N.E., Schreiber S. PopGen: population-based recruitment of patients and controls for the analysis of complex genotype-phenotype relationships. Community Genet. 2006;9:55–61. doi: 10.1159/000090694. [DOI] [PubMed] [Google Scholar]
- 14.Debette S., Metso T., Pezzini A., Abboud S., Metso A., Leys D., Bersano A., Louillet F., Caso V., Lamy C., Medeiros E., Samson Y., Grond-Ginsbach C., Engelter S.T., Thijs V., Beretta S., Béjot Y., Sessa M., Lorenza Muiesan M., Amouyel P., Castellano M., Arveiler D., Tatlisumak T., Dallongeville J., Cervical Artery Dissection and Ischemic Stroke Patients (CADISP) Group Association of vascular risk factors with cervical artery dissection and ischemic stroke in young adults. Circulation. 2011;123:1537–1544. doi: 10.1161/CIRCULATIONAHA.110.000125. [DOI] [PubMed] [Google Scholar]
- 15.Wang K., Li M., Hadley D., Liu R., Glessner J., Grant S.F., Hakonarson H., Bucan M. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007;17:1665–1674. doi: 10.1101/gr.6861907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ginsbach P., Chen B., Jiang X., Engelter S.T., Grond-Ginsbach C. Copy number studies in noisy datasets. Microarrays (Basel) 2013;2:284–303. doi: 10.3390/microarrays2040284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shaikh T.H., Gai X., Perin J.C., Glessner J.T., Xie H., Murphy K., O'Hara R., Casalunovo T., Conlin L.K., D'Arcy M., Frackelton E.C., Geiger E.A., Haldeman-Englert C., Imielinski M., Kim C.E., Medne L., Annaiah K., Bradfield J.P., Dabaghyan E., Eckert A., Onyiah C.C., Ostapenko S., Otieno F.G., Santa E., Shaner J.L., Skraban R., Smith R.M., Elia J., Goldmuntz E., Spinner N.B., Zackai E.H., Chiavacci R.M., Grundmeier R., Rappaport E.F., Grant S.F., White P.S., Hakonarson H. High-resolution mapping and analysis of copy number variations in the human genome: a data resource for clinical and research applications. Genome Res. 2009;19:682–1690. doi: 10.1101/gr.083501.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li J., Yang T., Wang L., Yan H., Zhang Y., Guo Y., Pan F., Zhang Z., Peng Y., Zhou Q., He L., Zhu X., Deng H., Levy S., Papasian C.J., Drees B.M., Hamilton J.J., Recker R.R., Cheng J., Deng H.W. Whole Genome Distribution and Ethnic Differentiation of Copy Number Variation in Caucasian and Asian Populations. PLoS One. 2009;4:e7958. doi: 10.1371/journal.pone.0007958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grond-Ginsbach C., Horstmann S., Hummel M., Wiest T., Honold C., Pfleger K., Hergenhahn M., Hollstein M., Weninger A., Knyazev Y., Mansmann U., Wagner S., Grau A.J. Increased expression of cell-cell signaling genes by stimulated mononuclear leukocytes in patients with previous atherothrombotic stroke. A whole genome expression profile study. Eur. Neurol. 2009;62:30–39. doi: 10.1159/000215878. [DOI] [PubMed] [Google Scholar]
- 20.Vissers L.E., Bhatt S.S., Janssen I.M., Xia Z., Lalani S.R., Pfundt R., Derwinska K., de Vries B.B., Gilissen C., Hoischen A., Nesteruk M., Wisniowiecka-Kowalnik B., Smyk M., Brunner H.G., Cheung S.W., van Kessel A.G., Veltman J.A., Stankiewicz P. Rare pathogenic microdeletions and tandem duplications are microhomology-mediated and stimulated by local genomic architecture. Hum. Mol. Genet. 2009;18:3579–3593. doi: 10.1093/hmg/ddp306. [DOI] [PubMed] [Google Scholar]
- 21.Fakhro K.A., Choi M., Ware S.M., Belmont J.W., Towbin J.A., Lifton R.P., Khokha M.K., Brueckner M. Rare copy number variations in congenital heart disease patients identify unique genes in left-right patterning. Proc. Natl. Acad. Sci. USA. 2011;108:2915–2920. doi: 10.1073/pnas.1019645108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holt R., Sykes N.H., Conceição I.C., Cazier J.B., Anney R.J., Oliveira G., Gallagher L., Vicente A., Monaco A.P., Pagnamenta A.T. CNVs leading to fusion transcripts in individuals with autism spectrum disorder. Eur. J. Hum. Genet. 2012;20:1141–1147. doi: 10.1038/ejhg.2012.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martin J.J., Hausser I., Lyrer P., Busse O., Schwarz R., Schneider R., Brandt T., Kloss M., Schwaninger M., Engelter S., Grond-Ginsbach C. Familial cervical artery dissections – clinical, morphologic and genetic studies. Stroke. 2006;37:2924–2929. doi: 10.1161/01.STR.0000248916.52976.49. [DOI] [PubMed] [Google Scholar]
- 24.Pezzini A., Drera B., Del Zotto E., Ritelli M., Carletti M., Tomelleri G., Bovi P., Giossi A., Volonghi I., Costa P., Magoni M., Padovani A., Barlati S., Colombi M. Mutations in TGFBR2 gene cause spontaneous cervical artery dissection. J. Neurol. Neurosurg. Psychiatry. 2011;82:1372–1374. doi: 10.1136/jnnp.2010.231902. [DOI] [PubMed] [Google Scholar]
- 25.Grozeva D., Conrad D.F., Barnes C.P., Hurles M., Owen M.J., O'Donovan M.C., Craddock N., Kirov G. WTCCC. Independent estimation of the frequency of rare CNVs in the UK population confirms their role in schizophrenia. Schizophr. Res. 2012;135:1–7. doi: 10.1016/j.schres.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Makrygiannis G., Loeys B., Defraigne J.O., Sakalihasan N. Cervical artery dissection and type A aortic dissecton in a family with a novel missense COL3A1 mutation of vascular type Ehlers-Danlos syndrome. Eur. J. Med. Genet. 2015;58:634–636. doi: 10.1016/j.ejmg.2015.10.009. [DOI] [PubMed] [Google Scholar]
- 27.Prakash S.K., LeMaire S.A., Guo D.C. Russel,l L.; Regalado, E.S.; Golabbakhsh, H.; Johnson R.J.; Safi H.J.; Estrera A.L.; Coselli J.S.; Bray M.S.; Leal S.M.; Milewicz D.M.; Belmont J.W. Rare copy number variants disrupt genes regulating vascular smooth muscle cell adhesion and contractility in sporadic thoracic aortic aneurysms and dissections. Am. J. Hum. Genet. 2010;87:743–756. doi: 10.1016/j.ajhg.2010.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fujita D., Takeda N., Morita H., Kato M., Nishimura H., Inuzuka R., Taniguchi Y., Nawata K., Hyodo H., Imai Y., Hirata Y., Komuro I. A novel mutation of TGFBR2 causing Loeys-Dietz syndrome complicated with pregnancy-related fatal cervical arterial dissections. Int. J. Cardiol. 2015;201:288–290. doi: 10.1016/j.ijcard.2015.07.109. [DOI] [PubMed] [Google Scholar]
- 29.Grond-Ginsbach C., Pjontek R., Aksay S.S., Hyhlik-Dürr A., Böckler D., Gross M-L. Spontaneous arterial dissection – phenotype and molecular pathogenesis. Cell. Mol. Life Sci. 2010;67:1799–1815. doi: 10.1007/s00018-010-0276-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bastida-Lertxundi N., López-López E., Piñán M.A., Puiggros A., Navajas A., Solé F., García-Orad A. Errors in the interpretation of copy number variations due to the use of public databases as a reference. Cancer Genet. 2014;207:164–167. doi: 10.1016/j.cancergen.2014.03.001. [DOI] [PubMed] [Google Scholar]
- 31.Grond-Ginsbach C., de Freitas G.R., Campos C.R., Thie A., Caso V., Machetanz J., Kloss M. Familial occurrence of cervical artery dissection - Coincidence or sign of familial predisposition? Cerebrovasc. Dis. 2012;33:466–470. doi: 10.1159/000337035. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material is available on the publisher’s web site along with the published article.