

Abstract
Pulmonary hypertension (PH) is defined by elevated mean pulmonary artery pressure following the pathological remodelling of small pulmonary arteries. An increase in right ventricular (RV) afterload results in RV hypertrophy and RV failure. The pathophysiology of PH, and RV remodelling in particular, is not well understood, thus explaining, at least in part, why current PH therapies have a limited effect. Existing therapies mostly target the pulmonary circulation. Because the remodelled RV fails to support normal cardiac function, patients eventually succumb from RV failure. Developing novel therapies that directly target the function of the RV may therefore benefit patients with PH. In the past decade, several promising studies have investigated novel cardioprotective strategies in experimental models of PH. This review aims to comprehensively discuss and highlight these novel experimental approaches to confer, in the long‐term, greater health benefit in patients with PH.
Abbreviations
- BMPR‐2
bone morphogenetic protein receptor‐2
- EUK‐134
superoxide dismutase and catalase mimetic
- HDACs
histone deacetylases
- HIF‐1α
hypoxia inducible factor‐1α
- LV
left ventricle
- miRs
micro‐RNAs
- NHE‐1
sodium hydrogen exchanger‐1
- NRF2
nuclear respiratory factor‐2
- PASMCs
pulmonary arterial smooth muscle cells
- PH
pulmonary hypertension
- RV
right ventricular
Tables of Links
| TARGETS | |
|---|---|
| Other protein targets a | Enzymes d |
| Bcl‐xl | Caspase‐3 |
| BMPR‐2 | eNOS, endothelial NO synthase |
| Notch 1–4 | HDAC |
| Survivin (BIRC5) | Pyruvate dehydrogenase kinase |
| Catalytic receptors b | Smad |
| NLRP‐3 | |
| Transporters c | |
| Glucose transporter 1 (GLUT1) | |
| NHE‐1, SLC9A1 |
These Tables list key protein targets and ligands in this article that are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,b,c,dAlexander et al., 2015a,b,c,d).
Introduction
Pulmonary hypertension (PH) is defined as a mean pulmonary arterial pressure ≥25 mm Hg at rest, as assessed with right heart catheterization (Galie et al., 2009; Simonneau et al., 2013). It is a growing health burden in both developed and developing countries (Mocumbi et al., 2015). The global prevalence of PH is not known, due to a lack of global PH registries (Thienemann et al., 2014). In Europe, the prevalence of PH is calculated to range from 0.3 to 6%, and in particular, idiopathic PH affects approximately six individuals per million people (Humbert et al., 2006; Mocumbi et al., 2015). A similar prevalence has been reported in Australia for PH secondary to left ventricular heart disease (0.33 to 6.6% per million people) (Lam et al., 2009; Strange et al., 2012). The prevalence of PH on the African continent is not known (Mocumbi et al., 2015), and therefore, a multinational multicentre registry of PH was recently established in Africa to describe PH presentation, severity, management, causes and comorbidities (Thienemann et al., 2014). In South Africa, the Heart of Soweto cohort study captured 2505 cases with heart failure, of which one‐third was diagnosed with right heart failure due to chronic lung disease (26%) and pulmonary artery hypertension (20%) (Sliwa et al., 2008; Stewart et al., 2011). This study demonstrated that the prevalence of PH may not be as rare as thought to be.
PH develops in many clinical conditions, including human immune virus/acquired immune deficiency syndrome, sickle cell disease, systemic sclerosis, schistosomiasis, congenital heart disease and chronic obstructive pulmonary disease (Graham et al., 2010; Papamatheakis et al., 2014; Simonneau et al., 2009; Wrobel et al., 2012). Based mainly on the aetiology of PH, it is classified into five groups (Task Force for D et al., 2009; Simonneau et al., 2013), namely, group‐1: pulmonary arterial hypertension; group‐2: PH due to left heart disease, pulmonary veno‐occlusive disease and/or pulmonary capillary hemangiomatosis; group‐3: PH associated with lung disease; group‐4: Chronic thromboembolic PH; and group‐5: PH with unclear multifactorial mechanisms (Task Force for Diagnosis and Treatment of Pulmonary Hypertension of European Society of Cardiology (ESC) et al., 2009; Simonneau et al., 2013).
The mortality of PH patients varies depending on the group of PH (Mocumbi et al., 2015). Current PH treatments as described in the 2016 European Society of Cardiology/European Respiratory Society guidelines (Galie et al., 2016) have limited effects on patient mortality (Wang et al., 2014) and quality of life (Lang et al., 2006; Oudiz et al., 2009; Gomberg‐Maitland et al., 2011; Hoeper, 2015; Speich et al., 2015). This has sparked a research interest in the potential use of pharmacological cardioprotective therapies to provide additional health benefits against PH. At this time, cardioprotection in PH is a fairly new and experimental approach, but recent animal studies show noticeable benefit with a number of cardioprotective therapies. In this review, we provide a brief overview of the pathophysiology of PH and right ventricle (RV) remodelling, and we comprehensively discuss various cardioprotective therapies that may provide benefit to PH patients, if administered in conjunction with current treatments.
The right ventricle in pulmonary hypertension
The pathophysiology of PH is complex, and many diverse factors have been implicated as trigger events in the pulmonary vascular remodelling displayed in PH (Rabinovitch, 2012; Magne et al., 2015) (Figure 1). This leads to (i) increased/disorganized proliferation of endothelial cells, increased endothelial cell apoptosis as well as (ii) increased proliferation of pulmonary arterial smooth muscle cells (PASMCs) and resistance of PASMCs to apoptosis (Farber and Loscalzo, 2004; Jurasz et al., 2010; Summer et al., 2011; Prins and Thenappan, 2016) (Figure. 2). A more in‐depth discussion of the pathophysiology of PH was recently reviewed elsewhere (see Prins and Thenappan, 2016). Due to the anatomical design of the cardiopulmonary system, PH is strongly correlated with the RV structure and function (Vonk Noordegraaf and Galie, 2011).
Figure 1.
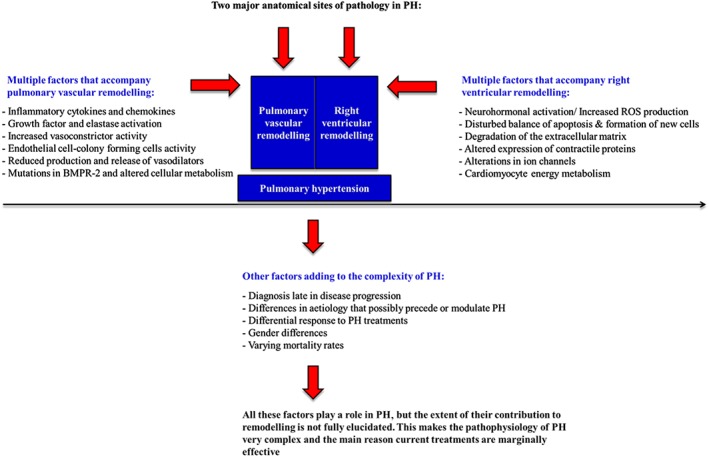
The complex nature of PH. A graphical representation of the two major anatomical sites of pathology in PH. This figure also outlines the range of factors that accompany both pulmonary vascular remodelling and RV remodelling.
Figure 2.
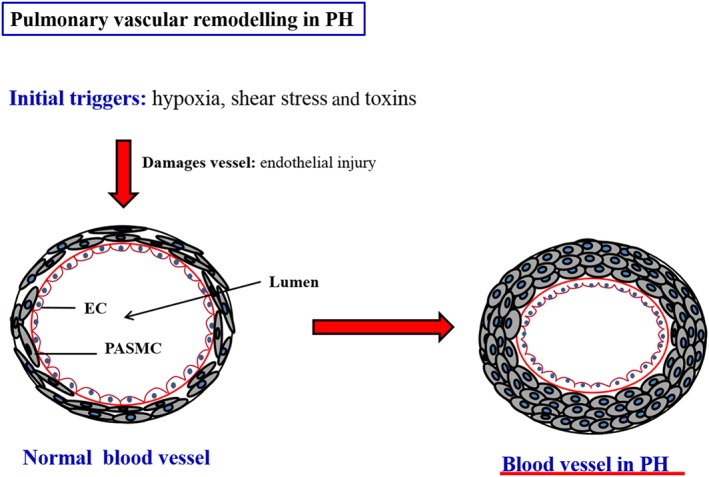
A simplified representation of the remodelling of the pulmonary vasculature in PH. Here, it is clear how the lumen of the vessels are constricted which ultimately results in increased pressure. Adapted from Wilkins MR. 2012 (Wilkins, 2012).
The RV is structurally, geometrically and mechanically distinct from the left ventricle (LV). It has a thinner wall than the LV, and it is half‐moon shaped, which is reflective of low pressures in the pulmonary circulation (Figure 3) (Opie et al., 2006; Voelkel et al., 2006, 2013b; Bogaard et al., 2009; Handoko et al., 2010; Umar et al., 2012; Vonk‐Noordegraaf et al., 2013). The thin wall of the RV renders it vulnerable to acute increases in ventricular wall stress (Naeije and Manes, 2014). During the initial stages of PH, increasing pulmonary vascular resistance and decreasing pulmonary vascular compliance increases afterload, but the RV can still cope (Vonk Noordegraaf and Galie, 2011; Vonk‐Noordegraaf et al., 2013). At this stage, RV systolic contraction is enhanced, and the RV undergoes concentric remodelling, while the right atrial pressure remains normal with a steep increase in mean pulmonary artery pressure and maintenance of cardiac index (Figure 3) (Vonk Noordegraaf and Galie, 2011; Vonk‐Noordegraaf et al., 2013). This is associated with increasing systolic and diastolic ventricular pressures, and increasing diastolic and systolic stretch on the RV wall, which leads initially to adaptive hypertrophy.
Figure 3.
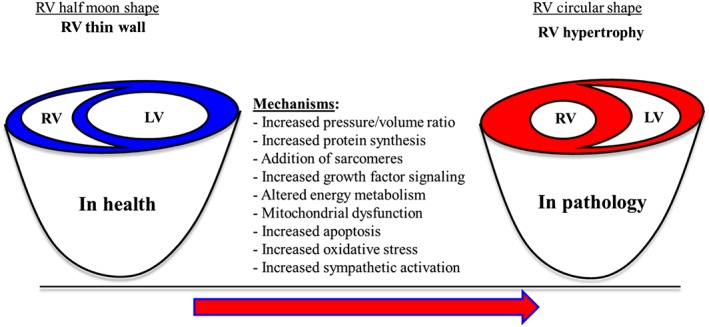
Collation of all the molecular and cellular mechanisms invovled in the developing of RV remodelling in PH.
However, when RV afterload becomes excessively elevated, ventricular dilation occurs (Vonk Noordegraaf and Galie, 2011; Vonk‐Noordegraaf et al., 2013). The RV contractility remains constant in spite of further increase in RV load, a phenomenon described as RV uncoupling. The molecular mechanisms that halt further adaptation of the RV are not fully elucidated but include an imbalance of oxygen supply and demand and changes in cardiomyocytes and the extracellular matrix (Vonk Noordegraaf and Galie, 2011; Vonk‐Noordegraaf et al., 2013). Nonetheless, the RV dilation increases wall tension, which increases myocardial oxygen demand and decreases RV perfusion, thus leading to compromised contractility and dilatation. Increased pulmonary vascular resistance (e.g. as caused by pulmonary arterial constriction to mimic massive pulmonary embolism) leads to dilation and rapid spike failure of the RV (Naeije and Manes, 2014). Furthermore, increased ventricular volume may lead to tricuspid regurgitation, caused by annular valve dilation and chordal traction that ultimately results in RV volume overload, further progressive annular dilation and RV remodelling (Vonk Noordegraaf and Galie, 2011; Vonk‐Noordegraaf et al., 2013). After the decline of RV function, increased RV contraction time and ventricular asynchrony and decreased RV stroke volume lead to suboptimal filling of the LV (Vonk Noordegraaf and Galie, 2011; Vonk‐Noordegraaf et al., 2013). The ventricular changes, together with systolic/diastolic RV dysfunction, contribute to the marked decline in cardiac output seen in severe PH (Vonk Noordegraaf and Galie, 2011; Vonk‐Noordegraaf et al., 2013). All together, these chains of events subsequently lead to RV failure if they occur without intervention (Vonk Noordegraaf and Galie, 2011; Vonk‐Noordegraaf et al., 2013).
Rationale for pharmacological cardioprotective therapies against pulmonary hypertension
Therapy for PH includes general measures, initial therapy with high‐dose calcium channel blockers in vasoactive patients followed by combination therapy with newer medical compounds (Galie et al., 2016). These compounds alleviate pulmonary vascular tone by targeting three major pathways (1) endothelin‐1 (ET‐1) receptor‐mediated vasoconstriction, (2) prostacyclin‐mediated vasodilation and (3) phosphodiesterase type‐5/guanylate cyclase‐mediated vasodilation (Benza et al., 2015; Humbert and Ghofrani, 2015). The current treatment regimen of PH is complex and varies depending on the group of PH (Galie et al., 2009; Task Force for Diagnosis and Treatment of Pulmonary Hypertension of European Society of Cardiology (ESC) et al., 2009). Evidence‐based therapeutic options including grade of recommendation and level of evidence have been published recently via the 2016 European Society of Cardiology/European Respiratory Society guidelines (Galie et al., 2016). Current PH treatments mainly target the pulmonary circulation, and considering the link between PH and the RV (Voelkel et al., 2006), cardioprotective therapies may have a role to play (Vonk‐Noordegraaf et al., 2013). RV function correlates well with the prognosis of patients with PH and is a good predictor of patient survival (Voelkel et al., 2006; Handoko et al., 2010; Sztrymf et al., 2010; Vonk Noordegraaf and Galie, 2011; Vonk‐Noordegraaf et al., 2013). Current PH treatments only modestly improve RV function in PH patients (Brittain et al., 2013), but cardioprotective therapies in addition to current treatments may provide greater cardioprotective benefit.
The purpose of cardioprotection in PH is not to stop the RV from remodelling, because this remodelling, as described earlier, is necessary for the heart to cope during PH. However, cardioprotection can be implemented in order to aid the RV and improve its functional properties in PH. If given as an adjunct, pharmacological cardioprotective treatment may provide the hope of improving quality of life and increasing survival of PH patients. In ventricular remodelling, a pathological stimulus (e.g. pressure/volume overload) induces cardiomyocyte growth/loss and activates cellular and molecular pathways that are associated with increased protein synthesis, addition of sarcomeres, cardiomyocyte apoptosis, cardiomyocyte energy metabolism, oxidative stress and sympathetic activation (Bogaard et al., 2009; Voelkel et al., 2012, 2013b; Ryan and Archer, 2014) (Figure 3). Most of the promising cardioprotective therapies, currently under testing in preclinical studies, target these main molecular and cellular pathways (Table 1).
Table 1.
Preclinical pharmacological cardioprotective therapies tested in animal models of pulmonary hypertension (PH)
| Pharmacological cardioprotective approach | Model of PH used to test the therapy | References | Disease category/subtype of PH targeted |
|---|---|---|---|
| ‐ Interference with the epigenetic control of cardiac gene transcription | ‐ Pulmonary artery banding | (Cho et al., 2010; Bogaard et al., 2011) | Groups 1 and 3 |
| ‐ Monocrotaline model | |||
| ‐ Interference with miRNAs | ‐ Chronic hypoxia model | (Pullamsetti et al., 2012; Brock et al., 2014) | Group 3 |
| ‐ Interference with nuclear factor κ‐B‐mediated modulation of Notch signalling | ‐ Monocrotaline model | (Kumar et al., 2012) | Group 1 |
| ‐ Interference with cardiomyocyte energy metabolism | ‐ Pulmonary artery banding | (Piao et al., 2010) | Group 1 |
| ‐ Monocrotaline model | (Piao et al., 2010) | – | |
| ‐ Interference with mitochondrial dynamics | ‐ Monocrotaline model | (Marsboom et al., 2012; Ryan et al., 2013) | Groups 1 and 3 |
| ‐ Chronic hypoxia model | |||
| ‐ Inhibition of the sodium‐hydrogen‐exchanger‐1 | ‐ Monocrotaline model | (Chen et al., 2001) | Group 1 |
| ‐ Inhibition of apoptosis and antioxidant therapy | ‐ Not tested in PH | – | – |
In the above table, the animal models of PH are categorized according to the group of PH they represent. Group‐1, pulmonary arterial hypertension and group‐3: PH associated with lung disease (Maarman et al., 2013).
Cardioprotective therapies targeting cardiomyocyte growth
Interference with the epigenetic control of cardiac gene transcription
A genetic approach in the treatment of ventricular failure is the pharmacological inhibition of histone deacetylases (HDACs) (McKinsey, 2011; Cavasin et al., 2012; Wang et al., 2015a). These HDACs are enzymes that remove acetyl groups from histone proteins and thus weaken the interaction between charged histones and the DNA backbone (Bush and McKinsey, 2010; McKinsey, 2011). By doing this, HDACs cause relaxation of the chromatin structure, which enhances the access to transcription factors (Bush and McKinsey, 2010; McKinsey, 2011). HDACs have a regulatory function in ventricular remodelling, and inhibitors such as trichostatin‐A, valproic acid and mocetinostat have been tested in RV remodelling, with significant success (Zhang et al., 2002; Cao et al., 2011; McKinsey, 2011; Nural‐Guvener et al., 2014). Bogaard et al. (2011), tested HDAC inhibitors in a rat model of pulmonary artery banding‐induced RV hypertrophy. Four weeks after surgery, when RV hypertrophy was overt, trichostatin‐A treatment was initiated (450 μg·kg−1 i.p., five times per week for a period of 2 weeks). Unfortunately, trichostatin‐A worsened RV remodelling and decreased endothelial nitric oxide synthase gene expression, which was attributed to RV dilatation and capillary rarefaction (Bogaard et al., 2011). The failure of trichostatin‐A to protect against RV remodelling is ascribed to the inhibitory effect that HDAC suppression had on myocardial angiogenesis (Bogaard et al., 2011). Although HDAC inhibition has positive effects on left ventricular remodelling secondary to pressure overload, the use of trichostatin‐A is not appropriate to RV remodelling.
Another HDAC inhibitor, valproic acid, had been shown to prevent RV remodelling in both the pulmonary artery banding and monocrotaline models (Cho et al., 2010). As previously mentioned, RV hypertrophy is necessary to compensate for high RV afterload in PH. Therefore, complete reduction of RV hypertrophy in PH may be detrimental. However, the inhibitor valproic acid was administered in drinking water, and therefore, the reduction in RV hypertrophy was not in isolation but possibly associated with improved PH. Therefore, the reduced RV hypertrophy afforded by valproic acid may be considered beneficial. However, valproic acid is a non‐specific inhibitor of HDACs that has a wide range of pharmaco‐physiological effects, such as the regulation of ion channel opening and the expression of glycogen synthase kinase‐3β and mitogen‐activated protein kinases (McKinsey, 2011). Due to the non‐specific nature of valproic acid, its impressive stunting of RV hypertrophy should be interpreted with caution as it may have other systemic effects (McKinsey, 2011). Therefore, it should be noted that more research is required before valproic acid can be implemented in a clinical setting.
On the other hand, selective class‐1 inhibition of HDACs, with benzamide, attenuated RV hypertrophy in the chronic hypoxia model of PH (Cavasin et al., 2012). Benzamide achieved its effects by decreasing caspase‐3/7 activity and interleukin mRNA expression (IL‐1β, IL‐2) (Cavasin et al., 2012). Benzamide reduced PH and RV hypertrophy and suppressed pathological gene expression, pro‐apoptotic caspase activity and pro‐inflammatory protein expression (Cavasin et al., 2012). This suggests that selective HDAC inhibition and/or targeted drug delivery to the myocardium or lungs may provide cardioprotective benefit in PH. Although promising, the use of HDAC inhibitors as a cardioprotective target for RV remodelling in PH requires further investigation (Wang et al., 2015b).
Interference with gene expression and micro‐RNAs
Micro‐RNAs (miRs) are short, non‐coding ribonucleic acids that regulate gene expression at the post‐transcriptional level (Latronico and Condorelli, 2011; Shah and Mann, 2011). They are activated by cellular stress signals, and their regulatory function is achieved by inhibiting protein translation or promoting degradation of target messenger‐RNAs (Wang et al., 2010; Latronico and Condorelli, 2011; Shah and Mann, 2011; van Rooij and Olson, 2012). MiRs are highly conserved and instrumental in biological processes such as cardiovascular development and pathology (Wang et al., 2010; Latronico and Condorelli, 2011; Shah and Mann, 2011; van Rooij and Olson, 2012). More specifically, they have important functions in the regulation of cardiomyocyte apoptosis, differentiation and proliferation (Pullamsetti et al., 2012). In heart failure, a number of miRs are either up‐ or down‐regulated including miR‐17, miR‐20a and miR‐24 (Naga Prasad et al., 2009; Latronico and Condorelli, 2011; Shah and Mann, 2011; Pullamsetti et al., 2012).
In chronic hypoxia and monocrotaline models of RV remodelling, inhibition of miR‐17 with the antagomir‐17 improved cardiac function (Pullamsetti et al., 2012). Also, antagomir‐20a significantly reduced RV hypertrophy in a mouse model of chronic hypoxia‐induced RV hypertrophy (Brock et al., 2014). Considering the shortfall of this model (see Maarman et al., 2013), future studies could investigate the efficacy of miRs as a cardioprotective therapy in PH in a more appropriate PH model such as a multiple‐pathological‐insult model including monocrotaline plus pneumonectomy and chronic hypoxia plus SUGEN‐5416 (see Maarman et al., 2013). Nevertheless, the miRs have drawn much attention as novel cardioprotective targets for cardiovascular disease due to their cardiac specific nature, and they may also afford cardioprotection in PH (Boucherat et al., 2015).
Interference with NFκ‐B and modulation of notch signalling
NF‐κB is a key modulator of the notch signalling pathway and is involved in the development of PH (Fan et al., 2016; Kumar et al., 2012). This complex juxtacrine signalling pathway is initiated by the interaction between notch transmembrane receptors (notch 1–4) and their ligands (jagged‐1 and 2) (Niessen and Karsan, 2008; Rusanescu et al., 2008). Notch/NF‐κB signalling is further involved in development of the cardiovascular system and cardiovascular pathology, and it regulates a wide range of cellular processes, including cell fate determination, development, differentiation, proliferation, apoptosis and regeneration (Gude et al., 2008; Niessen and Karsan, 2008; Rusanescu et al., 2008; Rizzo et al., 2013). Transgenic mice overexpressing the cardiac specific dominant‐negative‐IκB gene were injected with monocrotaline (60 mg·kg−1, s.c.) (Kumar et al., 2012). These mice displayed inhibited myocardial NF‐κB activation compared with the wildtype mice (Kumar et al., 2012). This inactivation of NF‐κB prevented RV hypertrophy in spite of the presence of and restored expression of BMPR‐2 and Smad signalling (Kumar et al., 2012). These changes were observed in both cardiac and lung tissues. In untreated mice, activation of NF‐κB decreased the expression of notch‐3, BMP‐2 and BMPR‐2, reduced inhibitory of differentiation proteins, Smad‐2 and 8, and increased Smad‐4 expression and RV hypertrophy. These observations were abolished in the transgenic mice where cardiac‐NF‐κB activation was inhibited (Kumar et al., 2012). These data strongly suggest that pharmacological modulation of the NF‐κB/notch signalling‐axis may be cardioprotective in PH.
Cardioprotective therapies targeting the sodium‐hydrogen‐exchanger 1 (NHE‐1)
The transporter protein NHE‐1 is an integral membrane glycoprotein, ubiquitously expressed in mammalian cells and responsible for the removal of intracellular protons from the cell at the expense of sodium ions (Karmazyn, 1999; Cingolani and Ennis, 2007; Karmazyn et al., 2008). It is regulated by hormonal, paracrine/autocrine regulators, ribosomal‐S‐6‐kinase and mechanical stretch (Wang et al., 1997; Moor and Fliegel, 1999; Karmazyn et al., 2008). The involvement of NHE‐1 in the cardiac hypertrophic response is thought to be initiated by mechanical stretch that stimulates NHE‐1 activity via the activation of kinases such as protein kinase C or MAP kinases (Karmazyn et al., 2008). The pro‐hypertrophic activity of NHE‐1 involves the activation of calcineurin, calmodulin‐dependent‐kinase‐II and ROS (Karmazyn et al., 2008), and the inhibition of phosphorylation‐dependent NHE‐1 activation in cardiomyocytes has been shown to occur via increased dephosphorylation by the catalytic subunit of protein phosphatase‐2A (Snabaitis et al., 2006; Karmazyn et al., 2008).
In monocrotaline‐induced RV failure, 7 days of treatment with the inhibitor of NHE‐1 cariporide attenuated RV hypertrophy (Chen et al., 2001). It also reduced the cardiac response to pulmonary vascular damage, and the cardioprotective effect was independent of the pulmonary vascular system (Chen et al., 2001). In general, the mechanism underlying the cardioprotective effects of NHE‐1 inhibition is based on two processes, prevention of calcium overload and opening of the mitochondrial permeability transition pore (Garciarena et al., 2008). Even though the exact mechanisms are not fully elucidated, there is strong evidence suggesting that inhibition of the NHE‐1 has therapeutic potential in PH‐induced RV protection.
Inhibition of cardiomyocyte apoptosis
Apoptosis plays an important role in cardiac remodelling (Yaoita et al., 1998; Das, 2007; Shah and Mann, 2011). In heart failure, apoptosis is triggered by activation of G‐protein‐coupled receptors, cytokines and increased ROS production (Shah and Mann, 2011). The regulated and ordered nature of apoptosis, allows intervention at an early stage to prevent ultimate cell death (Yaoita et al., 1998; Das, 2007; Shah and Mann, 2011). There are many proteins involved in apoptosis, with caspases being the most prominent proteins (Porter and Janicke, 1999). In the monocrotaline model of PH‐induced RV failure, cardiomyocyte apoptosis and the cell cycle are co‐activated in RV segments (Ecarnot‐Laubriet et al., 2002; Umar et al., 2012). In a study by Ecarnot‐Laubriet et al. (2002), RV‐cardiomyocyte apoptosis was associated with an increase of caspase‐3 expression and down‐regulation of the anti‐apoptotic protein, Bcl‐2. Anti‐apoptotic approaches with similar efficacy that have not been tested in PH would include ZVAD‐fmk (Yaoita et al., 1998), aurintricarboxylic acid (Zhao et al., 2003; Das, 2007), mitofusion‐2 (Shen et al., 2007; Yu et al., 2011) and survivin (Lee et al., 2014). In a recent study by Zungu‐Edmondson et al. (2016), rats were injected with SUGEN‐5416, an inhibitor of vascular endothelial growth factor, and thereafter exposed to 3 weeks of hypoxia to induce PH (Zungu‐Edmondson et al., 2016). These rats displayed elevated RV systolic pressure with significant RV myocyte apoptosis, associated with down‐regulation of the anti‐apoptotic protein Bcl‐xL, down‐regulated transcription factor GATA‐4 (transcriptional regulator of Bcl‐xL) and up‐regulated p‐53 (negative regulator of GATA‐4 gene transcription). The PH‐induced RV apoptosis was attenuated in p‐53 knockout animals (Zungu‐Edmondson et al., 2016). Despite a major loss of RV cardiomyocytes due to apoptosis, RV contractility was enhanced, suggesting that the remaining myocytes can perform improved contractile functions (Zungu‐Edmondson et al., 2016). These data suggest that RV decompensation is associated with cardiomyocyte apoptosis, and the remaining myocytes are capable of sustaining RV contractility (Zungu‐Edmondson et al., 2016).
Although apoptosis modulators have not been tested in patients with PH‐induced RV failure, their use may confer cardioprotection in PH as cardiomyocyte apoptosis is also instrumental in this type of RV failure (Ecarnot‐Laubriet et al., 2002; Umar et al., 2012). Caution should be taken with apoptosis inhibitors: If administered systemically, they may affect other organs, as apoptosis plays an important physiological role overall. Pharmacological inhibition of apoptosis may therefore be a long way from being tested in the clinical setting against PH, but its efficacy may be improved by targeting it towards the affected RV. However, taken together, current evidence suggest that inhibition of various steps in the apoptotic signalling pathway may be cardiopotective in PH, although further investigation is necessary.
Cardioprotective therapies targeting cardiomyocyte energy metabolism
When cardiomyocytes are exposed to decreased oxygen levels, they shift energy metabolism from oxidative phosphorylation to glycolysis, a shift which is reversed when oxygen levels return to normal (Stanley et al., 1997; Taegtmeyer, 2000; Stanley, 2004; Taegtmeyer, 2004; Stanley et al., 2005; Lopaschuk et al., 2010). In PH, the RV displays metabolic remodelling, which is associated with decreased angiogenesis and a transition from a compensated to a decompensated state (Sutendra et al., 2013). In response to pressure overload, cardiomyocytes develop a metabolic phenotype known as the ‘glycolytic shift’ that is characterized by increased glycolysis (Piao et al., 2010; Tuder et al., 2012). The general concept is that increased glucose oxidation might ameliorate cardiac function during cardiac pathology (Stanley, 2004; Stanley et al., 2005; Lopaschuk et al., 2010; Maarman et al., 2012). In the monocrotaline and pulmonary artery banding animal models of RV remodelling, dichloroacetate increased glucose oxidation by inhibiting pyruvate dehydrogenase kinase treatment and prevented RV hypertrophy (Piao et al., 2010). It also increased the expression of glucose transporter‐1 and pyruvate dehydrogenase and restored the expression of the voltage‐gated potassium channels (Kv1.5 and Kv4.2). This led to increased RV glucose oxidation, increased cardiac work, improved RV repolarization and improved RV function (Piao et al., 2010). These results have recently been corroborated by Sun et al. (2016) who administered dichloroacetate in monocrotaline‐induced PH (Sun et al., 2016). Furthermore, partial inhibition of fatty acid oxidation with trimetazidine and ranolazine increased cardiac output and exercise capacity in the pulmonary artery banding model of RV hypertrophy (Piao et al., 2010).
Abnormal fatty acid metabolism arising from increased fatty acid uptake (via fatty acid transporter, CD‐36) and disproportionate β oxidation may also cause RV dysfunction in PH (Sakao et al., 2015; Talati and Hemnes, 2015). Cytoplasmic lipids (triglycerides, ceramides and diacylglycerol) can then accumulate and lead to lipotoxicity (Talati and Hemnes, 2015). Although some experimental interventions aim to improve RV dysfunction by targeting fatty acid or glucose metabolism, there is a lack of a deeper understanding of the complex mechanisms involved in the metabolic remodelling of the RV in PH (Talati and Hemnes, 2015). Although these pharmacological treatments appear very effective, one should keep in mind that they are not cardiac specific and may have widespread systemic effects (Tuder et al., 2012). Nevertheless, these studies do suggest that dichloroacetate, trimetazidine and ranolazine may be considered as cardioprotective treatments for PH‐induced RV remodelling. Furthermore, pharmacological manipulation of cardiomyocyte energy metabolism has been shown effective in the clinical setting (Guarini et al., 2016). Therefore, compared with other experimental cardioprotective approaches, interventions targeting energy metabolism may be closer to clinical implementation.
Cardioprotective therapies targeting mitochondrial dynamics
Mitochondria consume oxygen in order to produce energy. During this process, ROS are produced at complex‐I and II of the electron transport chain (Weir et al., 2005). Mitochondria‐derived ROS can regulate membrane voltage‐gated potassium channels or components of the hypoxia inducible factor‐1 α (HIF‐1α) ‐signalling pathway in order to induce PASMC contraction (Sutendra and Michelakis, 2014). In recent years, mitochondria have increasingly received attention as important players in the pathogenesis of remodelling in PH as they can potentially regulate PASMC apoptosis/resistance to apoptosis and activation of the NLRP‐3 inflammasome (Kepp et al. (2011); Dromparis et al., 2010; Freund‐Michel et al., 2014; Ryan et al., 2015). Aside from its role in the lung, they are equally important in RV remodelling in response to PH (Tang et al., 2002; Vonk‐Noordegraaf et al., 2013; Ryan and Archer, 2015; Ryan et al., 2015). In PH, the RV displays decreased mitochondrial content, mitochondria with abnormal shape and size, impaired fatty acid oxidation and a glycolytic shift towards glycolysis (Enache et al., 2013; Gomez‐Arroyo et al., 2013; Vonk‐Noordegraaf et al., 2013; Balestra et al., 2015; Bruns et al., 2015; Sun et al., 2016). In PH, cardiomyocytes have a mitochondrial‐metabolic phenotype similar to cancer (Ryan and Archer, 2015). This phenotype includes increased energetic reliance on aerobic glycolysis; inhibition of mitochondrial respiration due to pathological activation of transcription factors (cMyc, fork head transcription factor and HIF‐1α) and pyruvate dehydrogenase kinase‐induced pyruvate dehydrogenase inhibition (Ryan and Archer, 2015). Furthermore, in the hypertrophied RV, cancer‐like metabolic changes, aerobic glycolysis and glutaminolysis reduced energy production and contractility (Ryan and Archer, 2015). Mitochondrial fragmentation contributes to the proliferative, apoptosis‐resistant phenotype of PH (Ryan and Archer, 2015). Therefore, RV remodelling in PH is strongly linked with changes in mitochondrial dynamics of the RV.
Experimental studies using animal models of PH have identified the therapeutic benefits of targeting mitochondrial dynamics with reduced mitofusin‐2 and dynamin‐related protein‐1 (Marsboom et al., 2012; Ryan et al., 2013). These interventions regressed pulmonary vascular obstruction and improved haemodynamic and RV contractility, increased cardiac output and reduced RV remodelling (Marsboom et al., 2012; Ryan et al., 2013). Other possible therapeutic targets could also be mitochondria‐specific drugs that aim to modulate mitochondrial function/efficiency (Enache et al., 2013; Gomez‐Arroyo et al., 2013; Balestra et al., 2015). There is a need for future studies that investigate the full scope of RV‐mitochondrial dysfunction in models of PH. This will provide better understanding of how PH affects the RV mitochondria, and this, in turn, will allow the development of cardioprotective therapies that target the mitochondrial milieu.
Cardioprotective therapies targeting oxidative stress
Oxidative stress is characterized by the increased production of oxidants such as ROS (including superoxide anion radical, hydrogen peroxide and hydroxyl radical) and insufficient levels/activities of antioxidant enzymes (including superoxide dismutase, catalase and glutathione peroxidase) (Bowers et al., 2004; Suzuki et al., 2013; Wong et al., 2013; Voelkel et al., 2013a) (Figure 4). It is well known that a disease such as PH enhances the production of ROS and reduces antioxidant enzyme activity (Demarco et al., 2010; Wong et al., 2013; Voelkel et al., 2013a). This results in an oxidative damage to crucial cellular components such as DNA, proteins and lipids (Demarco et al., 2010; Wong et al., 2013; Voelkel et al., 2013a). Oxidative damage elevates the levels of breakdown products during oxidative processes which can be detected in plasma, serum or urine samples of patients with PH (Demarco et al., 2010). Breakdown products and markers of oxidative stress include malondialdehyde, F2‐isoprostane, 8‐hydroxyguanosine, protein carbonyl content and nitrotyrosine (Figure 4).
Figure 4.
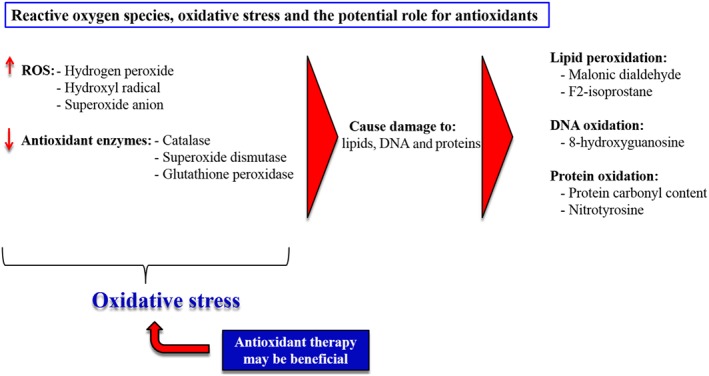
ROS and the damage they do to cellular lipids, DNA and proteins. A reduced expression and activity of antioxidant enzymes and increased ROS production lead to increased oxidative stress. Therefore, a potential role for antioxidant therapy in PH induced oxidative stress might be predicted.
As mentioned previously, oxidative stress plays an instrumental role in the pathogenesis of PH, as it can exacerbate endothelial dysfunction in the small pulmonary arteries (Bowers et al., 2004; Farber and Loscalzo, 2004; Farkas et al., 2009; Demarco et al., 2010; Summer et al., 2011). Furthermore, an array of pulmonary cells generates ROS mediated by alveolar macrophages via NADPH oxidase or mitochondrial electron transport chain (Farley et al., 2009). Another source of ROS in PH is endothelial NO synthase (eNOS) uncoupling caused by genetic mutations in the Alk‐1 gene (Jerkic et al., 2011; Dubois et al., 2013). Uncoupling between eNOS protein and NO production causes a switch in eNOS activity to generate superoxide rather than NO (Bouloumie et al., 1997). This uncoupling of eNOS and the consequent increase in ROS contribute to pulmonary vascular remodelling in PH (d'Uscio, 2011). Emerging evidence suggests that increased ROS production also contributes to RV hypertrophy and diastolic failure in PH (Rawat et al., 2014). In the monocrotaline model of PH, markers of oxidative stress (such as hydrogen peroxide and thioredoxin reductase activity) were increased in the RV but not the LV (Redout et al., 2010). These results were challenged when oxidative stress was also increased in the LVs of monocrotaline rats (Leichsenring‐Silva et al., 2011). At first, this may seem puzzling as it is known that pathological remodelling of the RV affects the LV with regard to contractility and ejection fraction (Vonk Noordegraaf et al., 1997). Taken together, these studies suggest that oxidative stress is central to the development of ventricular remodelling and builds a strong case for the use of antioxidant therapy as a RV protective treatment in PH. A number of studies have investigated the effects of various antioxidants in animal models of PH.
In a monocrotaline‐induced PH model, EUK‐134 (a superoxide dismutase and catalase mimetic) treatment given chronically on the 10th day after injection of monocrotaline attenuated RV hypertrophy and oxidative stress and prevented interstitial fibrosis (Redout et al., 2010). This suggests that EUK‐134 administration in PH at a progressed stage may provide health benefit. The attenuation of RV hypertrophy in this study was associated with improved mRNA expression of myosin heavy chain‐β. Ahmed et al. (2012) examined mice, with overexpressed extracellular superoxide dismutase that were subsequently exposed to hypoxia for 10 days (10% fraction of inspired oxygen). After the hypoxic protocol, mice had lower lung‐tissue levels of xanthine oxidase and ROS as well as improved RV hypertrophy (Ahmed et al., 2012). Resveratrol (a polyphenol compound with strong antioxidant, anti‐inflammatory and endothelial protective properties) treatment started 1 day after monocrotaline injection (25 mg·kg−1·day−1 i.p.) attenuated RV remodelling as well as oxidative stress levels in lung tissue after 21 days (Csiszar et al., 2009).
In a different study, intragastric resveratrol treatment (10 and 30 mg·kg−1 twice daily for 21 days) improved PH and RV remodelling in monocrotaline‐induced PH (50 mg·kg−1 i.p.) (Yang et al., 2010). This was associated with a reduction in cardiomyocyte apoptosis (Yang et al., 2010). Oral resveratrol treatment (3 mg·kg−1) also attenuated monocrotaline‐induced PH and RV remodelling (Paffett et al., 2012). What makes the latter study so relevant is the fact that resveratrol treatment was started on day 28 after the monocrotaline injection (50 mg·kg−1 i.p.). This is important, because at this time point, the disease is at a late stage and it therefore suggests that pharmacological antioxidant treatment may be effective in late‐stage PH. Although the authors of both studies described above did not directly measure markers of oxidative stress, it is appropriate to speculate that the beneficial effects of resveratrol may be due to its potent antioxidant properties (Suzuki et al., 2013; Wong et al., 2013; Voelkel et al., 2013a).
Melatonin, a hormone synthesized in the pineal gland and the gastrointestinal system, is a strong antioxidant. Our group recently administered melatonin to rats with monocrotaline‐induced PH and RV remodelling (Maarman et al., 2015). We were able to show that melatonin (6 mg·kg−1 in drinking water) reduced RV hypertrophy, improved cardiac function, reduced cardiac interstitial fibrosis and reduced plasma oxidative stress (Maarman et al., 2015), and this effect was still significant even if the treatment with melatonin started once the disease had already developed. These data strongly suggest that melatonin should be considered as a safe cardioprotective therapy against RV failure in PH.
It is important to mention that even though oxidative stress is detrimental in the pulmonary vasculature and in cardiac remodelling, its actions in the heart are somewhat complex (Suzuki et al., 2013; Wong et al., 2013; Voelkel et al., 2013a). ROS play two major roles in the heart, which include its ability to cause injury to cell components as well as antioxidant defence (Suzuki et al., 2013; Wong et al., 2013; Voelkel et al., 2013a). Piantadosi et al. showed that ROS play a crucial role in antioxidant‐induced cardiac mitochondrial biogenesis by liberating, stabilizing and translocating the transcription factor, nuclear respiratory factor‐2 (NRF2), to the nucleus (Piantadosi et al., 2008). Upon activation by ROS, NRF2 subsequently binds to many antioxidant‐response element motifs, which bring about regulation of mitochondrial biogenesis in conjunction with PPAR‐γ coactivator‐1‐α (Piantadosi et al., 2008). This suggests that ROS enables the cardiomyocyte to ‘sense’ damage and counteract ROS‐induced damage as an adaptive response (Piantadosi et al., 2008; Wong et al., 2013). Therefore, although antioxidant therapy in experimental PH and RV remodelling is strikingly beneficial, it presents a potential challenge as ROS play an important physiological role in cardiovascular health and pathology (Suzuki et al., 2013; Wong et al., 2013; Voelkel et al., 2013a). This physiological balance should be kept in mind when ROS scavenging or antioxidant treatments are administered, as complete depletion of ROS may be detrimental. Nevertheless, antioxidant therapy remains an attractive therapeutic target to protect the RV in PH.
Interrogation of PH animal models and their clinical translatability
The findings discussed in this review were generated in various animal models of PH, including monocrotaline, transgenic mice, pulmonary artery banding and chronic hypoxia. These findings show that the abovementioned treatment strategies are able to protect the RV in PH. However, these models do not fully develop PH or RV remodelling to the same extent humans do, and this should be considered when inferences are drawn from the data.
The monocrotaline model displays severe PH, in addition to a direct toxin effect on the myocardium resembling myocarditis, which is not observed in humans with severe PH (Gomez‐Arroyo et al., 2012). This suggests that the pathophysiology of monocrotaline‐induced PH and its effects on the RV are somewhat different to the clinical disease profile. In the light of this, it could be argued that one should carefully interpret the clinical applicability of RV protective agents tested in the monocrotaline model. However, whether or not monocrotaline induces myocarditis‐like features, it still causes severe PH and overt RV remodelling. Therefore, there is no reasonable argument against the clinical applicability of cardioprotective agents tested in the model. That being said, as with most animal studies, their findings can only be implemented in the clinical setting if supported by clinical data from well‐designed clinical trials.
Transgenic mouse models are useful in the investigation of signalling pathways that underlie PH. However, in the clinical setting, PH patients mostly display mutations in the BMPR‐2 gene or related genes. As discussed in this paper, transgenic mice were used in which a cardiac specific dominant‐negative‐IκB gene was overexpressed (Kumar et al., 2012). Considering the spectra of gene mutations involved in human PH, a model such as the IκB gene overexpression mice does not allow clinical applicability or translatability. On the contrary, findings made in these mice are not to advocate clinical implementation of experimental drugs, but they are rather used to further investigate and understand isolated molecular pathways in PH. Additionally, the model provides important information regarding the cardioprotective effect generated when pharmacological agents are used to modulate the NF‐κB/notch signalling axis.
The pulmonary artery banding model develops RV remodelling but not PH (see Maarman et al., 2013). Therefore, although it is a preferred model to study RV remodelling in isolation, it does not provide the advantage of studying RV remodelling as induced by PH. Understandably so, the lack of PH in this model may suggest that different molecular pathways are activated to bring about RV remodelling. Moreover, it could be speculated that the absence of PH may cause RV remodelling that is not as pronounced as in the presence of PH. If this speculation holds true, it could mean that cardioprotective pharmacological agents tested in this model may not have the same efficacy when tested in the clinical setting. Further research should therefore be done to assess these agents in more clinical relevant models, such as the multiple pathological‐insult models reviewed elsewhere (Maarman et al., 2013).
The chronic hypoxia‐induced PH model has been widely criticized because its PH and RV hypertrophy are reversible. Whereas this is not the case for patients with PH and affected RV (Voelkel and Tuder, 2000). This casts doubt on the true efficacy of cardioprotective pharmacological agents if returning these rats to normal oxygen can just as efficiently reverse their PH and RV remodelling. Therefore, we propose that future studies investigate the efficacy of cardioprotective pharmacological agents in rats with RV remodelling induced by both chronic hypoxia and SUGEN‐5416 (Maarman et al., 2013). It is crucial that the promising cardioprotective effects of the discussed pharmacological strategies be considered with caution. Furthermore, their safety, toxicity and tolerability should be tested to ensure that long‐term use does not cause adverse effects.
Conclusions
Preclinical research in PH has broadened its scope with a new focus on cardioprotective treatments, which could be given adjunctively to current PH pulmonary‐targeted treatments. The hope is to provide greater health benefit as these cardioprotective treatments may protect the RV, in conjunction with treatments targeted at the pulmonary circulation. We have discussed the available studies that tested cardioprotective treatments and were able to demonstrate improved cardiac function and PH symptoms. There is an important role for cardioprotective treatments in PH, but more studies are needed to confirm the benefit of these treatments in preclinical studies before translation can be considered.
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
The authors would like to thank the following entities for the financial support of their research: National Research Foundation of South Africa, South African Medical Research Council, University of Cape Town, Canon Collins Educational Trust, Pulmonary Vascular Research Institute, Harry Oppenheimer Memorial Trust and Winetech.
Maarman, G. J. , Schulz, R. , Sliwa, K. , Schermuly, R. T. , and Lecour, S. (2017) Novel putative pharmacological therapies to protect the right ventricle in pulmonary hypertension: a review of current literature. British Journal of Pharmacology, 174: 497–511. doi: 10.1111/bph.13721.
References
- Ahmed MN, Zhang Y, Codipilly C, Zaghloul N, Patel D, Wolin M et al (2012). Extracellular superoxide dismutase overexpression can reverse the course of hypoxia induced pulmonary hypertension. Mol Med 18: 38–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Overview. Br J Pharmacol 172: 5729–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Catalytic receptors. Br J Pharmacol 172: 5979–6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Transporters. Br J Pharmacol 172: 6110–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015d). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balestra GM, Mik EG, Eerbeek O, Specht PA, van der Laarse WJ, Zuurbier CJ (2015). Increased in vivo mitochondrial oxygenation with right ventricular failure induced by pulmonary arterial hypertension: mitochondrial inhibition as driver of cardiac failure? Respir Res 16: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benza RL, Gomberg‐Maitland M, Demarco T, Frost AE, Torbicki A, Langleben D et al. (2015). ET‐1 Pathway polymorphisms affect outcome in pulmonary arterial hypertension. Am J Respir Crit Care Med 192: 1345–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogaard HJ, Abe K, Vonk Noordegraaf A, Voelkel NF (2009). The right ventricle under pressure: cellular and molecular mechanisms of right‐heart failure in pulmonary hypertension. Chest 135: 794–804. [DOI] [PubMed] [Google Scholar]
- Bogaard HJ, Mizuno S, Hussaini AA, Toldo S, Abbate A, Kraskauskas D et al. (2011). Suppression of histone deacetylases worsens right ventricular dysfunction after pulmonary artery banding in rats. Am J Respir Crit Care Med 183: 1402–1410. [DOI] [PubMed] [Google Scholar]
- Boucherat O, Potus F, Bonnet S (2015). microRNA and pulmonary hypertension. Adv Exp Med Biol 888: 237–252. [DOI] [PubMed] [Google Scholar]
- Bouloumie A, Bauersachs J, Linz W, Scholkens BA, Wiemer G, Fleming I et al. (1997). Endothelial dysfunction coincides with an enhanced nitric oxide synthase expression and superoxide anion production. Hypertension 30: 934–941. [DOI] [PubMed] [Google Scholar]
- Bowers R, Cool C, Murphy RC, Tuder RM, Hopken MW, Flores SC et al. (2004). Oxidative stress in severe pulmonary hypertension. Am J Respir Crit Care Med 169: 764–769. [DOI] [PubMed] [Google Scholar]
- Brittain EL, Pugh ME, Wheeler LA, Robbins IM, Loyd JE, Newman JH et al. (2013). Prostanoids but not oral therapies improve right ventricular function in pulmonary arterial hypertension. JACC Heart Fail 1: 300–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brock M, Samillan VJ, Trenkmann M, Schwarzwald C, Ulrich S, Gay RE et al. (2014). AntagomiR directed against miR‐20a restores functional BMPR2 signalling and prevents vascular remodelling in hypoxia‐induced pulmonary hypertension. Eur Heart J 35: 3203–3211. [DOI] [PubMed] [Google Scholar]
- Bruns DR, Brown RD, Stenmark KR, Buttrick PM, Walker LA (2015). Mitochondrial integrity in a neonatal bovine model of right ventricular dysfunction. Am J Physiol Lung Cell Mol Physiol 308: L158–L167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush EW, McKinsey TA (2010). Protein acetylation in the cardiorenal axis: the promise of histone deacetylase inhibitors. Circ Res 106: 272–284. [DOI] [PubMed] [Google Scholar]
- Cao DJ, Wang ZV, Battiprolu PK, Jiang N, Morales CR, Kong Y et al. (2011). Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proc Natl Acad Sci U S A 108: 4123–4128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavasin MA, Demos‐Davies K, Horn TR, Walker LA, Lemon DD, Birdsey N et al. (2012). Selective class I histone deacetylase inhibition suppresses hypoxia‐induced cardiopulmonary remodeling through an antiproliferative mechanism. Circ Res 110: 739–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Gan XT, Haist JV, Feng Q, Lu X, Chakrabarti S et al. (2001). Attenuation of compensatory right ventricular hypertrophy and heart failure following monocrotaline‐induced pulmonary vascular injury by the Na + −H+ exchange inhibitor cariporide. J Pharmacol Exp Ther 298: 469–476. [PubMed] [Google Scholar]
- Cho YK, Eom GH, Kee HJ, Kim HS, Choi WY, Nam KI et al. (2010). Sodium valproate, a histone deacetylase inhibitor, but not captopril, prevents right ventricular hypertrophy in rats. Circ J 74: 760–770. [DOI] [PubMed] [Google Scholar]
- Cingolani HE, Ennis IL (2007). Sodium‐hydrogen exchanger, cardiac overload, and myocardial hypertrophy. Circulation 115: 1090–1100. [DOI] [PubMed] [Google Scholar]
- Csiszar A, Labinskyy N, Olson S, Pinto JT, Gupte S, Wu JM et al. (2009). Resveratrol prevents monocrotaline‐induced pulmonary hypertension in rats. Hypertension 54: 668–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d'Uscio LV (2011). eNOS uncoupling in pulmonary hypertension. Cardiovasc Res 92: 359–360. [DOI] [PubMed] [Google Scholar]
- Das M (2007). Apoptosis as a therapeutic target in heart failure. Am J Physiol Heart Circ Physiol 293: H1322–H1323. [DOI] [PubMed] [Google Scholar]
- Demarco VG, Whaley‐Connell AT, Sowers JR, Habibi J, Dellsperger KC (2010). Contribution of oxidative stress to pulmonary arterial hypertension. World J Cardiol 2: 316–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dromparis P, Sutendra G, Michelakis ED (2010). The role of mitochondria in pulmonary vascular remodeling. J Mol Med 88: 1003–1010. [DOI] [PubMed] [Google Scholar]
- Dubois M, Delannoy E, Duluc L, Closs E, Li H, Toussaint C et al. (2013). Biopterin metabolism and eNOS expression during hypoxic pulmonary hypertension in mice. PLoS One 8: e82594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ecarnot‐Laubriet A, Assem M, Poirson‐Bichat F, Moisant M, Bernard C, Lecour S et al. (2002). Stage‐dependent activation of cell cycle and apoptosis mechanisms in the right ventricle by pressure overload. Biochim Biophys Acta 1586: 233–242. [DOI] [PubMed] [Google Scholar]
- Enache I, Charles AL, Bouitbir J, Favret F, Zoll J, Metzger D et al. (2013). Skeletal muscle mitochondrial dysfunction precedes right ventricular impairment in experimental pulmonary hypertension. Mol Cell Biochem 373: 161–170. [DOI] [PubMed] [Google Scholar]
- Fan J, Fan X, Li Y, Ding L, Zheng Q, Guo J et al. (2016). Chronic normobaric hypoxia induces pulmonary hypertension in rats: role of NF‐kappaB. High Alt Med Biol 17: 43–49. [DOI] [PubMed] [Google Scholar]
- Farber HW, Loscalzo J (2004). Pulmonary arterial hypertension. N Engl J Med 351: 1655–1665. [DOI] [PubMed] [Google Scholar]
- Farkas L, Farkas D, Ask K, Moller A, Gauldie J, Margetts P et al. (2009). VEGF ameliorates pulmonary hypertension through inhibition of endothelial apoptosis in experimental lung fibrosis in rats. J Clin Invest 119: 1298–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farley KS, Wang L, Mehta S (2009). Septic pulmonary microvascular endothelial cell injury: role of alveolar macrophage NADPH oxidase. Am J Physiol Lung Cell Mol Physiol 296: L480–L488. [DOI] [PubMed] [Google Scholar]
- Freund‐Michel V, Khoyrattee N, Savineau JP, Muller B, Guibert C (2014). Mitochondria: roles in pulmonary hypertension. Int J Biochem Cell Biol 55: 93–97. [DOI] [PubMed] [Google Scholar]
- Galie N, Hoeper MM, Humbert M, Torbicki A, Vachiery JL, Barbera JA et al. (2009). Guidelines for the diagnosis and treatment of pulmonary hypertension: the task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Heart J 30: 2493–2537. [DOI] [PubMed] [Google Scholar]
- Galie N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A et al. (2016). 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: the joint task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J 37: 67–119. [DOI] [PubMed] [Google Scholar]
- Garciarena CD, Caldiz CI, Correa MV, Schinella GR, Mosca SM, Chiappe de Cingolani GE et al. (2008). Na+/H+ exchanger‐1 inhibitors decrease myocardial superoxide production via direct mitochondrial action. J Appl Physiol 105: 1706–1713. [DOI] [PubMed] [Google Scholar]
- Gomberg‐Maitland M, Dufton C, Oudiz RJ, Benza RL (2011). Compelling evidence of long‐term outcomes in pulmonary arterial hypertension? A clinical perspective. J Am Coll Cardiol 57: 1053–1061. [DOI] [PubMed] [Google Scholar]
- Gomez‐Arroyo J, Mizuno S, Szczepanek K, Van Tassell B, Natarajan R, dos Remedios CG et al. (2013). Metabolic gene remodeling and mitochondrial dysfunction in failing right ventricular hypertrophy secondary to pulmonary arterial hypertension. Circ Heart Fail 6: 136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez‐Arroyo JG, Farkas L, Alhussaini AA, Farkas D, Kraskauskas D, Voelkel NF et al. (2012). The monocrotaline model of pulmonary hypertension in perspective. Am J Physiol Lung Cell Mol Physiol 302: L363–L369. [DOI] [PubMed] [Google Scholar]
- Graham BB, Bandeira AP, Morrell NW, Butrous G, Tuder RM (2010). Schistosomiasis‐associated pulmonary hypertension: pulmonary vascular disease: the global perspective. Chest 137: 20S–29S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarini G, Huqi A, Morrone D, Marzilli M (2016). pharmacological agents targeting myocardial metabolism for the management of chronic stable angina: an update. Cardiovasc Drugs Ther 30: 379–391. [DOI] [PubMed] [Google Scholar]
- Gude NA, Emmanuel G, Wu W, Cottage CT, Fischer K, Quijada P et al. (2008). Activation of Notch‐mediated protective signaling in the myocardium. Circ Res 102: 1025–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handoko ML, de Man FS, Allaart CP, Paulus WJ, Westerhof N, Vonk‐Noordegraaf A (2010). Perspectives on novel therapeutic strategies for right heart failure in pulmonary arterial hypertension: lessons from the left heart. Eur Respir Rev 19: 72–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeper MM (2015). Pharmacological therapy for patients with chronic thromboembolic pulmonary hypertension. Eur Respir Rev 24: 272–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humbert M, Ghofrani HA (2015). The molecular targets of approved treatments for pulmonary arterial hypertension. Thorax 0: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V et al. (2006). Pulmonary arterial hypertension in France: results from a national registry. Am J Respir Crit Care Med 173: 1023–1030. [DOI] [PubMed] [Google Scholar]
- Jerkic M, Kabir MG, Davies A, Yu LX, McIntyre BA, Husain NW et al. (2011). Pulmonary hypertension in adult Alk1 heterozygous mice due to oxidative stress. Cardiovasc Res 92: 375–384. [DOI] [PubMed] [Google Scholar]
- Jurasz P, Courtman D, Babaie S, Stewart DJ (2010). Role of apoptosis in pulmonary hypertension: from experimental models to clinical trials. Pharmacol Ther 126: 1–8. [DOI] [PubMed] [Google Scholar]
- Karmazyn M (1999). The role of the myocardial sodium‐hydrogen exchanger in mediating ischemic and reperfusion injury. From amiloride to cariporide. Ann N Y Acad Sci 874: 326–334. [DOI] [PubMed] [Google Scholar]
- Karmazyn M, Kilic A, Javadov S (2008). The role of NHE‐1 in myocardial hypertrophy and remodelling. J Mol Cell Cardiol 44: 647–653. [DOI] [PubMed] [Google Scholar]
- Kepp O, Galluzzi L, Kroemer G (2011). Mitochondrial control of the NLRP3 inflammasome. Nat Immunol 12: 199–200. [DOI] [PubMed] [Google Scholar]
- Kumar S, Wei C, Thomas CM, Kim IK, Seqqat R, Kumar R et al. (2012). Cardiac‐specific genetic inhibition of nuclear factor‐kappaB prevents right ventricular hypertrophy induced by monocrotaline. Am J Physiol Heart Circ Physiol 302: H1655–H1666. [DOI] [PubMed] [Google Scholar]
- Lam CS, Borlaug BA, Kane GC, Enders FT, Rodeheffer RJ, Redfield MM (2009). Age‐associated increases in pulmonary artery systolic pressure in the general population. Circulation 119: 2663–2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang I, Gomez‐Sanchez M, Kneussl M, Naeije R, Escribano P, Skoro‐Sajer N et al. (2006). Efficacy of long‐term subcutaneous treprostinil sodium therapy in pulmonary hypertension. Chest 129: 1636–1643. [DOI] [PubMed] [Google Scholar]
- Latronico MV, Condorelli G (2011). microRNAs in hypertrophy and heart failure. Exp Biol Med 236: 125–131. [DOI] [PubMed] [Google Scholar]
- Lee PJ, Rudenko D, Kuliszewski MA, Liao C, Kabir MG, Connelly KA et al. (2014). Survivin gene therapy attenuates left ventricular systolic dysfunction in doxorubicin cardiomyopathy by reducing apoptosis and fibrosis. Cardiovasc Res 101: 423–433. [DOI] [PubMed] [Google Scholar]
- Leichsenring‐Silva F, Tavares AM, Mosele F, Berger B, Llesuy S, Bello‐Klein A (2011). Association of the time course of pulmonary arterial hypertension with changes in oxidative stress in the left ventricle. Clin Exp Pharmacol Physiol 38: 804–810. [DOI] [PubMed] [Google Scholar]
- Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC (2010). Myocardial fatty acid metabolism in health and disease. Physiol Rev 90: 207–258. [DOI] [PubMed] [Google Scholar]
- Maarman G, Blackhurst D, Thienemann F, Blauwet L, Butrous G, Davies N et al. (2015). Melatonin as a preventive and curative therapy against pulmonary hypertension. J Pineal Res 59: 343–353. [DOI] [PubMed] [Google Scholar]
- Maarman G, Lecour S, Butrous G, Thienemann F, Sliwa K (2013). A comprehensive review: the evolution of animal models in pulmonary hypertension research; are we there yet? Pulm Circ 3: 739–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maarman G, Marais E, Lochner A, du Toit EF (2012). Effect of chronic CPT‐1 inhibition on myocardial ischemia–reperfusion injury (I/R) in a model of diet‐induced obesity. Cardiovasc Drugs Ther 26: 205–216. [DOI] [PubMed] [Google Scholar]
- Magne J, Pibarot P, Sengupta PP, Donal E, Rosenhek R, Lancellotti P (2015). Pulmonary hypertension in valvular disease: a comprehensive review on pathophysiology to therapy from the HAVEC Group. JACC Cardiovasc Imaging 8: 83–99. [DOI] [PubMed] [Google Scholar]
- Marsboom G, Toth PT, Ryan JJ, Hong Z, Wu X, Fang YH et al. (2012). Dynamin‐related protein 1‐mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ Res 110: 1484–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinsey TA (2011). The biology and therapeutic implications of HDACs in the heart. Handb Exp Pharmacol 206: 57–78. [DOI] [PubMed] [Google Scholar]
- Mocumbi AO, Thienemann F, Sliwa K (2015). A global perspective on the epidemiology of pulmonary hypertension. Can J Cardiol 31: 375–381. [DOI] [PubMed] [Google Scholar]
- Moor AN, Fliegel L (1999). Protein kinase‐mediated regulation of the Na(+)/H(+) exchanger in the rat myocardium by mitogen‐activated protein kinase‐dependent pathways. J Biol Chem 274: 22985–22992. [DOI] [PubMed] [Google Scholar]
- Naeije R, Manes A (2014). The right ventricle in pulmonary arterial hypertension. Eur Respir Rev 23: 476–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naga Prasad SV, Duan ZH, Gupta MK, Surampudi VS, Volinia S, Calin GA et al. (2009). Unique microRNA profile in end‐stage heart failure indicates alterations in specific cardiovascular signaling networks. J Biol Chem 284: 27487–27499. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Niessen K, Karsan A (2008). Notch signaling in cardiac development. Circ Res 102: 1169–1181. [DOI] [PubMed] [Google Scholar]
- Nural‐Guvener HF, Zakharova L, Nimlos J, Popovic S, Mastroeni D, Gaballa MA (2014). HDAC class I inhibitor, mocetinostat, reverses cardiac fibrosis in heart failure and diminishes CD90+ cardiac myofibroblast activation. Fibrogenesis Tissue Repair 7: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opie LH, Commerford PJ, Gersh BJ, Pfeffer MA (2006). Controversies in ventricular remodelling. Lancet 367: 356–367. [DOI] [PubMed] [Google Scholar]
- Oudiz RJ, Galie N, Olschewski H, Torres F, Frost A, Ghofrani HA et al. (2009). Long‐term ambrisentan therapy for the treatment of pulmonary arterial hypertension. J Am Coll Cardiol 54: 1971–1981. [DOI] [PubMed] [Google Scholar]
- Paffett ML, Lucas SN, Campen MJ (2012). Resveratrol reverses monocrotaline‐induced pulmonary vascular and cardiac dysfunction: a potential role for atrogin‐1 in smooth muscle. Vascul Pharmacol 56: 64–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papamatheakis DG, Mocumbi AO, Kim NH, Mandel J (2014). Schistosomiasis‐associated pulmonary hypertension. Pulm Circ 4: 596–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piantadosi CA, Carraway MS, Babiker A, Suliman HB (2008). Heme oxygenase‐1 regulates cardiac mitochondrial biogenesis via Nrf2‐mediated transcriptional control of nuclear respiratory factor‐1. Circ Res 103: 1232–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piao L, Fang YH, Cadete VJ, Wietholt C, Urboniene D, Toth PT et al. (2010). The inhibition of pyruvate dehydrogenase kinase improves impaired cardiac function and electrical remodeling in two models of right ventricular hypertrophy: resuscitating the hibernating right ventricle. J Mol Med 88: 47–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter AG, Janicke RU (1999). Emerging roles of caspase‐3 in apoptosis. Cell Death Differ 6: 99–104. [DOI] [PubMed] [Google Scholar]
- Prins KW, Thenappan T (2016). World Health Organization group I pulmonary hypertension: epidemiology and pathophysiology. Cardiol Clin 34: 363–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pullamsetti SS, Doebele C, Fischer A, Savai R, Kojonazarov B, Dahal BK et al. (2012). Inhibition of microRNA‐17 improves lung and heart function in experimental pulmonary hypertension. Am J Respir Crit Care Med 185: 409–419. [DOI] [PubMed] [Google Scholar]
- Rabinovitch M (2012). Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest 122: 4306–4313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawat DK, Alzoubi A, Gupte R, Chettimada S, Watanabe M, Kahn AG et al. (2014). Increased reactive oxygen species, metabolic maladaptation, and autophagy contribute to pulmonary arterial hypertension‐induced ventricular hypertrophy and diastolic heart failure. Hypertension 64: 1266–1274. [DOI] [PubMed] [Google Scholar]
- Redout EM, van der Toorn A, Zuidwijk MJ, van de Kolk CW, van Echteld CJ, Musters RJ et al. (2010). Antioxidant treatment attenuates pulmonary arterial hypertension‐induced heart failure. Am J Physiol Heart Circ Physiol 298: H1038–H1047. [DOI] [PubMed] [Google Scholar]
- Rizzo P, Miele L, Ferrari R (2013). The notch pathway: a crossroad between the life and death of the endothelium. Eur Heart J 34: 2504–2509. [DOI] [PubMed] [Google Scholar]
- Rusanescu G, Weissleder R, Aikawa E (2008). Notch signaling in cardiovascular disease and calcification. Curr Cardiol Rev 4: 148–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan J, Dasgupta A, Huston J, Chen KH, Archer SL (2015). Mitochondrial dynamics in pulmonary arterial hypertension. J Mol Med 93: 229–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan JJ, Archer SL (2014). The right ventricle in pulmonary arterial hypertension: disorders of metabolism, angiogenesis and adrenergic signaling in right ventricular failure. Circ Res 115: 176–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan JJ, Archer SL (2015). Emerging concepts in the molecular basis of pulmonary arterial hypertension: part I: metabolic plasticity and mitochondrial dynamics in the pulmonary circulation and right ventricle in pulmonary arterial hypertension. Circulation 131: 1691–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan JJ, Marsboom G, Fang YH, Toth PT, Morrow E, Luo N et al. (2013). PGC1alpha‐mediated mitofusin‐2 deficiency in female rats and humans with pulmonary arterial hypertension. Am J Respir Crit Care Med 187: 865–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakao S, Miyauchi H, Voelkel NF, Sugiura T, Tanabe N, Kobayashi Y et al. (2015). Increased right ventricular fatty acid accumulation in chronic thromboembolic pulmonary hypertension. Ann Am Thorac Soc 12: 1465–1472. [DOI] [PubMed] [Google Scholar]
- Shah AM, Mann DL (2011). In search of new therapeutic targets and strategies for heart failure: recent advances in basic science. Lancet 378: 704–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen T, Zheng M, Cao C, Chen C, Tang J, Zhang W et al. (2007). Mitofusin‐2 is a major determinant of oxidative stress‐mediated heart muscle cell apoptosis. J Biol Chem 282: 23354–23361. [DOI] [PubMed] [Google Scholar]
- Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, Ghofrani A et al. (2013). Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 62: D34–D41. [DOI] [PubMed] [Google Scholar]
- Simonneau G, Robbins IM, Beghetti M, Channick RN, Delcroix M, Denton CP et al. (2009). Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 54: S43–S54. [DOI] [PubMed] [Google Scholar]
- Sliwa K, Wilkinson D, Hansen C, Ntyintyane L, Tibazarwa K, Becker A et al. (2008). Spectrum of heart disease and risk factors in a black urban population in South Africa (the Heart of Soweto Study): a cohort study. Lancet 371: 915–922. [DOI] [PubMed] [Google Scholar]
- Snabaitis AK, D'Mello R, Dashnyam S, Avkiran M (2006). A novel role for protein phosphatase 2A in receptor‐mediated regulation of the cardiac sarcolemmal Na+/H+ exchanger NHE1. J Biol Chem 281: 20252–20262. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44 (Database Issue): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speich R, Ulrich S, Domenighetti G, Huber LC, Fischler M, Treder U et al. (2015). Efficacy and safety of long‐term imatinib therapy for pulmonary arterial hypertension. Respiration 89: 515–524. [DOI] [PubMed] [Google Scholar]
- Stanley WC (2004). Myocardial energy metabolism during ischemia and the mechanisms of metabolic therapies. J Cardiovasc Pharmacol Ther 9 (Suppl 1): S31–S45. [DOI] [PubMed] [Google Scholar]
- Stanley WC, Lopaschuk GD, McCormack JG (1997). Regulation of energy substrate metabolism in the diabetic heart. Cardiovasc Res 34: 25–33. [DOI] [PubMed] [Google Scholar]
- Stanley WC, Recchia FA, Lopaschuk GD (2005). Myocardial substrate metabolism in the normal and failing heart. Physiol Rev 85: 1093–1129. [DOI] [PubMed] [Google Scholar]
- Stewart S, Mocumbi AO, Carrington MJ, Pretorius S, Burton R, Sliwa K (2011). A not‐so‐rare form of heart failure in urban black Africans: pathways to right heart failure in the Heart of Soweto Study cohort. Eur J Heart Fail 13: 1070–1077. [DOI] [PubMed] [Google Scholar]
- Strange G, Playford D, Stewart S, Deague JA, Nelson H, Kent A et al. (2012). Pulmonary hypertension: prevalence and mortality in the Armadale echocardiography cohort. Heart 98: 1805–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summer R, Walsh K, Medoff BD (2011). Obesity and pulmonary arterial hypertension: is adiponectin the molecular link between these conditions? Pulm Circ 1: 440–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun XQ, Zhang R, Zhang HD, Yuan P, Wang XJ, Zhao QH et al. (2016). Reversal of right ventricular remodeling by dichloroacetate is related to inhibition of mitochondria‐dependent apoptosis. Hypertens Res 39: 302–311. [DOI] [PubMed] [Google Scholar]
- Sutendra G, Dromparis P, Paulin R, Zervopoulos S, Haromy A, Nagendran J et al. (2013). A metabolic remodeling in right ventricular hypertrophy is associated with decreased angiogenesis and a transition from a compensated to a decompensated state in pulmonary hypertension. J Mol Med 91: 1315–1327. [DOI] [PubMed] [Google Scholar]
- Sutendra G, Michelakis ED (2014). The metabolic basis of pulmonary arterial hypertension. Cell Metab 19: 558–573. [DOI] [PubMed] [Google Scholar]
- Suzuki YJ, Steinhorn RH, Gladwin MT (2013). Antioxidant therapy for the treatment of pulmonary hypertension. Antioxid Redox Signal 18: 1723–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sztrymf B, Souza R, Bertoletti L, Jais X, Sitbon O, Price LC et al. (2010). Prognostic factors of acute heart failure in patients with pulmonary arterial hypertension. Eur Respir J 35: 1286–1293. [DOI] [PubMed] [Google Scholar]
- Taegtmeyer H (2000). Metabolism – the lost child of cardiology. J Am Coll Cardiol 36: 1386–1388. [DOI] [PubMed] [Google Scholar]
- Taegtmeyer H (2004). Cardiac metabolism as a target for the treatment of heart failure. Circulation 110: 894–896. [DOI] [PubMed] [Google Scholar]
- Talati M, Hemnes A (2015). Fatty acid metabolism in pulmonary arterial hypertension: role in right ventricular dysfunction and hypertrophy. Pulm Circ 5: 269–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Iqbal M, Cawthon D, Bottje WG (2002). Heart and breast muscle mitochondrial dysfunction in pulmonary hypertension syndrome in broilers (Gallus domesticus). Comp Biochem Physiol A Mol Integr Physiol 132: 527–540. [DOI] [PubMed] [Google Scholar]
- Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT) , Galie N et al. (2009). Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J 34: 1219–1263. [DOI] [PubMed] [Google Scholar]
- Thienemann F, Dzudie A, Mocumbi AO, Blauwet L, Sani MU, Karaye KM et al. (2014). Rationale and design of the Pan African pulmonary hypertension Cohort (PAPUCO) study: implementing a contemporary registry on pulmonary hypertension in Africa. BMJ Open 4: e005950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuder RM, Davis LA, Graham BB (2012). Targeting energetic metabolism: a new frontier in the pathogenesis and treatment of pulmonary hypertension. Am J Respir Crit Care Med 185: 260–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umar S, Lee JH, de Lange E, Iorga A, Partow‐Navid R, Bapat A et al. (2012). Spontaneous ventricular fibrillation in right ventricular failure secondary to chronic pulmonary hypertension. Circ Arrhythm Electrophysiol 5: 181–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rooij E, Olson EN (2012). MicroRNA therapeutics for cardiovascular disease: opportunities and obstacles. Nat Rev Drug Discov 11: 860–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voelkel NF, Bogaard HJ, Al Husseini A, Farkas L, Gomez‐Arroyo J, Natarajan R (2013a). Antioxidants for the treatment of patients with severe angioproliferative pulmonary hypertension? Antioxid Redox Signal 18: 1810–1817. [DOI] [PubMed] [Google Scholar]
- Voelkel NF, Gomez‐Arroyo J, Abbate A, Bogaard HJ (2013b). Mechanisms of right heart failure – a work in progress and a plea for failure prevention. Pulm Circ 3: 137–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voelkel NF, Gomez‐Arroyo J, Abbate A, Bogaard HJ, Nicolls MR (2012). Pathobiology of pulmonary arterial hypertension and right ventricular failure. Eur Respir J 40: 1555–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voelkel NF, Quaife RA, Leinwand LA, Barst RJ, McGoon MD, Meldrum DR et al. (2006). Right ventricular function and failure: report of a National Heart, Lung, and Blood Institute working group on cellular and molecular mechanisms of right heart failure. Circulation 114: 1883–1891. [DOI] [PubMed] [Google Scholar]
- Voelkel NF, Tuder RM (2000). Hypoxia‐induced pulmonary vascular remodeling: a model for what human disease? J Clin Invest 106: 733–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonk‐Noordegraaf A, Haddad F, Chin KM, Forfia PR, Kawut SM, Lumens J et al. (2013). Right heart adaptation to pulmonary arterial hypertension: physiology and pathobiology. J Am Coll Cardiol 62: D22–D33. [DOI] [PubMed] [Google Scholar]
- Vonk Noordegraaf A, Galie N (2011). The role of the right ventricle in pulmonary arterial hypertension. Eur Respir Rev 20: 243–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonk Noordegraaf A, Marcus JT, Roseboom B, Postmus PE, Faes TJ, de Vries PM (1997). The effect of right ventricular hypertrophy on left ventricular ejection fraction in pulmonary emphysema. Chest 112: 640–645. [DOI] [PubMed] [Google Scholar]
- Wang H, Silva NL, Lucchesi PA, Haworth R, Wang K, Michalak M et al. (1997). Phosphorylation and regulation of the Na+/H+ exchanger through mitogen‐activated protein kinase. Biochemistry 36: 9151–9158. [DOI] [PubMed] [Google Scholar]
- Wang J, Hu X, Jiang H (2015a). HDAC inhibition: a novel therapeutic target for attenuating myocardial ischemia and reperfusion injury by reversing cardiac remodeling. Int J Cardiol 190: 126–127. [DOI] [PubMed] [Google Scholar]
- Wang J, Saren G, Jiang H (2015b). HDAC inhibition: a novel therapeutic target for attenuating pulmonary hypertension by regulating Tregs. Int J Cardiol 198: 176–177. [DOI] [PubMed] [Google Scholar]
- Wang RC, Jiang FM, Zheng QL, Li CT, Peng XY, He CY et al. (2014). Efficacy and safety of sildenafil treatment in pulmonary arterial hypertension: a systematic review. Respir Med 108: 531–537. [DOI] [PubMed] [Google Scholar]
- Wang X, Zhang X, Ren XP, Chen J, Liu H, Yang J et al. (2010). MicroRNA‐494 targeting both proapoptotic and antiapoptotic proteins protects against ischemia/reperfusion‐induced cardiac injury. Circulation 122: 1308–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir EK, Lopez‐Barneo J, Buckler KJ, Archer SL (2005). Acute oxygen‐sensing mechanisms. N Engl J Med 353: 2042–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins MR (2012). Pulmonary hypertension: the science behind the disease spectrum. Eur Respir Rev 21: 19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong CM, Bansal G, Pavlickova L, Marcocci L, Suzuki YJ (2013). Reactive oxygen species and antioxidants in pulmonary hypertension. Antioxid Redox Signal 18: 1789–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrobel JP, Thompson BR, Williams TJ (2012). Mechanisms of pulmonary hypertension in chronic obstructive pulmonary disease: a pathophysiologic review. J Heart Lung Transplant 31: 557–564. [DOI] [PubMed] [Google Scholar]
- Yang DL, Zhang HG, Xu YL, Gao YH, Yang XJ, Hao XQ et al. (2010). Resveratrol inhibits right ventricular hypertrophy induced by monocrotaline in rats. Clin Exp Pharmacol Physiol 37: 150–155. [DOI] [PubMed] [Google Scholar]
- Yaoita H, Ogawa K, Maehara K, Maruyama Y (1998). Attenuation of ischemia/reperfusion injury in rats by a caspase inhibitor. Circulation 97: 276–281. [DOI] [PubMed] [Google Scholar]
- Yu H, Guo Y, Mi L, Wang X, Li L, Gao W (2011). Mitofusin 2 inhibits angiotensin II‐induced myocardial hypertrophy. J Cardiovasc Pharmacol Ther 16: 205–211. [DOI] [PubMed] [Google Scholar]
- Zhang CL, McKinsey TA, Chang S, Antos CL, Hill JA, Olson EN (2002). Class II histone deacetylases act as signal‐responsive repressors of cardiac hypertrophy. Cell 110: 479–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao ZQ, Morris CD, Budde JM, Wang NP, Muraki S, Sun HY et al. (2003). Inhibition of myocardial apoptosis reduces infarct size and improves regional contractile dysfunction during reperfusion. Cardiovasc Res 59: 132–142. [DOI] [PubMed] [Google Scholar]
- Zungu‐Edmondson M, Shults NV, Wong CM, Suzuki YJ (2016). Modulators of right ventricular apoptosis and contractility in a rat model of pulmonary hypertension. Cardiovasc Res 110: 30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]