

ABSTRACT
Extracellular vesicles (EVs) have been the focus of great interest, as they appear to be involved in numerous important cellular processes. They deliver bioactive macromolecules such as proteins, lipids, and nucleic acids, allowing intercellular communication in multicellular organisms. EVs are secreted by all cell types, including immune cells such as natural killer cells (NK), and they may play important roles in the immune system. Currently, a large-scale procedure to obtain functional NK EVs is lacking, limiting their use clinically. In this report, we present a simple, robust, and cost-effective method to isolate a large quantity of NK EVs. After propagating and activating NK cells ex vivo and then incubating them in exosome-free medium for 48 h, EVs were isolated using a polymer precipitation method. The isolated vesicles contain the tetraspanin CD63, an EV marker, and associated proteins (fibronectin), but are devoid of cytochrome C, a cytoplasmic marker. Nanoparticle tracking analysis showed a size distribution between 100 and 200 nm while transmission electron microscopy imaging displayed vesicles with an oval shape and comparable sizes, fulfilling the definition of EV. Importantly, isolated EV fractions were cytotoxic against cancer cells. Furthermore, our results demonstrate for the first time that isolated activated NK (aNK) cell EVs contain the cytotoxic proteins perforin, granulysin, and granzymes A and B, incorporated from the aNK cells. Activation of caspase -3, -7 and -9 was detected in cancer cells incubated with aNK EVs, and caspase inhibitors blocked aNK EV-induced cytotoxicity, suggesting that aNK EVs activate caspase pathways in target cells. The ability to isolate functional aNK EVs on a large scale may lead to new clinical applications.
Abbreviations: NK: natural killer cells; activated NK (aNK) cells; EVs: extracellular vesicles; ALL: acute lymphoblastic leukaemia; aAPC: artificial antigen-presenting cell; TEM: transmission electron microscope; PBMC: peripheral blood mononuclear cells; FBS: foetal bovine serum.
KEYWORDS: Scale-up isolation, natural killer cells, extracellular vesicles, cytotoxicity, caspases, cancer treatment
Introduction
Extracellular vesicles (EVs) such as exosomes, microvesicles and apoptotic bodies are released by cells in vitro into culture medium. They also are released naturally in vivo, and have been found in blood, urine, amniotic fluid, breast milk, seminal fluid, saliva and malignant effusions.[1,2] Released vesicles are heterogeneous in size and intracellular in origin. Exosomes are generated through fusion of the plasma membrane with specific endosomal compartments called multivesicular bodies, whereas microvesicles are formed by outward budding from the plasma membrane. Further differentiation between these two types of vesicles is difficult; thus, we have used the term “EV” throughout this report. They have been implicated in cell–cell communication, for delivery of genes, and as disease biomarkers.[3,4] All EVs, whatever their cellular origin, share a characteristic composition of proteins and lipids. They may have some common markers, like major histocompatibility complex (MHC), flotillin, and heat shock 70-kDa proteins. They are also enriched in tetraspanins (CD9, CD63, CD81), cytoskeletal proteins (actin, tubulin), and certain lipids such as phosphatidylserine, ceramide, and cholesterol.[5] EVs also contain specific proteins depending on the cell of origin; for example, EVs from tumour cells contain tumour antigens, platelet-derived exosomes contain coagulation factors, and exosomes from dendritic cells express MHC II–peptide complexes.[6] In addition, EVs can contain microRNAs and mRNAs in different proportions than in their cell of origin.[7,8] EV can be used by immune cells and other cell types as devices for communication via the direct exchange of proteins and RNA between cells at a distance,[9] thereby exerting numerous functions in physiology and pathology.
Isolation and analysis of EV are essential for understanding their biological roles and for investigating their potential clinical use. Several methods have been developed thus far. Ultracentrifugation is currently the most commonly used method to obtain EV.[10] It is simple and straightforward, but has its limitations, which include a low yield, and the possible aggregation, fusion and/or disruption of the vesicles due to the centrifugal force. Buoyant density ultracentrifugation is a more stringent isolation procedure that yields EV of higher purity; however, it not well suited to scale-up.[11] Filtration has often been used in combination with ultracentrifugation-based methods. It is fast, simple and scalable, but it also presents limitations such as non-specific binding of abundant soluble proteins and vesicles to the membrane surfaces and clogging of the filters.[12] The more recently developed methods, tangential flow filtration [13] and microfluidic filtration,[14] may overcome some of these drawbacks. However, the resulting EV will be diluted and will need to be concentrated for most applications, a process that may result in considerable loss. Precipitation with agglutinating agents has been applied for EV isolation.[15] The yield is high, yet the resulting EV fraction is not as pure, due to co-isolation of other components. Commercial kits utilising polymers or related agglutinating agents are available to precipitate EV from sample specimens, but these usually are for small-scale isolation. Chromatography-based methods are a rapidly growing field in EV purification. A variety of affinity ligands (antibodies, synthetic peptides, heparins and lectins) and surfaces have been used to immobilise vesicles,[16] and the use of immunoaffinity chromatography with antibodies may result in the isolation of different tissue-specific EV subtypes.[17] The quality of the antibody is critical for this method because non-specific binding is often observed. Size exclusion chromatography has become widely used, as the process is gentle and reproducible,[18] and it allows the purification of biologically active EV. However, the resolution of the size exclusion columns is low and the resulting sample may be diluted several fold, which becomes problematic when applied to large-scale isolations. Thus, each current methodology has its own advantages and limitations, and a combination of more than one method is commonly used to isolate EVs. Nevertheless, a simple, robust, and high throughput method to isolate sufficient amounts of bioactive EVs that could be used for clinical applications is still lacking.
Natural killer (NK) cells are innate immune effector cells that contain lytic granules that are released to induce cellular cytotoxicity upon stimulation. NK cells are currently being evaluated for cell-based cancer treatment. Nevertheless, despite the potency of NK cells in pre-clinical studies, clinical trials have produced mixed results, and significant side effects have been reported. For example, allogeneic activated NK (aNK) cells were associated with severe graft-versus-host disease in paediatric cancer patients despite HLA matching.[19] Human NK cells release EVs that contain typical NK cell markers (e.g. CD56) and proteins (e.g. FASL, perforin) that can induce target cell death.[20] This raises the possibility that EV-based delivery of perforin and/or other cytotoxic molecules might be exploited as an entirely new approach to treat cancer.[21] Recent studies have shown that co-culturing peripheral blood mononuclear cells (PBMC) with artificial antigen-presenting cells (aAPC) can induce significant propagation (>2000-fold) and activation of NK cells (aNK).[22,23] We have demonstrated that these aNK cells are highly cytotoxic, either alone or with anti-disialoganglioside (GD2) mAb ch14.18/dinutuximab, against multidrug sensitive and resistant neuroblastoma cell lines in vitro and secrete an array of cytokines and chemokines with antitumour potential while mediating antibody-dependent cellular cytotoxicity (ADCC). These aNK cells maintain their functional activities after viable cryopreservation, and, most importantly, retain potent antitumour activity with mAb ch14.18 when infused intravenously immediately after thawing into non-obese diabetic/severe combined immunodeficient (NOD/SCID) mice with disseminated human neuroblastoma. We hypothesised that large-scale propagation and activation of human NK cells could be used to produce cytotoxic EVs.
In this report, we show for the first time that a large quantity of functional aNK cell-derived EVs can be obtained by scaling-up the growth of aNK cells in the presence of the artificial aAPC, K562-mbIL21. Moreover, these EVs contain several cytotoxic molecules and exhibit significant cytotoxicity against cancer cell lines. Furthermore, caspase-related pathways are activated in target cells after incubation with aNK cell-derived EVs. The large-scale isolation of cytotoxic aNK EVs should lead to new strategies for clinical exploitation.[24]
Materials and methods
Reagents and materials
Histopaque-1077, polyethylene glycol-8000, and OptiPrep™ Density Gradient medium (60% iodixanol solution) were purchased from Sigma-Aldrich Chem. Co. (Saint Louis, MO, USA). Interleukin-2 was obtained from PeproTech (Rocky Hill, NJ, USA). AlbuRx 25 (25% solution of human albumin) was obtained from CSL Behring (King of Prussia, PA, USA) (NDC number 44206-251-10). Protein concentration was determined by the BioRad Bradford assay. The G-Rex cell culture device was purchased from Wilson Wolf Manufacturing Corporation (New Brighton, MN, USA).
Isolation of activated NK EVs from ex vivo NK cell culture
Thirty to 50 ml of blood was drawn from each donor under a protocol approved by the Institutional Review Board at Children’s Hospital Los Angeles (Los Angeles, CA, USA). Peripheral blood mononuclear cells (PBMC) were isolated by density separation using Histopaque-1077. The K562 Clone 9.mbIL21 cells (clinical-grade master cell bank designated CJLCKT64.86.41BBL.CD19.mbIL21) that were used for NK cell propagation and activation express a membrane-bound variant of IL-21.[25] Before initiating the K562 Clone 9.mbIL-21 aAPC and PBMC co-cultures on day 0, the aAPC were γ-irradiated (100 G) and then suspended in RPMI-1640 and 10% foetal bovine serum (FBS) supplemented with 50 IU ml–1 recombinant human IL-2 (PeproTech, Rocky Hill, NJ, USA). On day 0, PBMC from normal donors were incubated with aAPC at a 1:1 ratio (2 × 107 of each cell type) in 400 ml of RPMI-1640 with 10% FBS in a G-Rex100 culture device. After seven days of co-culture, cells were counted; remaining T cells were removed using a human CD3 positive selection kit (STEMCELLTM (Cambridge, MA, USA), cat. #18051); and new γ-irradiated aAPC were added (total cell:aAPC ratio 1:0.5). Cells were grown for a total of 14 days, at which time they were phenotyped by flow cytometry, demonstrating that >95% of the cells in the cultures were NK cells (CD56+/CD16+/CD3ˉ) (Figure 1(b)). Aliquots of these cells were viably frozen in a mixture of 50% Cryoprotective Medium (Lonza, Allendale, NJ, USA), 25% RPMI-1640, and 25% FBS as NK cell stocks, while the remainder of the NK cells continued to grow with irradiated APC (1:0.5). At day 19, the culture medium was replaced with exosome-free FBS, and the culture supernatant was harvested 48 h later and filtered with 0.8 μm pore size membrane (cat # A080A047A) from Advantec, Inc. (Dublin, CA, USA) An equal volume of 50% sterile PEG8000 was added to precipitate the EV derived from aNK cells at 4°C overnight, followed by centrifugation to pellet the vesicles, and then by dialysis with PBS-5% glycerol and a 100 KDa cut-off dialysis bag (Spectrum Labs, Com (Rancho Dominguez, CA, USA); part #131420) at 4°C overnight. The isolated aNK EVs are stable at −80°C for at least 12 months under this protocol (Figure S1(b)).
Figure 1.
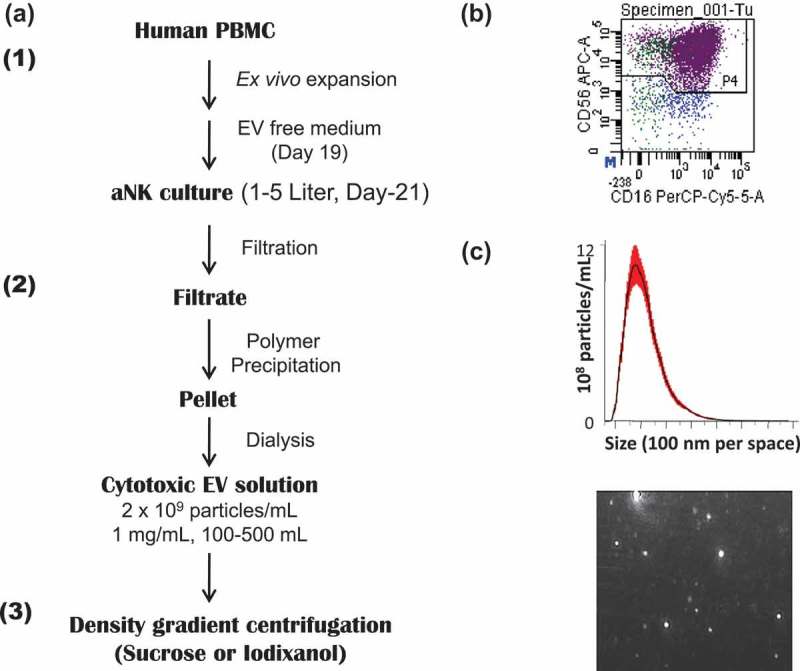
Schema for large-scale propagation and isolation of aNK cell-derived extracellular vesicles. (a) Schema for isolation of EVs from 48-h conditioned medium of activated NK (aNK) cells. The flow chart for EV isolation using NK cells expanded ex vivo and our PEG8000 precipitation method is shown. The detailed procedure is described under Materials and methods. The procedure is divided into three steps: first, ex vivo expansion of NK cells through the use of IL-2 and of artificial antigen presenting cell K562 with membrane bound IL21. The EVs were harvested from the aNK cell culture on day 21. The collected culture supernatant was filtered and precipitated by PEG8000 (Figure S1(a)). The pellet was suspended and dialysed in PBS-5% glycerol with a cut-off size of 100 KDa. The resulting EVs are cytotoxic and cryo-stable for one year at −80°C (Figure S1(b)). The EVs can be further purified by density gradient centrifugation or other methods. (b) aAPC selectively propagate NK cells in co-culture. The CD56 and CD16 positive cells represent NK cells, resulting in 95% aNK cells on day 19 (n = 25). (c) Nanoparticle tracking analysis of EVs. Aliquots of isolated aNK EVs were diluted 1:100 in PBS and analysed using a Nanosight NS300 in light scatter mode. The NTA software defined the number and size range of the vesicles within the sample, plotting the particle size versus particle number per ml. A representative chart of the mean particle size at 155 nm (top) and an image of light reflective particles (bottom) are shown.
OptiPrepTM buoyant density gradient centrifugation
A gradient former was used, containing 4.5 ml PBS and 45% iodixanol solution in the front and back chambers, respectively, with a stirrer bar mixing the solution in the front chamber. A continuous gradient from 0 to 45% was pumped into a 12 ml SW40 centrifuge tube, and then 1 ml EV sample was loaded at the top of the gradient. The tubes were spun at 100,000 x g in an SW40 rotor for 3 h at 4°C. Nine iodixanol fractions (1 ml each) were collected from the top to the bottom of the centrifuge tubes. The density of each fraction was determined by the GeneMate ultrasensitive scale. Forty microlitres of each fraction were used for Western blots with antibodies against CD63 (BD Bioscience (San Jose, CA, USA), H5C6, 1:500) under non-reducing condition (Figure 5(c)): granulysin (Antibodies-online, Inc. (Atlanta, GA, USA), ABON1107438, 1:500), Perforin 1 (Santa Cruz Biotech (Dallas, TX, USA), sc-373943, 1:250), granzyme-A (GeneTex (Irvine, CA, USA), GTX114461, 1:400), granzyme-B (Biolegend (San Diego, CA, USA), #674301, 1:400), or fibronectin (GeneTex, GTX72724, 1:1,000).
Figure 5.
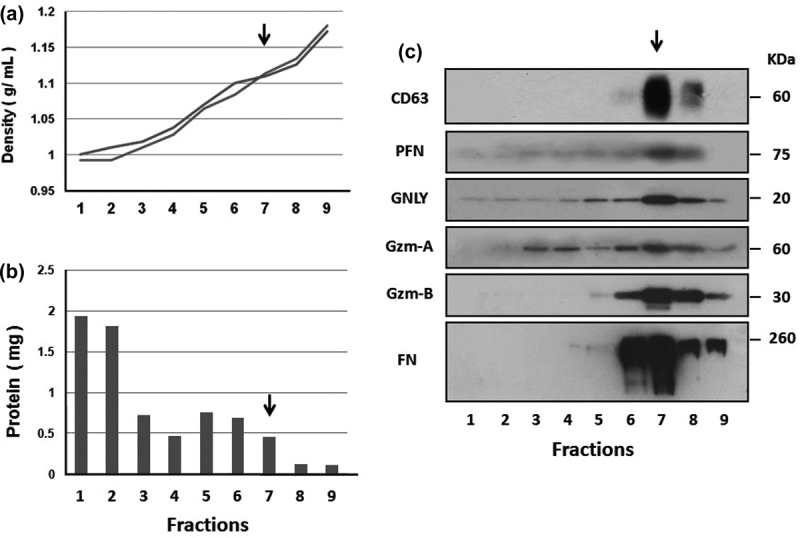
aNK extracellular vesicles contain cytotoxic proteins. A 0% to 45% iodixanol continuous density gradient centrifugation was performed. Samples were loaded in duplicate ultracentrifuge tubes. After centrifugation at 100,000 × g for 3 h, nine iodixanol fractions were collected from the top. The density of each fraction was determined from two balanced tubes, as shown in (a). The total protein content represents the average of each fraction (b). Fractions were analysed by Western blotting (c) using antibodies to CD63 (BD Bioscience, H5C6) under non-reducing condition, PFN (perforin), granulysin (GNLY), granzyme A (Gzm-A), granzyme B (Gzm-B), and fibronectin (FN). The arrow points to fraction 7, which has a density of 1.08–1.12 g ml–1 and contains most of the analysed proteins. The un-cropped images with size markers are shown in Figure S3.
Nanoparticle tracking analysis (NTA)
The number and size of the isolated NK-derived EV (10 μl, 1:100 dilution with PBS) were assessed by nanoparticle tracking analysis (NTA) (NanoSight Model NS300, NanoSight Ltd, Salisbury, UK). The parameters for NTA capture setting were as follows: camera type (sCMO8), camera level,[9] shutter setting (600), camera gain (350) and capture duration (60 s). Each sample was read in triplicate. Particle sizes and numbers were recorded. The “Mean” is the average size of all particles measured, and the “Mode” is the size of particles that occurred most often, i.e. the size of the particles at the peak.
Western blotting analysis
Cell lysates and isolated EV from NK cells, neuroblastoma cells (CHLA-255 cell line) and leukaemia cells (HL60 cell line) were prepared in parallel. Thirty µg of total protein per lane or 5 µg for fibronectin due to its abundance were separated by SDS-PAGE and proteins were electrotransferred to PVDF membranes (Millipore, Billerica, MA, USA). The blots were probed with antibodies to CD63 (GeneTex, GTX37555) (Figure 2(a)), or from BD Bioscience, H5C6 under non-reducing condition (Figure 5(c)), Rab5A (Aviva Systems Biology (San Diego, CA, USA), ARP56563-P050), cytochrome C (Santa Cruz Biotech, sc-13560, 1:500), fibronectin (GeneTex, GTX72724, 1:1,000), and granulysin (Antibodies-online, Inc., ABON1107438, 1:500). The blots were probed with anti-mouse- or anti-rabbit-HRP secondary antibody conjugates (1:5000 dilutions) and then were developed with a chemi-luminescent substrate (cat. # 34080) from ThermoFisher Scientific (Waltham, MA, USA). A protein size ladder was used to determine molecular weight (Tri-color prestained protein ladder I; Bioland Scientific (Paramount, CA, USA), cat. #PM01-01).
Figure 2.
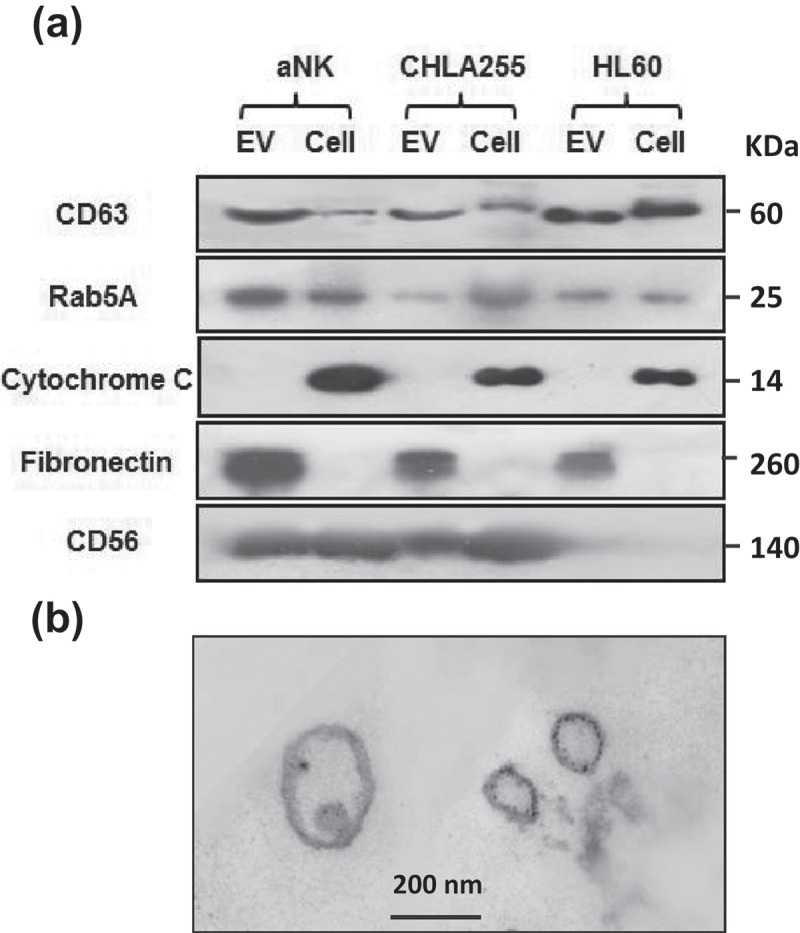
Markers and TEM image of isolated aNK extracellular vesicles. (a) Western blot analysis of isolated EVs and their parental cells. Thirty μg protein were loaded in each lane and probed with antibodies to CD63 (GeneTex, GTX37555, 1:500), Rab5A (Aviva Systems Biology, ARP56563-P050), cytochrome C (Santa Cruz Biotech, sc-13560, 1:500), granulysin (Antibodies-online, Inc. ABON1107438, 1:500), CD56 (EMD Millipore, MAB2120Z, 1:500). For fibronectin, only 5 μg of protein was loaded (GeneTex, GTX72724, 1:1,000). The un-cropped images with size markers are shown in Figure S2. (b) Transmission electron microscopy of EVs. aNK EVs were isolated as described in Figure 1. Aliquots of samples were subjected to TEM imaging as described in Materials and methods. A representative image is shown. Bar: 200 nm
Transmission electron microscopy (TEM)
TEM was used to examine the morphology of the isolated aNK EVs. Isolated EVs were embedded in 1% agarose and then fixed with 4% paraformaldehyde and 2.5% glutaraldehyde in PBS for 1 h. The samples were then centrifuged for 10 min at 7000 x g and post-fixed with 2% buffered OsO4 for 1 h, dehydrated through graded ethanol solutions and propylene oxide, and embedded in Epon solution. Ultrathin sections were cut, stained with uranyl acetate and lead citrate, and examined with a Philips CM TEM.
Cytotoxicity of aNK EVs
Two cytotoxicity assays were performed. In the first, firefly luciferase labelled target cells were used, and the flux of live cells was measured after adding D-luciferin. CHLA255-Luc or SupB15-Luc cells (104 cells per well) were incubated for 6 h in 96-well plates with either aNK cells (effector:target cell ratio 1:1) or different amounts of purified EVs, as indicated. The luciferase substrate, D-luciferin (potassium salt solution; 0.175 mg ml–1), was added to the wells, and flux was quantified with the Promega Glomax multi-detection system (Madison, WI, USA) with a one second read per well following the manufacturer’s luminescence protocol. The second cytotoxicity assay was performed using the Luna™ Cell Counter (Logos Biosystems, Inc., Annandale, VA, USA). NALM-6, SupB15, CHLA-255, CHLA-136, and MCF-7 (104 cells) were seeded into Costar 96-well black plates (Corning, Manassas, VA, USA) in 0.1 ml RPMI-1640 and 2% FBS, and isolated aNK exosomes were added in triplicates. At various time-points, the AO/PI (acridine orange and propidium iodide) dual fluorescence staining method was used to examine total (green, AO) and dead (red, PI) cells. Total, live, and dead cell number and concentration, percentage of viable/dead, cell size and distribution were all displayed on the LCD monitor of the Luna™ Cell Counter, and the instrument software provided data analyses, histograms, and image storage. The ratio of dead cells versus total cells was calculated (n = 5). PBS was used in place of the EVs as a negative control.
Caspase pathways in target cells
The Caspase Inhibitor Sample Pack was purchased from R&D Systems (Minneapolis, MN, USA), and it contains the tripeptide fluoromethyl ketone (FMK)-derivative inhibitors Z-VAD-FMK (pan-caspase), Z-WEHD-FMK (caspase 1), Z-VDVAD-FMK (caspase 2), Z-DEVD-FMK (caspase 3), Z-YVAD-FMK (caspase 4), Z-VEID-FMK (caspase 6), Z-IETD-FMK (caspase 8), Z-LEHD-FMK (caspase 9), Z-AEVD-FMK (caspase 10), and Z-LEED-FMK (caspase 13). Caspase-9 inhibitor (ALX-260-079-M001) was ordered from ENZO life Sciences, Inc. (Farmingdale, NY, USA). Cleaved Caspase Antibody Sampler Kit (cat. #9929) was purchased from Cell Signaling Technology (Danvers, MA, USA). Western blot analyses were performed as above. BD PharmingenTM Annexin V-PE Apoptosis Detection Kit (cat. #: 559703), which includes 7-AAD staining, was used for flow cytometry quantification of apoptotic cells under different treatments.
Statistical analysis
Analysis of variance (ANOVA) was used for statistical analysis of the data. The dependent variables were the percentage of cell survival or cell death, whereas the independent variables were the treatment (different cell lines, different treatments in the presence or absence of EVs, etc.). Raw data were entered into Microsoft Excel (Redmond, WA, USA) files and converted automatically into the statistical packages. ANOVA and co-variates were followed by a multiple comparison test, such as the Newmann–Keuls test, to calculate the statistical significance between the control and treatment groups. P < 0.05 was considered significant.
Results
Large-scale isolation of extracellular vesicles from ex vivo propagated and activated human NK cells
We previously demonstrated that co-culturing PBMC from human blood with K562-mbIL21 aAPC induces major expansion (>2000-fold) and activation of NK cells (CD56+ CD3−) by two weeks.[22] Activated NK (aNK) cells were cultured for an additional seven days to generate EVs (Figure 1(a)). On day 19, the cultures contained >95% NK cells (CD56+/CD16+/CD3ˉ) (Figure 1(b)). At this time, aNK cells were cultured in RPMI-1640 with 10% exosome-free FBS, and aNK cell conditioned medium was collected after 48 h (day 21). After filtration, the cell-free conditioned medium was mixed with an equal volume of 50% PEG8000 to precipitate the aNK EVs. Comparison of different final concentrations of PEG8000 demonstrated that 25% precipitated the most particles based on Nanosight nanoparticle tracking analysis (NTA) (Figure S1(a)). After dialysis of the pellet, isolated aNK EVs are stored at −80°C (Figure 1(a)). The isolated samples were analysed for particle number and size by the NTA, which revealed that the majority of the aNK EVs were in the range of 50–200 nm, with a peak around 155 nm (Figure 1(c)). The aNK EVs can be further enriched by buoyant density gradient centrifugation (Figures 1(a), 5). We have used this protocol to isolate EVs from 50 ml to 5 l of conditioned medium (Figure 3, Tables 1, 2).
Figure 3.
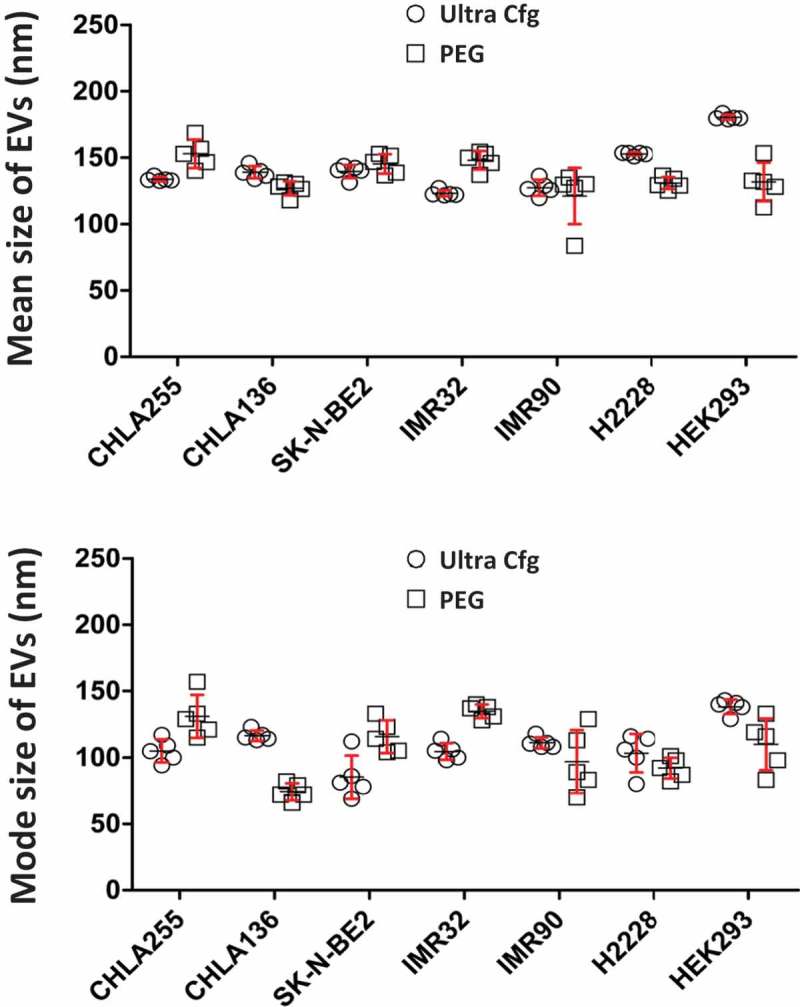
Size comparison of isolated extracellular vesicles from different cancer cell lines. Nanoparticle tracking analysis was performed to compare the sizes of EVs isolated from different cell lines. All cell lines were grown in RPMI medium with 10% FBS until log phase. Cell media were replaced with 10% exosome-free FBS with cells at a density of 0.5 × 106 ml–1. After 48 h, the culture supernatants were filtered, PEG8000-precipitated, dialysed, and suspended in one-tenth of the original volume of PBS containing 10% glycerol. The resulting EV preparations were subjected to nanoparticle tracking analysis. Dot plot were graphed with statistical software GraphPad Prime version 6.0 from five readings of samples prepared by ultracentrifugation (Ↄ) or PEG precipitation (□) side by side from different cell lines. CHLA-255, CHLA-136, SK-N-BE2, IMR-32 are neuroblastoma cell lines; H2228 is a non-small cell lung cancer adenocarcinoma cell line; HEK293 is a kidney embryonic cell line; and IMR-90 cells are normal fibroblasts.
Table 1.
Size comparison of aNK extracellular vesicles obtained by different isolation methods. aNK cell supernatant (150 ml) obtained from exosome-free culture was divided into three parts, and aNK EVs were isolated with three methods. The resulting EV preparations were recovered in a volume of 5 ml, and aliquots of samples were analysed by nanoparticle tracking analysis to determine their size and particle concentrations. Analysis of cell culture medium (non-conditioned) is also shown.
| Particle Size (nm) |
Particle # |
||
|---|---|---|---|
| Protocol | Mean | Mode | (108/mL) |
| ExoQuick | 158+/-1.7 | 126 +/- 5.2 | 19.2 +/- 2.09 |
| Ultra-Cfg | 173+/-13.6 | 149 +/- 11.6 | 12.4 +/- 0.05 |
| Precipitation | 155+/-5.9 | 120 +/- 6.4 | 20.6 +/- 0.09 |
| Culture Medium | .89+/-5.0 | .78 +/- 8.0 | 0.12 +/- 0.15 |
Table 2.
Nanoparticle tracking analysis of isolated extracellular vesicles by polymer precipitation method from various cell lines. All cell lines were grown in RPMI medium, except D54MG in Ham’s F-12, MCF7 in DMEM, and DU145 in IMDM, with 10% FBS until log phase. Cell media were replaced with 10% exosome-free FBS and plated at a density of 0.5 × 106 cells ml–1. After 48 h, the culture supernatants were prepared and subjected to nanoparticle tracking analysis.
| Cell line | Cell type | Mean (nm) | Mode (nm) | Particles # (x 108/ mL) |
|||
|---|---|---|---|---|---|---|---|
| CHLA255 | Neuroblastoma | 153 | ± 28.42 | 131 | ± 42 | 33.48 | ± 1.72 |
| SK-N-BE(2) | Neuroblastoma | 145.3 | ± 51.38 | 115.8 | ± 29 | 6.43 | ± 1.38 |
| SupB15 | ALL | 146 | ± 22.6 | 101.6 | ± 9 | 13.64 | ± 1.07 |
| D54MG | Glioma | 194.5 | ± 15.98 | 166.4 | ± 53 | 5.91 | ± 1.17 |
| McF7 | Breast Carcinoma | 120.6 | ± 6.31 | 82.4 | ± 11 | 20.48 | ± 1.85 |
| DU145 | Prostate | 131.1 | ± 30.37 | 63.4 | ± 76 | 20.48 | ± 4.61 |
| Hela | Cervical cancer | 224.5 | ± 54.79 | 210.6 | ± 65 | 3.1 | ± 1.11 |
| HepG2 | Hepatoma | 134.6 | ± 30.32 | 85.2 | ± 63 | 25.99 | ± 3.51 |
| HEK293 | Embronyic Kidney | 131.6 | ± 40.64 | 109.8 | ± 50 | 18.41 | ± 6.87 |
| MSC | Mesenchymal Stroma Cell | 175.2 | ± 12.29 | 147.4 | ± 22 | 9.375 | ± 1.97 |
Characteristics of isolated aNK extracellular vesicles
The EVs derived from human aNK cells were compared to those prepared from two cell lines, CHLA-255 and HL60, using the same isolation methods. Western blot analyses show that common markers of exosomes (CD63, Rab5A) and associated fibronectin were detected in the EVs isolated from the three cell types, which were all devoid of cytochrome C, a cellular marker (Figure 2(a)). CD56, also known as neural cell adhesion molecule (NCAM), was detected in both cell lysate and EV preparations of NK and neuroblastoma CHLA-255. TEM imaging of isolated EVs demonstrated membrane-enclosed structures with a translucent inside, an oval shape, and different sizes, which are typical for EVs (Figure 2(b)). Therefore, according to their size, markers, and morphology, the vesicles isolated from different cell types by our protocol are enriched EVs.[26]
Comparison of PEG8000 precipitation and other methods for isolating extracellular vesicles
We used NTA to examine the EVs isolated by our PEG8000 method and other accepted methods. Using aNK cell conditioned medium, there were no differences in particle size or number of EVs isolated by ExoQuick (System Biosciences, Inc., Mountain View, CA, USA), ultracentrifugation (100,000 × g for 1 h), and our PEG8000 precipitation method. As expected, non-conditioned culture medium had a small number of particles with a mean diameter of <90 nm (Table 1).
EVs were isolated from cultures of different cancer cell lines using our PEG8000 protocol or ultracentrifugation (Figure 3). All cells produced EVs of similar sizes, as analysed by NTA (Figure 3). Our PEG8000 protocol also was used to isolate EVs released from 10 different types of cultured cells on a small or large scale (Table 2). Analysis with NTA revealed some variation in size and particle number among the cell lines, most likely representing their individual uniqueness. Thus, our protocol could isolate EVs from various sources of culture medium, could be scaled up or down, and could isolate EVs that were similar to those obtained with other methods.
aNK extracellular vesicles are cytotoxic for cancer cell lines
To determine if isolated EVs were functional, we performed two different cytotoxicity assays. We used SupB15 acute lymphoblastic leukaemia (ALL) and CHLA-255 neuroblastoma cells that were transfected with the firefly luciferase gene (SupB15-fLuc and CHLA-255-fLuc, respectively) to quantify target cell survival using bioluminescence. After 6 h of incubation with aNK cell-derived EVs, survival was decreased in both ALL and neuroblastoma cells in a dose-dependent manner (Figure 4(a)). In the second method, cytotoxicity was assessed by propidium iodide staining, which showed a time-dependent cytotoxicity induced by aNK cell-derived EVs in the SupB15 and CHLA-255 cell lines. When EVs isolated from CHLA-255 cells were used as the control, no cytotoxicity was detected (Figure 4(b)). To further examine aNK cell-derived EVs, cytotoxicity was also observed in other tested cancer cell lines including ALL (NALM-6), neuroblastoma (CHLA-136), and breast carcinoma (MCF-7) (Figure 4(c)).
Figure 4.
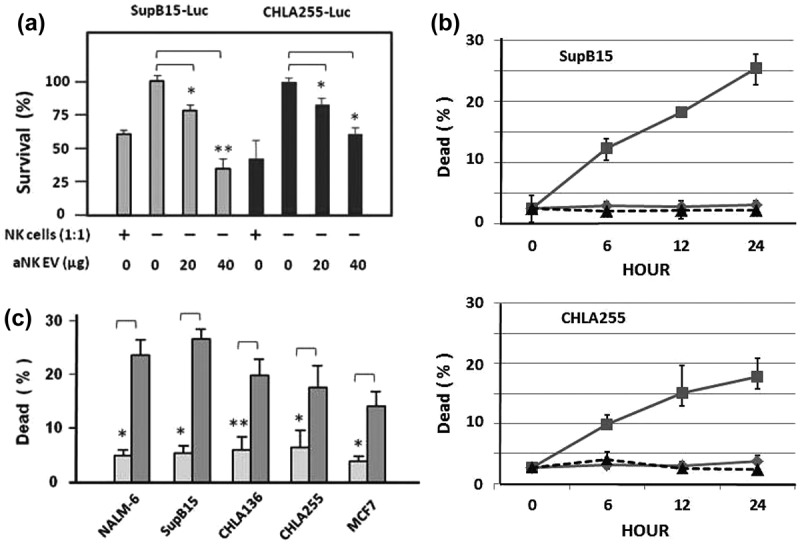
Cytotoxicity of isolated aNK extracellular vesicles. Luciferase assay. EVs were isolated from 48 h conditioned medium of aNK cells as in Figure 1. ALL SupB15-fLuc cells (104 cells) or neuroblastoma CHLA255-fLuc cells (104 cells) were incubated in 96-well plates with aNK cells (104 cells; effector:target cell ratio 1:1) (columns 1 and 5) as the positive control, or different amounts of purified exosomes as indicated. After 6 h incubation, the luciferase substrate Beetle Luciferin (Promega, E1605) was added, and bioluminescence was quantified. Untreated samples were designated as 100% survival for CHLA255-fLuc or SupB15-fLuc (lanes 2 or 6, respectively) and results were expressed as per cent survival. fLuc: firefly luciferase labelled cells. *p < 0.05, **p < 0.01 (a) Acridine orange/propidium iodide fluorescence assay: 104 cells (SupB15 or CHLA-255) from log phase cell culture were transferred to 96-well dark plates in the presence (∎, solid line) or absence (△, dotted line) of EVs (40 μg) in a final volume of 100 μl. Similarly, 40 μg EVs isolated from neuroblastoma CHLA-255 cells was used as the negative control (⋄, solid line). The suspensions were incubated at 37°C, and the percentage of dead cells was calculated by the LUNA cell counter at various times (0, 6, 12, 24 h). (b) The acridine orange/propidium iodide fluorescence assay was performed using different cancer cell lines, as indicated. The percentage of dead cells, without (light box) or with EVs (40 µg) (grey box), was determined by the LUNA cell counter at 24 h. The error bars on the graphs are generated by the Excel software based on 4–8 readings in each test. *p < 0.05, **p < 0.01.
aNK extracellular vesicles contain cytotoxic molecules
We examined the aNK EV protein composition by Western blot analysis to determine whether they contained active cytotoxic proteins derived from NK cells. Buoyant density gradient centrifugation was performed to further purify aNK EVs. The density and total protein content of the nine fractions were analysed (Figure 5(a), 5(b)). CD63, a universal marker, was used to locate the exosomes (Figure 5(c)), which were mainly found at a density of 1.08–1.12 g ml–1. About 5–10% of the total proteins were recovered in fraction 7 (Figure 5(b)), which not only contained CD63 and fibronectin, but also perforin, granulysin, granzyme A, and granzyme B (Figure 5(c)). Fraction 8 contained the same proteins to a lesser degree, and a second peak of granzyme A could also be detected in the lighter fractions (fractions 3 and 4). These cytotoxic proteins may contribute to the cytotoxicity of aNK cell-derived EVs for the studied cancer cell lines (Figure 4).
aNK extracellular vesicles induce caspase-mediated cytotoxicity
To gain insight into the killing process, we examined whether aNK EV-induced cytotoxicity was caspase-mediated by incubating caspase inhibitors with CHLA-255 and NALM-6 cell lines prior to adding aNK EVs. The pan-caspase inhibitor (Figure 6, column 3) significantly reduced cytotoxicity in both cell lines. Human caspases can be subdivided into three functional groups: inflammatory or cytokine activation (caspase-1, -4, and -13), apoptosis initiation (caspase-2, -8, -9, and -10), and apoptosis execution (caspase-3, -6, -7, and -12), and specific inhibitors for each group are available. Inhibitors of the inflammatory group had no detectable effect on cytotoxicity. However, inhibitors of the apoptosis initiation group (caspase-2, -8, -9, and -10) effectively blocked EV-induced cytotoxicity, as did the inhibitors of caspase-3 (apoptosis execution group), and to a lesser extent those of caspase-6 and -12 (Figure 6). To complement the above experiments, we used flow cytometry analysis of PE Annexin V staining to quantify the percentage of apoptotic cells (Figure S4). Overall, our results suggest that the caspase members in the initiation and execution groups are involved, at least in part, in the aNK EV killing of cancer cells.
Figure 6.
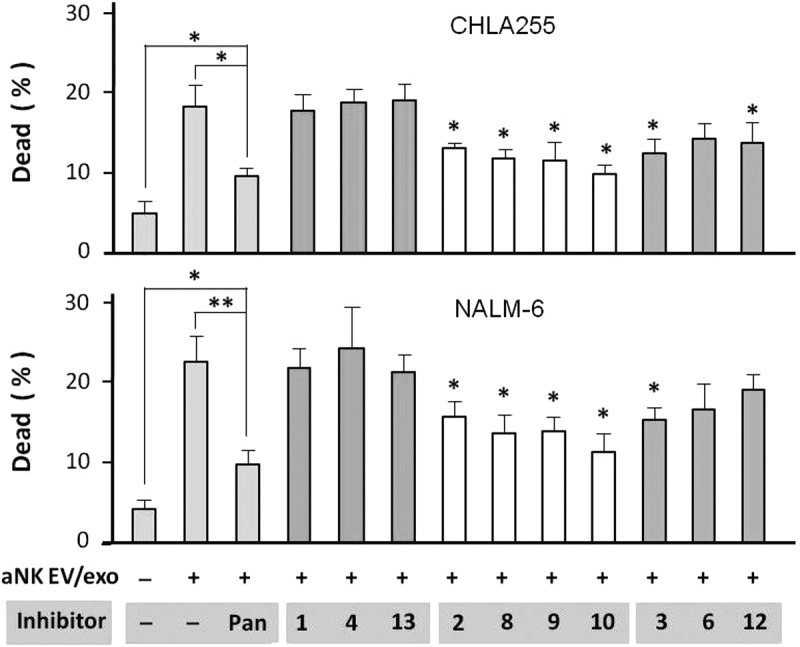
Caspase inhibitors partially block aNK extracellular vesicle-induced cytotoxicity. Neuroblastoma CHLA-255 cells (top panel) or ALL NALM-6 cells (bottom panel) were pre-incubated with different caspase inhibitors (100 μM) for 30 min, prior to the addition of aNK EVs. The AO/PI cytotoxicity assay was performed as described in Materials and methods. The inhibitors alone had no effect on these cells. After 24 h, the percentage of dead cells was calculated by the LUNA Cell Counter. The percentage of dead cells under different treatments are shown (N = 6; *p < 0.05, **p < 0.01). The error bars on the graphs are generated by Excel software, based on 4–8 readings in each test. Independent testing of caspase inhibitors 3, 8, 10, and 12 by Annexin V flow cytometry is shown in Figure S4.
We then utilised antibodies to specifically probe for cleaved caspase protein fragments in aNK EV-treated cells. Target cells were incubated with isolated aNK EVs for 0, 2, 4, 6, 8, 10 and 12 h. At the initial time-point (0 h), no cleaved caspases could be detected, but thereafter the cleaved forms of different caspases could be detected (Figure 7). In addition, degradation products of the common caspase substrate protein, PARP, could be detected after a short period of time (2 h) and were found at a high level throughout the tested time-points (4–12 h). These results clearly demonstrate that apoptotic pathways can be activated in target cells by the EVs isolated from aNK cells and that cytotoxicity is mediated at least in part by caspases.
Figure 7.
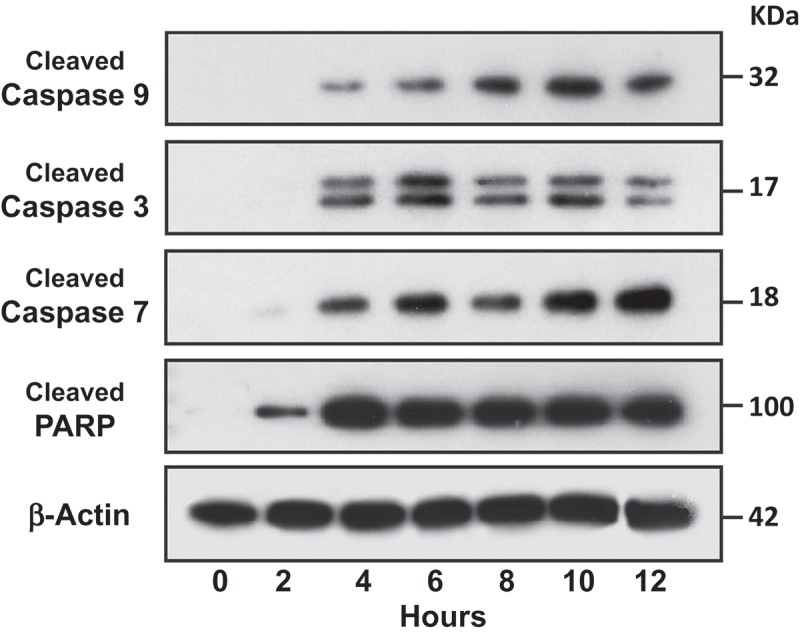
Activation of caspases in target cells by aNK extracellular vesicles. ALL NALM-6 cells were treated with aNK EVs for different time points as indicated. Cells were then harvested and lysed in SDS-PAGE sample buffer. Each lane contains 30 μg cell lysate proteins as determined by the Orbit® protein assay (Life Technologies). Antibodies against cleaved caspase -3, -7, and -9, and PARP from the Cleaved Caspase Antibody Sampler Kit were used to probe the blots according to the manufacturer’s protocol. β-Actin was used as the loading control. The un-cropped images with size markers are shown in Figure S5.
Discussion
Many techniques have been developed to isolate EVs from cell culture, body fluids, or other sources. However, an easy and inexpensive process for large-scale isolation of EVs that preserves their biological activity has not been previously reported. We have developed a simple, robust, and cost-effective method to isolate a large quantity of EVs from NK cells expanded in culture. Importantly, the resulting EV preparations exhibit cytotoxic properties towards ALL and neuroblastoma cancer cells lines, opening the possibility of their use for clinical applications. One key element was the highly efficient ex vivo propagation and activation of NK cells from human PBMC using co-culture with K562-mbIL21 aAPCs as previously described [22,27] (Figure 1). Interleukin-2 was included to activate cultured NK cells through the expansion period. This protocol was originally developed to obtain cytotoxic NK cells for cancer immunotherapy. We adapted the protocol for EV isolation at day 21, resulting in large amounts of NK cells that released abundant EVs into the conditioned medium. Using the G-Rex culture system, 1–3 billion aNK cells can be grown per 100 cm2 of surface area, which increases aNK cell-derived EV production while decreasing the complexity and cost of NK cultures.
The polymer PEG8000 was used to precipitate EVs from the aNK cell conditioned medium (Figure 1(a)), concentrating the EVs to a small volume. PEG molecules have been used for several clinical applications, and the presence of PEG may stabilise EVs. The advantages of this method are that it is scalable, that it has essentially no volume limitation, and that no special instrumentation is required. One drawback of using PEG precipitation to isolate EVs is that the polymer can also precipitate other protein components from the culture medium, despite using exosome-free serum. The problem may be overcome by additional purification using ultrafiltration and size exclusion chromatography. With this modification, our procedure could be used to isolate aNK EVs for human clinical trials.
The isolated vesicles were identified as EVs by Western blotting with EV markers and TEM imaging (Figure 2). CD63 is a glycoprotein which may show smear/multiple bands, depending on the sample sources and preparations. Our NK EVs contain a band around 60 KDa in a regular reduced SDS-PAGE (Figure 2(a)). Some small bands were observed, possibly due to its degradation forms (Figure S2). Under a non-reducing condition, a single smear band around 60 KDa band was observed (Figure 5(c)). OptiPrep centrifugation may remove the degraded forms (Figure S3). In any case, the presence of CD63 verifies the NK EVs that were isolated from our ex vivo expansion cultures. Our protocol results in EVs that are comparable to those obtained with other isolation methods (Tables 1, 2, Figure 3), and it can be scaled up or down as needed. This method might also be applied to isolate EVs released by other immune cells.
NK cells contain lytic granules that are released upon stimulation and induce cytotoxicity.[28,29] We provide evidence that isolated aNK EVs are functional and have retained the NK cell killing function through incorporation of cytotoxic proteins including granulysin, perforin, granzyme A, and granzyme B (Figure 5). A previous study showed that isolated aNK exosomes were cytotoxic for cell lines derived from haematologic malignancies (Jurkat, K562, and DAUDI cell lines) but not for one from a solid tumour (breast carcinoma SKBR3).[20] The discrepancy between this report and our results may be due to the fact that we used different tumour cell lines (i.e. neuroblastoma and breast cancer). Additionally, our protocol used highly activated NK cells for production of EVs.
It is well known that activated human cytotoxic T lymphocytes and NK cells secrete cytotoxic proteins as the major mechanism for their cytotoxicity,[30–33] which involves multiple cell death pathways. Interestingly, our isolated aNK EVs also contain cytotoxic proteins perforin (PFN), granzymes (Gzm-A and Gzm-B), and granulysin (GNLY) (Figure 5). Through the action of PFN (pore formation), granzymes and other cytotoxins may enter into the cytoplasm of the target cells.[34] Gzm-A is a tryptase that induces caspase independent cell death through histone digestion and by facilitating endogenous DNAse access to DNA during granule-mediated apoptosis.[33] Gzm-B is a serine protease that triggers activation of caspases -8, -10, and then the caspases -9, -3, -7 cascade, which leads to apoptosis.[32] Granulysin can disrupt membranes, damage mitochondria, and activate caspase-9 and -12 to induce apoptosis.[31] Our isolated NK EVs contain significant amounts of these cytotoxic molecules (Figure 5), and our results suggest that they are functional in EVs activating multiple caspase pathways in target cells (Figures 6, 7).
aNK EVs have multiple features that might make them suitable for clinical applications. They can be prepared from individual patients for autologous use. EV preparations are stable at −80°C for at least one year (Figure S1(b)), making them readily available “off-the-shelf”. They are cytotoxic to cancer cells and may not be affected by chemotherapy resistance mechanisms. EVs may have reduced toxicity in comparison to whole cell therapy (e.g. T or NK cells), as the latter may also release cytokines that cause severe immune responses (e.g. cytokine release syndrome, graft-versus-host disease).[35] Notably, aNK EVs may be altered to improve targeting and cytotoxicity via engineering of the aNK cells.[36] Finally, it is known that EVs can cross the blood–brain barrier [37], and thus they may be able to enter cancer sanctuary sites such as the central nervous system. Thus, the scale-up isolation procedure resulting in aNK EVs with efficient cytotoxic activity lays the foundation for the future development of new anti-cancer therapeutics.
Supplementary Material
Acknowledgements
The authors thank Dr Martine Torres for reviewing and editing. We thank Dr Petra Wise for assistance with NanoSight® tracking analysis and Dr Michael Sheard for assistance with flow cytometry. This study was supported in part by awards from the Alex’s Lemonade Stand Foundation, the Team Connor Foundation, the William Lawrence and Blanche Hughes Foundation, and the Whittier Foundation.
Responsible Editor Dr. Aled Clayton Cardiff University, United Kingdom
Funding Statement
This work was supported by the Alex’s Lemonade Stand Foundation; the Team Connor Foundation; the William Lawrence and Blanche Hughes Foundation; the Whittier Foundation.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental data
Supplemental data for this article can be accessed here.
References
- Colombo M, Raposo G, Théry C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol. 2014;30:255–12. doi: 10.1146/annurev-cellbio-101512-122326. PubMed PMID: 25288114. [DOI] [PubMed] [Google Scholar]
- Bobrie A, Colombo M, Raposo G. Exosome secretion: molecular mechanisms and roles in immune responses. Traffic. 2011 Jun 08;12(12):1659–1668. doi: 10.1111/j.1600-0854.2011.01225.x. Epub. 2011. PubMed PMID: 21645191. [DOI] [PubMed] [Google Scholar]
- Ludwig AK, Giebel B. Exosomes small vesicles participating in intercellular communication. Int J Biochem Cell Biol. 2012 Oct 26;44(1):11–15. doi: 10.1016/j.biocel.2011.10.005. Epub. 2011. PubMed PMID: 22024155. [DOI] [PubMed] [Google Scholar]
- Théry C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol. 2009;9(8):581–593. doi: 10.1038/nri2567. PubMed PMID: 19498381. [DOI] [PubMed] [Google Scholar]
- Raimondo F, Morosi L, Chinello C. Advances in membranous vesicle and exosome proteomics improving biological understanding and biomarker discovery. Proteomics. 2011;11(4):709–720. doi: 10.1002/pmic.201000422. PubMed PMID: 21241021. [DOI] [PubMed] [Google Scholar]
- Choi D-S, Kim D-K, Kim Y-K. Proteomics, transcriptomics and lipidomics of exosomes and ectosomes. Proteomics. 2013;13(10–11):1554–1571. doi: 10.1002/pmic.201200329. PubMed PMID: 23401200. [DOI] [PubMed] [Google Scholar]
- Challagundla KB, Wise PM, Neviani P. Exosome-mediated transfer of microRNAs within the tumor microenvironment and neuroblastoma resistance to chemotherapy. J Natl Cancer Inst. 2015;107:7. doi: 10.1093/jnci/djv135. PubMed PMID: 25972604; PMCID: PMC4651042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valadi H, Ekström K, Bossios A. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9(6):654–659. doi: 10.1038/ncb1596. PubMed PMID: 17486113. [DOI] [PubMed] [Google Scholar]
- Camussi G, Deregibus MC, Bruno S. Exosomes/microvesicles as a mechanism of cell-to-cell communication. Kidney Int. 2010 Aug 13;78(9):838–848. doi: 10.1038/ki.2010.278. Epub. 2010. PubMed PMID: 20703216. [DOI] [PubMed] [Google Scholar]
- Greening DW, Xu R, Ji H. A protocol for exosome isolation and characterization: evaluation of ultracentrifugation, density-gradient separation, and immunoaffinity capture methods. Methods Mol Biol. 2015;1295:179–209. doi: 10.1007/978-1-4939-2550-6_15. PubMed PMID: 25820723. [DOI] [PubMed] [Google Scholar]
- Lobb RJ, Becker M, Wen SW. Optimized exosome isolation protocol for cell culture supernatant and human plasma. J Extracell Vesicles. 2015;4:27031. doi: 10.3402/jev.v4.27031. PubMed PMID: 26194179; PMCID: PMC4507751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinemann ML, Ilmer M, Silva LP. Benchtop isolation and characterization of functional exosomes by sequential filtration. J Chromatogr A. 2014;1371:125–135. doi: 10.1016/j.chroma.2014.10.026. PubMed PMID: 25458527. [DOI] [PubMed] [Google Scholar]
- Tan CY, Lai RC, Wong W. Mesenchymal stem cell-derived exosomes promote hepatic regeneration in drug-induced liver injury models. Stem Cell Res Ther. 2014;5(3):76. doi: 10.1186/scrt465. PubMed PMID: 24915963; PMCID: PMC4229780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies RT, Kim J, Jang SC. Microfluidic filtration system to isolate extracellular vesicles from blood. Lab on a Chip. 2012;12(24):5202–5210. doi: 10.1039/c2lc41006k. PubMed PMID: 23111789. [DOI] [PubMed] [Google Scholar]
- Kordelas L, Rebmann V, Ludwig AK. MSC-derived exosomes: a novel tool to treat therapy-refractory graft-versus-host disease. Leukemia. 2014;28(4):970–973. doi: 10.1038/leu.2014.41. PubMed PMID: 24445866. [DOI] [PubMed] [Google Scholar]
- Balaj L, Atai NA, Chen W. Heparin affinity purification of extracellular vesicles. Sci Rep. 2015;5:10266. doi: 10.1038/srep10266. PubMed PMID: 25988257; PMCID: 4437317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowal J, Arras G, Colombo M. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc Natl Acad Sci U S A. 2016;113(8):E968–E977. doi: 10.1073/pnas.1521230113. PubMed PMID: 26858453; PMCID: PMC4776515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böing AN, Van Der Pol E, Grootemaat AE. Single-step isolation of extracellular vesicles by size-exclusion chromatography. J Extracell Vesicles. 2014;3 doi: 10.3402/jev.v3.23430. PubMed PMID: 25279113; PMCID: PMC4159761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carotta S. Targeting NK cells for anticancer immunotherapy: clinical and preclinical approaches. Front Immunol. 2016;7:152. doi: 10.3389/fimmu.2016.00152. PubMed PMID: 27148271; PMCID: PMC4838611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugini L, Cecchetti S, Huber V. Immune surveillance properties of human NK cell-derived exosomes. J Immunol. 2012 Aug 21;189(6):2833–2842. doi: 10.4049/jimmunol.1101988. Epub. 2012. PubMed PMID: 22904309. [DOI] [PubMed] [Google Scholar]
- Fais S. NK cell-released exosomes: natural nanobullets against tumors. Oncoimmunology. 2013 Mar 14;2(1):e22337. doi: 10.4161/onci.22337. Epub. 2013. PubMed PMID: 23482694; PMCID: 3583913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Wu HW, Sheard MA. Growth and activation of natural killer cells ex vivo from children with neuroblastoma for adoptive cell therapy. Clin Cancer Res. 2013 Feb 05;19(8):2132–2143. doi: 10.1158/1078-0432.CCR-12-1243. Epub. 2013. PubMed PMID: 23378384; PMCID: PMC3658308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Cui Y, Voong N. Activating signals dominate inhibitory signals in CD137L/IL-15 activated natural killer cells. J Immunother. 2011 Feb 10;34(2):187–195. doi: 10.1097/CJI.0b013e31820d2a21. Epub. 2011. PubMed PMID: 21304401; PMCID: 3128544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fais S, Logozzi M, Lugini L. Exosomes: the ideal nanovectors for biodelivery. Biol Chem. 2013 Dec 18;394(1):1–15. doi: 10.1515/hsz-2012-0236. Epub. 2012. PubMed PMID: 23241589t. [DOI] [PubMed] [Google Scholar]
- Denman CJ, Senyukov VV, Somanchi SS. Membrane-bound IL-21 promotes sustained ex vivo proliferation of human natural killer cells. Plos One. 2012 Jan 27;7(1):e30264. doi: 10.1371/journal.pone.0030264. Epub. 2012. PubMed PMID: 22279576; PMCID: 3261192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lötvall J, Hill AF, Hochberg F. Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the international society for extracellular vesicles. J Extracell Vesicles. 2014;3:26913. doi: 10.3402/jev.v3.26913. PubMed PMID: 25536934; PMCID: PMC4275645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei F, Lim M, George AA. Cytotoxicity of CD56-positive lymphocytes against autologous B-cell precursor acute lymphoblastic leukemia cells. Leukemia. 2015;29(4):788–797. doi: 10.1038/leu.2014.246. PubMed PMID: 25134458; PMCID: PMC4334757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzewski K, Coligan JE. Human NK cell lytic granules and regulation of their exocytosis. Front Immunol. 2012;3:335. doi: 10.3389/fimmu.2012.00335. PubMed PMID: 23162553; PMCID: PMC3494098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu HT, Orange JS. Distinct integrin-dependent signals define requirements for lytic granule convergence and polarization in natural killer cells. Sci Signal. 2014;7(346):pe24. doi: 10.1126/scisignal.2005816. PubMed PMID: 25292212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaspar AA, Okada S, Kumar J. A distinct pathway of cell-mediated apoptosis initiated by granulysin. J Immunol. 2001;167(1):350–356. doi: 10.4049/jimmunol.167.1.350. PubMed PMID: 11418670. [DOI] [PubMed] [Google Scholar]
- Saini RV, Wilson C, Finn MW. Granulysin delivered by cytotoxic cells damages endoplasmic reticulum and activates caspase-7 in target cells. J Immunol. 2011 Feb 08;186(6):3497–3504. doi: 10.4049/jimmunol.1003409. Epub. 2011. PubMed PMID: 21296981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewen CL, Kane KP, Bleackley RC. A quarter century of granzymes. Cell Death Differ. 2012;19(1):28–35. doi: 10.1038/cdd.2011.153. PubMed PMID: 22052191; PMCID: PMC3252830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman J. Granzyme A activates another way to die. Immunol Rev. 2010;235(1):93–104. doi: 10.1111/j.0105-2896.2010.00902.x. PubMed PMID: 20536557; PMCID: PMC2905780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podack ER, Hengartner H, Lichtenheld MG. A central role of perforin in cytolysis? Annu Rev Immunol. 1991;9:129–157. doi: 10.1146/annurev.iy.09.040191.001021. PubMed PMID: 1910674. [DOI] [PubMed] [Google Scholar]
- Shah NN, Baird K, Delbrook CP. Acute GVHD in patients receiving IL-15/4-1BBL activated NK cells following T-cell-depleted stem cell transplantation. Blood. 2015;125(5):784–792. doi: 10.1182/blood-2014-07-592881. PubMed PMID: 25452614; PMCID: PMC4311226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y, Li S, Song J. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials. 2014 Dec 19;35(7):2383–2390. doi: 10.1016/j.biomaterials.2013.11.083. Epub. 2013. PubMed PMID: 24345736. [DOI] [PubMed] [Google Scholar]
- Zhuang X, Xiang X, Grizzle W. 2011. pp. 19:1769–1779. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.