

Abstract
Background and Purpose
Iridoid glycosides containing the double bond scaffold of cyclopentapyran are reversible and orthosteric agonists of glucagon‐like peptide‐1 (GLP‐1) receptors and exert anti‐nociceptive and neuroprotective actions. Morroniside, derived from the medicinal herb Cornus officinalis , is an atypical secoiridoid containing a six‐membered cyclic inner ether fragment. Here we investigated whether morroniside was an orthosteric GLP‐1 receptor agonist and had anti‐hypersensitivity activities in a model of neuropathic pain.
Experimental Approach
We used a model of neuropathic pain, induced by tight ligation of L5/L6 spinal nerves in rats. Hydrogen peroxide‐induced oxidative damage was also assayed in N9 microglial cells and human HEK293 cells stably expressing GLP‐1 receptors.
Key Results
Morroniside protected against hydrogen peroxide‐induced oxidative damage in N9 microglial and HEK293 cells that expressed mouse or human GLP‐1 receptors, but not in HEK293T cells without GLP‐1 receptors. The GLP‐1 receptor orthosteric antagonist exendin(9‐39) also concentration‐dependently shifted the concentration‐protective response curves of morroniside and exenatide to the right without affecting maximal protection, with similar pA2 values. Furthermore, morroniside given by oral gavage or intrathecally in neuropathic rats dose‐dependently attenuated mechanical allodynia, with comparable Emax values and ED50s of 335 mg·kg−1 and 7.1 μg and completely blocked thermal hyperalgesia. Daily intrathecal injections of morroniside over 7 days did not induce anti‐allodynic tolerance. Pretreatment with intrathecal exendin(9‐39) completely blocked systemic and intrathecal morroniside‐induced mechanical anti‐allodynia.
Conclusion and Implications
Our data demonstrated that morroniside was an orthosteric agonist of GLP‐1 receptors and produced antihypersensitivity in a neuropathic pain model by activation of spinal GLP‐1 receptors.
Abbreviations
- ED50 or EC50
half‐effective dose or half‐effective concentration
- Emax
maximum effect
- GLP‐1
glucagon‐like peptide‐1
- MTT
3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyl tetrazolium bromide
Tables of Links
| TARGETS |
|---|
| GLP‐1 receptors |
| LIGANDS |
|---|
| Exenatide |
| Exendin(9‐39) |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Introduction
Glucagon‐like peptide‐1 (GLP‐1) receptors, a B class of G‐protein‐coupled receptors, are widely distributed in humans in tissues such as the pancreatic islets, lungs, cardiovascular system and the CNS, although they are absent from skeletal muscle (Baggio and Drucker, 2007; Koole et al., 2013; Gong et al., 2014c). GLP‐1 receptors expressed in the pancreatic islet beta cells represent the main target for treatment of type 2 diabetes mellitus (Baggio and Drucker, 2007; Koole et al., 2013). Beside its clinical utility against diabetes mellitus, activation of GLP‐1 receptors also exhibits neuroprotection in several animal models of neurodegenerative disorders, such as Alzheimer's disease, Parkinson's disease, amyotrophic lateral sclerosis, peripheral neuropathy, multiple sclerosis, ischaemia and stroke (Kim et al., 2009; Harkavyi and Whitton, 2010; Holscher, 2012; Hansen et al., 2015; Jia et al., 2015). Moreover, activation of spinal GLP‐1 receptors by the peptide agonists GLP‐1(7‐37) and exenatide and the non‐peptide agonist WB4‐24 was shown to produce antinociception in a variety of rodent models of pain hypersensitivity, including neuropathic pain, inflammatory pain, bone cancer pain and diabetic neuropathic pain, through spinal β‐endorphin expression and secretion (Zhu et al., 2014; Gong et al., 2014a; Gong et al., 2014c; Fan et al., 2015; Fan et al., 2016).
The therapeutic success of peptide agonists of GLP‐1 receptors against type 2 diabetes mellitus and identification of GLP‐1 receptors as a potential target in the treatment of pain have inspired efforts to develop orally available low MW GLP‐1 receptor agonists (Wang et al., 2010; Willard et al., 2012). Iridoid glycosides are compounds that have a scaffold of cyclopentapyran with double bond between C7 and C8, and the iridoid glycosides, including geniposide, geniposidic acid, loganin, catalpol, shanzhiside methylester and 8‐O‐acetyl shanzhiside methylester, are reversible and orthosteric agonists of GLP‐1 receptors, presumably acting at the same binding site as exendin(9‐39) (Liu et al., 2007; Zhu et al., 2014; Gong et al., 2014a; Jia et al., 2015; Fan et al., 2016). However, it is not clear whether secoiridoids with the broken double bond at the C7 and C8 in the five‐membered carbon ring have the same activity.
The medicinal herb Cornus officinalis (C. officinalis) Sieb. Et Zucc. (Shan Zhu Yu, Figure 1A) belongs to the Cornaceae and is indigenous in Henan, Gansu and Sichuan of China. Preparations of this herb show a neuroprotective ability and are widely used to treat ischaemic cerebrovascular and neurodegenerative diseases, in addition to immunological diseases and vascular complications of diabetes mellitus, such as diabetic angiopathy and the early stage of diabetic nephropathy (Xu et al., 2004; Li et al., 2005; Xu et al., 2006; Li et al., 2007). Iridoid glycosides, particularly morroniside and loganin, are major active ingredients in C. officinalis (Wang et al., 2003). Morroniside is an atypical secoiridoid with a broken double bond at C7 and C8 in the five‐membered carbon ring and replaced by a six‐membered cyclic inner ether fragment (Figure 1B). Administration, by oral gavage, of the C. officinalis extracts or total iridoid glycosides, including morroniside, promoted neurogenesis (Li et al., 2005; Li et al., 2007; Yao et al., 2009) and improved scopolamine‐induced memory loss in mice (Lee et al., 2009). Specifically, morroniside also protected against hydrogen peroxide‐induced cytotoxicity in human neuroblastoma SH‐SY5Y cells (Ai et al., 2008; Wang et al., 2008; Wang et al., 2009a; Wang et al., 2009b). Similar administration of morroniside to rats significantly improved focal ischaemia/reperfusion‐induced infarction and neurological behaviours, which was related to its antioxidant and anti‐apoptotic properties in brain (Wang et al., 2010). However, it remains to be determined whether morroniside has antinociceptive activities in peripheral nerve injury‐induced neuropathic pain.
Figure 1.
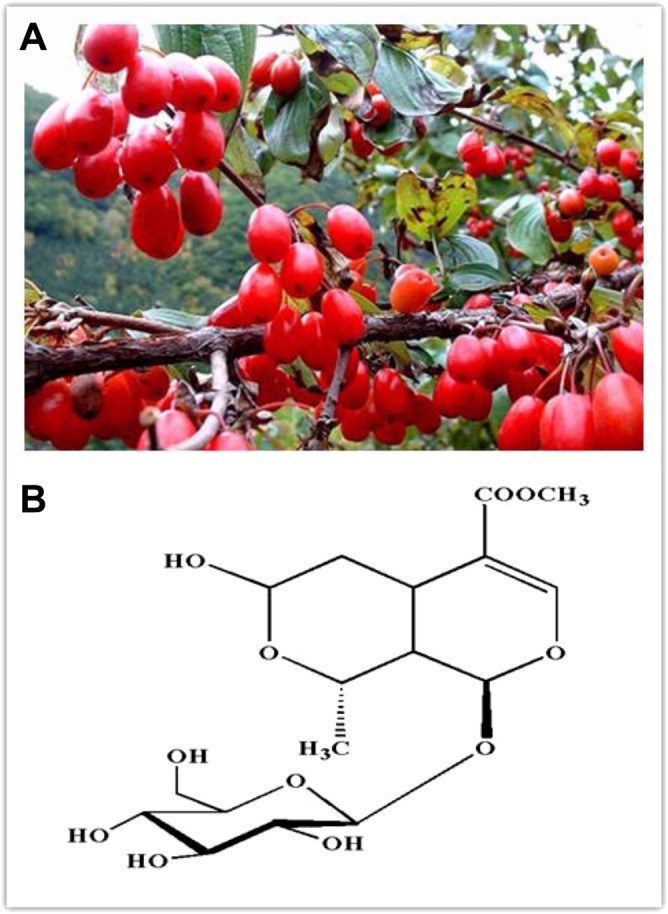
Flowers of Cornus officinalis (A) and the chemical structure of morroniside (B).
Thus, in this study, we aimed to assess (1) whether morroniside could activate GLP‐1 receptors; (2) whether it was effective in attenuation of peripheral nerve injury‐induced neuropathic pain; and (3) whether it was able to produce antinociception by activation of spinal GLP‐1 receptors.
Methods
Cell culture and MTT bromide assay
HEK 293 cells with stable expression of human GLP‐1 receptors (a generous gift of Dr Bao‐Hong Zhang from BD Bioscience, Shanghai, China) and the parental HEK293T cell line (Applied StemCell, Sunnyvale, CA, USA) were cultured in high‐glucose DMEM containing 10% fetal bovine serum, 2.0 mM L‐glutamine, 100 μg·mL−1 streptomycin B, 100 U·mL−1 penicillin and 100 mg·mL−1 streptomycin. N9 cells, originally derived from mouse brain microglia, were purchased from the Cell Bank of Shanghai Institute for Cell Biology (Shanghai, China) and routinely cultured in Iscove's modified Dulbecco's medium containing 10% fetal bovine serum, 100 U·mL−1 penicillin and 100 mg·mL−1 streptomycin at 37°C in a humidified atmosphere of 95 and 5% CO2 and were passaged, using trypsinization, every 2 to 3 days.
Cells were seeded into 96‐well plates at a density of 6 × 103 cells per well and grown for 24 h. Hydrogen peroxide (Shanghai Lingfeng Chemical Reagent Co., Shanghai, China) was then added to a final concentration of 600 μM for N9 cells for 15 min incubation, or 400 μM for HEK293 cells and 800 μM for HEK298T cells for 5 min incubation. Next, cells were washed with PBS and treated with morroniside and exenatide at different concentrations in the presence or absence of exendin(9‐39) for 12 h. After that, 10 μL of 0.5 mg·mL−1 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyl tetrazolium bromide (MTT; Amresco, Solon, OH, USA) were added to each well and cells were incubated at 37°C for additional 4 h. Cell culture medium was then removed, and 200 μL of DMSO was added to each well and mixed using a microplate shaker to dissolve the blue MTT‐formazan crystals. Finally, the optical density was read at 570 nm against a reference wavelength of 630 nm using a Multiskan MK3 microplate reader (Thermo Labsystems, Vantaa, Finland). Changes in cell viability induced by hydrogen peroxide were expressed as a percentage of the controls. The MTT assay was performed in triplicate.
Experimental animals
All animal care and experimental procedures were approved and performed in accordance with the Animal Care and Welfare Committee of the Shanghai Jiao Tong University (Shanghai, China). Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
Adult male Wistar rats (160 to 240 g) were purchased from the Shanghai Experimental Animal Institute for Biological Sciences (Shanghai, China) and housed in plastic cages (three to four per cage) with soft bedding and free access to food and water with a temperature of 22 ± 2°C and humidity of 60%. The light was controlled with a 12 h light/dark cycle (with lights on at 0700 h). All animals were acclimatised for 3 to 7 days before surgical and experimental procedures. Experimental groups (n = 5–6 in each group) were randomly assigned and the investigator was blinded to the treatments in the behaviour testing.
Immunofluorescent staining
Double immunofluorescent labelling of GLP‐1 receptors and the microglial biomarker was performed on cultured microglial N9 cells and observed on a TCS SP8 confocal microscope (Leica Microsystem, Buffalo Grove, IL, USA). In brief, cells were placed onto poly‐L‐lysine‐coated coverslips (5 × 104 cells per well) in six‐well plates. At the end of treatments, cells were fixed in 4% paraformaldehyde and subsequently incubated in 10% goat serum (v/v) and 0.5% Triton X‐100 (v/v) in PBS for 1 h. Cells were then incubated with a rabbit polyclonal anti‐GLP‐1 receptor antibody (Ab119287, Abcam, Cambridge, UK) at a dilution of 1:200 and a mouse monoclonal anti‐OX42 antibody (Ab1211, Abcam) at a dilution of 1:200 at 4°C overnight. The GLP‐1 receptor signal was visualized with the Alexa‐555‐conjugated goat anti‐rabbit secondary antibody (1:200, Invitrogen, Grand Island, NY, USA). The optimal dilution and non‐specific binding of the anti‐GLP‐1 receptor and anti‐OX42 antibodies had been previously assessed (Avila‐Martin et al., 2011; Gong et al., 2014c).
Intrathecal catheterization and injection in rats
Surgical procedures were performed under isoflurane (4% for induction and 1% for maintenance) in oxygen anaesthesia according to protocols described previously (Gong et al., 2014a). A 18 cm PE‐10 polyethylene catheter of 0.28 mm i.d. per 0.61 mm o.d. (Clay Adams, Parsippany, NJ, USA) with volume of approximately 13 μL was inserted into the rat spinal cord at the lumbar level. After a 2 day recovery from anaesthesia, correct intrathecal cannula placement was verified by administering 4% lidocaine. Only those rats without motor impairment before lidocaine injection but with bilateral paralysis of the hindlimbs after intrathecal administration of lidocaine were selected for the experiments (such an exclusion rate was usually under 5% in our laboratory). For intrathecal administration of control and test articles, the drugs were microinjected with a 50 μL microinjector (Shanghai Anting Micro‐Injector Factory, Shanghai, China) in a volume of 10 μL followed by a normal saline flush in a volume of 15 μL.
Rat model of neuropathic pain
Rats were anaesthetized with isoflurane (4% for induction and 1% for maintenance) provided by an anesthesiameter (Ugo Basile Gas Anesthesia System). The back was shaved, and the left lumbar paravertebral region was exposed. After subperiosteal removal of sixth lumbar transverse process, both left L5 and L6 spinal nerves were isolated and tightly ligated with 6‐0 silk suture. After ligation, the lumbar fascia was closed by using the 4‐0 silk suture and the skin was then dusted with penicillin powder and sutured, and the rats were allowed to recover. Of the nerve‐ligated rats, only those with marked unilateral hypersensitivity to mechanical stimulation (hindlimb withdrawal thresholds in the operated side less than 8 g) and without major motor impairment were selected for further experiments. Drug administration was started 1–3 weeks after spinal nerve ligation.
Behavioural assessments of mechanical allodynia and thermal hyperalgesia
To evaluate mechanical allodynia, rats were acclimatised for approximately half an hour to the test environment on a plexiglass box on a metal grid (0.5 × 0.5 cm). The hindpaw withdrawal threshold was measured with a 2450 CE Electronic Von Frey (IITC Life Science Inc, Woodland Hill, CA, USA). The electronic hand‐held transducer with the number 15 monofilament (with forces ranging between 0.1 and 90 g) was applied perpendicularly to the medial surface of the hindpaws, with the force increasing until the rat suddenly withdrew or licked the paw. The lowest force producing a sudden withdrawal response was considered the nociceptive threshold; this was based on three repeated measurements within a 10 min interval and the mean of the three threshold values for each hindpaw, at each time point.
Hindpaw thermal hyperalgesia was measured according to a previous study (Li et al., 2015). After being trained at least three times, rats were placed in plexiglass boxes on an elevated glass plate, and, after 30 min adaptation, the withdrawal latency was measured by turning on a radiant heat emitted by a 390G Plantar Test Analgesia Meter (IITC Life Science Inc.). The cut‐off was set at 30 s to prevent tissue injury. The latency was recorded as the time from heat beam light turning on to hindpaw withdrawal. Both contralateral and ipsilateral hindpaws were subsequently assessed three times with a 5 min interval. The value was averaged from the three repeated measurements.
Rat rotarod test
Motor coordination performance was assessed by a YLS‐4C Rota Rod with automatic timers and falling sensors (Yiyan Scientific Ltd., Shandong, China). The rats were trained and tested by accelerating the rotarod speed from 5 to 25 rpm within 1 min followed by 25 rpm for two extra minutes. The accumulated time (s·3 min−1) for animals to spend on the rotarod was recorded during the 3 min observation period after the animals were trained once a day each for 9 min for 3 days. The accumulated time spent on the rod had to be at least 120 s to allow inclusion in the study, and the cut‐off of the accumulated time was 5 min. For the final testing, the accumulated time (s·5 min−1) spent on the rotarod was recorded after receiving control or test articles (Zhu et al., 2014).
Data and statistical analyses
The data and statistical analysis in this study comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). For the dose–response curve analysis, the minimum effect, maximum effect (Emax), half‐effective dose (ED50) or half‐effective concentration (EC50) and Hill coefficient (n) were calculated. The values of a response (Y) were fitted by nonlinear least squares curves to the relationship Y = a + bx, where x = [D]n/(ED50 n + [D]n) or [C]n/(EC50 n + [C]n), to give the value of ED50 or EC50 and b (Emax), yielding a minimum residual sum of squares of deviations from the theoretical curve according to the previous study (Wang and Pang, 1993).
For the modified Schild plot and pA2 analysis, the ratio of the concentration of an agonist (A') required to produce the EC50 in the presence of an antagonist (B) to the concentration required to produce the same response in the absence of the antagonist (A) was calculated using [A]/([A] + KD) + [A']/([A'] + KD(1 + [B]/KB)), where KD and KB are the equilibrium dissociation constants for A and B respectively (Wang et al., 1993). The pA2 was determined by applying at least three different concentrations of antagonist and plotting log ((A'/A) − 1) against the negative log B. If the regression of log ((A'/A) − 1) on −log B was linear with a slope of 1, the antagonism was competitive, and, by definition, the agonist and antagonist acted at the same recognition sites. The x‐intercept of the fitted regression line was an estimate of the pA2, which was the negative logarithm of the molar concentration of the antagonist required for a twofold increase of the agonist concentration (Gong et al. 2014a).
The data are expressed as the SEM, and there were no missing data. The statistical significance was evaluated with a two‐tailed and unpaired Student's t‐test or a two‐way ANOVA using Prism (version 5.01, GraphPad Software Inc., San Diego, CA, USA). A post hoc Student–Newman–Keuls test was applied when a statistically significant drug (dose), time and their interaction were observed. The probability values were two‐tailed, and the statistical significance criterion P value was 0.05.
Materials
Morroniside was purchased from Chengdu Push BioTechnology Co. (Chengdu, China) with a purity of greater than 98%, as determined by the manufacturer using high‐performance liquid chromatography. Exenatide and exendin(9‐39), with peptide content of greater than 98%, were obtained from Kaijie Bio‐Pharmaceuticals (Chendu, China) and Shanghai TASH Biotechnology Co. (Shanghai, China) respectively. All tested drugs were freshly dissolved in normal saline (0.9% NaCl) solution and used in the experiments immediately.
Results
Morroniside activated mouse and human GLP‐1 receptors
Microglial N9 cells are originally derived from mouse brain (Walker et al., 1995) and share many phenotypical characteristics with primary microglia (Hickman et al., 2008). Double immunofluorescence staining with the anti‐GLP‐1 receptor antibody and anti‐OX42 antibody confirmed the presence of GLP‐1 receptors in all of N9 cells (Figure 2A). Activation of GLP‐1 receptors is known to protect against hydrogen peroxide‐induced oxidative damage (Liu et al., 2007; Oeseburg et al., 2010). In order to determine whether morroniside activated GLP‐1 receptors at the cellular level, the protective effects of morroniside and exenatide against oxidative damage induced by hydrogen peroxide were assessed in N9 cells using the MTT assay for cell viability. Treatment with hydrogen peroxide (600 μM) in N9 cells for 15 min induced significant cellular oxidative damage and loss of cell viability (considered to be 0% cell viability), compared with control cells without hydrogen peroxide treatment (considered to be 100% cell viability), whereas treatment with morroniside (up to 1 × 10−4 M) and exenatide (up to 1 × 10−7 M) over 12 h was not associated with any cellular dysfunction. As shown in Figure 2B, C, treatment with exenatide (1 × 10−11, 1 × 10−10, 1 × 10−9, 1 × 10−8 and 1 × 10−7 M) and morroniside (1 × 10−8, 1 × 10−7, 1 × 10−6, 1 × 10−5 and 1 × 10−4 M) for 12 h inhibited hydrogen peroxide‐induced loss of cell viability in a concentration‐dependent manner. Dose–response analyses showed that the maximal protection was 100% for both morroniside and exenatide, and the EC50s were 3.6 × 10−7 M for morroniside and 4.5 × 10−10 M for exenatide. Co‐treatment with the specific GLP‐1 receptor orthosteric antagonist exendin(9‐39) (1 × 10−7, 3 × 10−7 and 1 × 10−6 M) concentration‐dependently shifted the concentration response curves of both morroniside and exenatide to the right, without affecting maximal protection. The modified Schild plots analyses showed that the pA2 values were 7.78 and 7.17 for morroniside and exenatide respectively.
Figure 2.
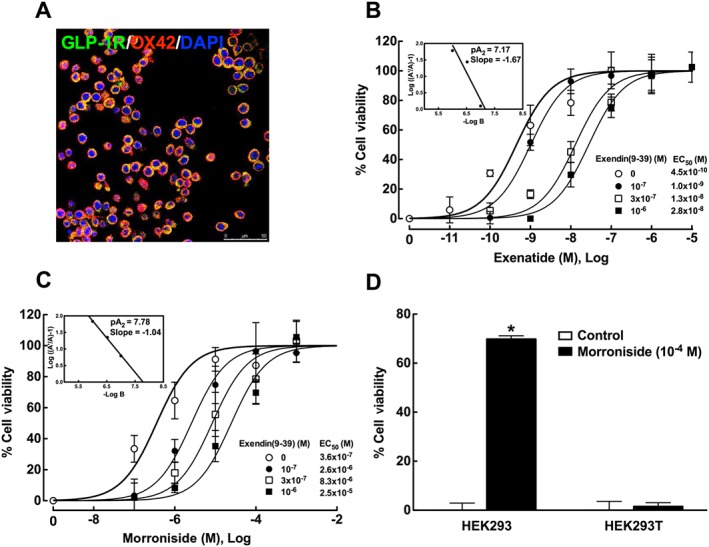
Expression of GLP‐1 receptors (A) and the protective effects of exenatide and morroniside against hydrogen peroxide‐induced loss of cell viability in N9 microglial cells (B, C) and HEK293 cells (D). Double immunofluorescence staining was conducted using anti‐GLP‐1 receptor and anti‐OX42 antibodies. Hydrogen peroxide‐induced loss of cell viability was assessed using the MTT assay. The concentration–response curves of morroniside and exenatide were constructed in the presence and absence of the GLP‐1 receptor antagonist exendin(9‐39). Schild plots of exendin(9‐39) on morroniside and exenatide are inserted in B and C. Data are presented as means ± SEM ;n = 6 with triplicates at each concentration. *P < 0.05, significantly different from control groups; two‐tailed and unpaired Student's t‐test.
We previously showed the presence and absence of human GLP‐1 receptors on HEK293 cells and HEK293T cells using immunofluorescence staining, and the protective effect of exenatide against loss of cell viability induced by hydrogen peroxide in HEK293 cells but not in HEK293T cells (Fan et al., 2015). Morroniside was further employed to test whether it could activate GLP‐1 receptors in HEK293 cells. Treatment with hydrogen peroxide in HEK293 (400 μM) and HEK293T cells (800 μM) for 5 min induced significant loss of cell viability (0% cell viability), compared with control cells (100% cell viability). Treatment with morroniside (10−4 M) alone did not significantly affect cellular functions in HEK293 or HEK293T cells. However, morroniside markedly protected against hydrogen peroxide‐induced loss of cell viability in HEK293 cells expressing GLP‐1 receptors (P < 0.05 using the two‐tailed and unpaired Student's t‐test), whereas it was not effective in altering cellular dysfunctions induced by hydrogen peroxide in HEK293T cells that did not express GLP‐1 receptors (Figure 2D).
Morroniside exhibited mechanical anti‐allodynia and thermal hyperalgesia in a rat model of neuropathic pain
Peripheral nerve injury causes clinically relevant painful peripheral neuropathy (Kim and Chung, 1992). Tight ligation of L5 and L6 spinal nerves produced immediate and long‐lasting mechanical allodynia and thermal hyperalgesia in the ipsilateral hindpaws. Five groups of neuropathic rats received oral gavage of saline (3 mL·kg−1) and a range of doses of morroniside (30, 100, 300 and 600 mg·kg−1). Paw withdrawal thresholds were measured before and 0.5, 1, 2 and 4 h after oral gavage. After normal saline, paw withdrawal thresholds to mechanical stimuli in both contralateral and ipsilateral paws remained unchanged during the 4 h observation period. Intragastric administration of morroniside dose‐dependently suppressed mechanical allodynia in the ipsilateral paws but did not significantly alter withdrawal thresholds in the contralateral paws (Figure 3A). The mechanical antiallodynic effect was time‐dependent, with peak effect at 1 h or earlier after oral gavage and a duration of longer than 4 h. Dose–response analysis showed that the ED50 was 335 mg·kg−1 and the Emax was 75% inhibition, as calculated from the values at 1 h after oral gavage (Figure 3B).
Figure 3.
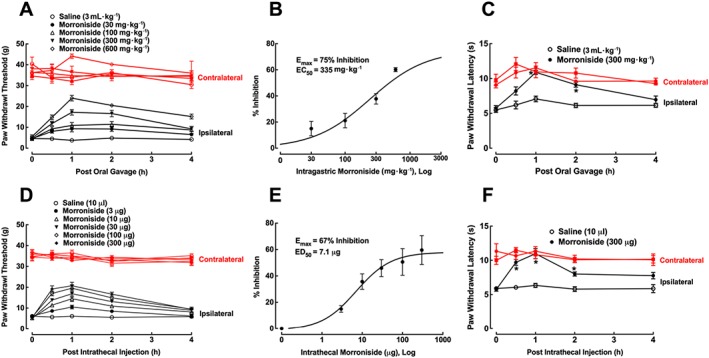
Mechanical antiallodynic and thermal antihyperalgesic effects of oral gavage (A–C) and intrathecal (D–F) morroniside administration in a rat model of neuropathic pain. Peripheral neuropathy was induced by tight ligation of L5/L6 spinal nerves and the paw withdrawal responses to electronic von Frey filaments and radiant heat were measured (B and E). Dose–response analyses of morroniside on mechanical hypersensitivity in the ipsilateral paw were calculated using the thresholds measured 1 h after drug administration, as projected by the non‐linear least‐squares method. Data are presented as means ± SEM; n = 6 per group. *P < 0.05, significantly different from saline control group; repeated measures two‐way ANOVA followed by post hoc Student–Newman–Keuls test.
An additional two groups of rats with neuropathic pain received oral gavage of normal saline (3 mL·kg−1) or morroniside (300 mg·kg−1). In normal saline‐treated rats, hindpaw withdrawal latencies to thermal stimuli in both contralateral and ipsilateral paws remained unchanged during the 4 h observation period. Intragastric administration of morroniside time‐dependently suppressed thermal hyperalgesia in the ipsilateral paws, with a peak effect of 100% inhibition at 1 h after gavage (P < 0.05, repeated measures two‐way ANOVA followed by the post hoc Student–Newman–Keuls test) but did not significantly alter withdrawal latencies in the contralateral paws (Figure 3C).
In six groups of neuropathic rats with intrathecal catheters and receiving intrathecal injection of normal saline (10 μL) or morroniside (3, 10, 30, 100 and 300 μg) the paw withdrawal thresholds were measured before and 0.5, 1, 2 and 4 h after intrathecal injection. Paw withdrawal thresholds in control rats remained unchanged during the observation period of time. Morroniside given intrathecally attenuated mechanical allodynia in the ipsilateral paws in a time‐ and dose‐dependent manner, without affecting paw withdrawal thresholds in the contralateral paws (Figure 3D). Dose–response analysis showed an Emax of 67% inhibition and ED50 of 7.1 μg, derived from the mechanical values at 1 h after injection (Figure 3E).
Two additional groups of neuropathic rats received intrathecal injection of normal saline (10 μL) or morroniside (300 μg), and paw withdrawal latencies to radiant heat were measured before and 0.5, 1, 2 and 4 h after intrathecal injection. Paw withdrawal latencies in saline control rats remained unchanged during the observation period of time. Intrathecal morroniside time‐dependently attenuated thermal hyperalgesia in the ipsilateral paws, with a peak effect of 100% at 1 h after intrathecal injection (P < 0.05; repeated measures two‐way ANOVA followed by the post hoc Student–Newman–Keuls test), without affecting paw withdrawal thresholds in the contralateral paws (Figure 3F).
There was no apparent sedation or motor side effects of morroniside, given either by oral gavage or intrathecally, observed during the experimental period of time. The rotarod test was conducted to examine further the possible motor side effects of morroniside. Two groups of normal rats (n = 5 in each group) received oral gavage of normal saline (3 mL·kg−1) or morroniside (600 mg·kg−1). The rotarod test (25 rpm) was undertaken 1 h after gavage. The accumulated time spent on the rotarod was 296 ± 8.9 and 287 ± 29.1 s·per 5 min in normal saline‐ and morroniside‐treated rats respectively. In addition, two groups of normal rats with intrathecal catheters (n = 5 in each group) received intrathecal injection of saline (10 μL) or morroniside (300 μg) and were subjected to the rotarod test. The accumulated time was 288 ± 26.8 and 298 ± 4.5 s·per 5 min in normal saline‐ and morroniside‐treated rats respectively.
Tolerance to analgesia is one of the major disadvantages of clinical application of opiates, especially morphine. Given its wide clinical use in China in treatment of different diseases, we assessed whether long‐term treatment with morroniside showed tolerance to mechanical antiallodynia in neuropathic rats. Two groups of neuropathic rats received daily intrathecal injection of saline (10 μL·day−1) or morroniside (100 μg·day−1) for 7 days. Both contralateral and ipsilateral paw withdrawal thresholds were measured 1 h after each intrathecal injection over 7 days. Normal saline did not significantly affect withdrawal thresholds in either contralateral or ipsilateral paws over the observation period of time. In contrast, intrathecal injection with morroniside significantly increased paw withdrawal thresholds in the ipsilateral paws by 58%. The marked mechanical antiallodynic effect persisted for the 7 day intrathecal injections without an obvious decline (P < 0.05 using the repeated measures two‐way ANOVA followed by the post hoc Student–Newman–Keuls test; Figure 4).
Figure 4.
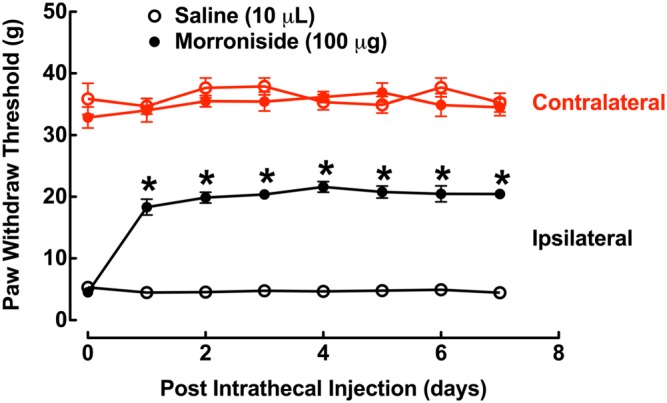
Mechanical antiallodynic effects of repeated daily intrathecal injections of morroniside (100 μg) in a rat model of neuropathic pain. Peripheral neuropathy was induced by tight ligation of L5/L6 spinal nerves and the paw withdrawal thresholds were measured using an electronic von Frey filaments 1 h after each drug administration. Data are presented as means ± SEM; n = 6 for each group. *P < 0.05, significantly different from saline control group; repeated measures two‐way ANOVA followed by post hoc Student–Newman–Keuls test.
Morroniside produced mechanical antiallodynia by activation of spinal GLP‐1 receptors
To explore whether the antiallodynic effect of morroniside was due to activation of spinal GLP‐1 receptors, two groups of neuropathic rats received intrathecal injection of saline (10 μL) or exendin(9‐39) (2 μg), 30 min followed by morroniside (300 mg·kg−1) by oral gavage. Paw withdrawal thresholds were measured before and 0.5, 1, 2 and 4 h after morroniside administration. As shown in Figure 5A, 300 mg·kg−1 oral morroniside produced time‐dependent mechanical antiallodynia in the ipsilateral paws. Although intrathecal injection of 2 μg exendin(9‐39) was previously shown not to significantly alter withdrawal responses in either contralateral or ipsilateral paws (Gong et al., 2014a; Gong et al., 2014c), it completely blocked systemic morroniside‐induced mechanical antiallodynia in the ipsilateral paws (P < 0.05, repeated measures two‐way ANOVA followed by the post hoc Student–Newman–Keuls test).
Figure 5.
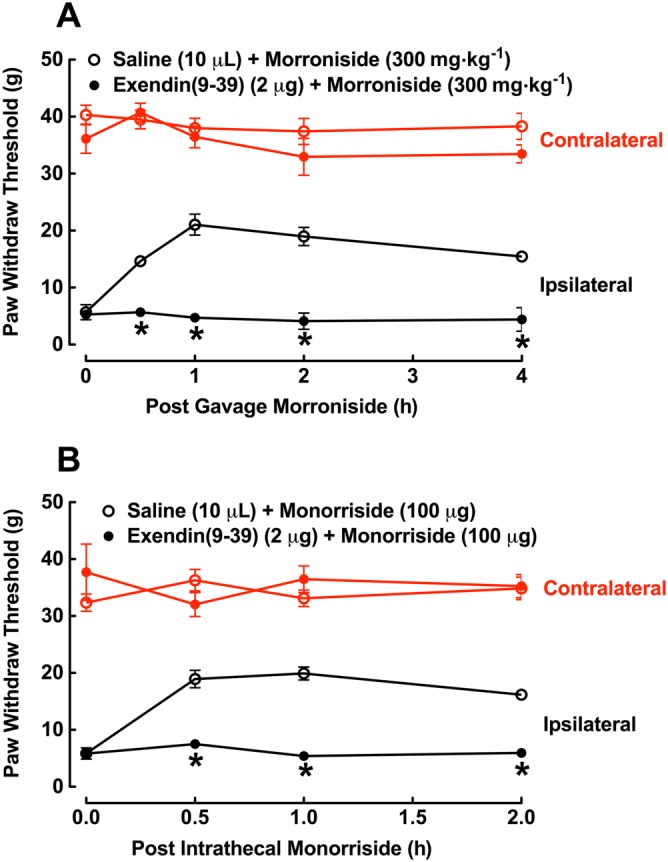
Blockade of intrathecal injection of GLP‐1 receptor antagonist exendin(9‐39) on the mechanical antiallodynic effects of oral (A) and intrathecal (B) morroniside in a rat model of neuropathic pain, induced by tight ligation of L5/L6 spinal nerves. Rats received intrathecal injection of saline (10 μL) or exendin(9‐39), 30 min followed by morroniside administered by oral gavage or intrathecal injection. Hindpaw withdrawal thresholds were measured using an electronic von Frey filaments before and after morroniside administration. Data are presented as means ± SEM; n = 6 of each group. *P < 0.05, significantly different from saline control group; repeated measures two‐way ANOVA followed by post hoc Student–Newman–Keuls test.
In addition, two groups of neuropathic rats with intrathecal catheters also received intrathecal injection of saline (10 μL) or exendin(9‐39) (2 μg), 30 min followed by intrathecal injection of morroniside (100 μg). Paw withdrawal thresholds were measured before and 0.5, 1, 2 and 4 h after intrathecal morroniside. Morroniside in the ipsilateral paws produced time‐dependent mechanical antiallodynia, which was completely inhibited by pretreatment with intrathecal exendin(9‐39) (P < 0.05, repeated measures two‐way ANOVA followed by the post hoc Student–Newman–Keuls test; Figure 5B).
Discussion
The protective effect of morroniside against hydrogen peroxide‐induced oxidative cytotoxicity has been extensively studied in human neuroblastoma SH‐SY5Y cells (Ai et al., 2008; Wang et al., 2008; Wang et al., 2009a; Wang et al., 2009b). We extended this finding to assess its protection in mouse microglial N9 cells and explore the mechanisms underlying morroniside antihypersensitivity, as GLP‐1 receptors are specifically expressed on microglia in the spinal dorsal horn but not in neurons or astrocytes, while GLP‐1 receptor agonists induced expression/release of β‐endorphin in microglia but not in neurons or astrocytes (Gong et al., 2014c; Fan et al., 2015; Fan et al., 2016). Our double immunofluorescence staining on microglial N9 cells revealed the expression of GLP‐1 receptors, which are known to induce protection against oxidative damage induced by hydrogen peroxide in different types of cells (Liu et al., 2007; Oeseburg et al., 2010). Although it was much less potent than exenatide, morroniside reversed hydrogen peroxide‐induced oxidative damage in a concentration‐dependent manner, with maximum protection of 100%. Furthermore, the pretreatment with exendin(9‐39) in microglial N9 cells concentration‐dependently shifted the concentration response curves of both morroniside and exenatide to the right without affecting their maximal protection, indicating a competitive blockade effect. Their similar pA2 values (7.8 vs. 7.2) suggest that morroniside, like exenatide, is a reversible and orthosteric agonist of GLP‐1 receptors with full efficacy and probably acts at the same binding site as exenatide and exendin(9‐39) (Osswald, 1975; Doxey et al., 1977; Wang et al., 1993). The GLP‐1 receptor‐dependent protection by morroniside was further shown by the nearly complete protection against hydrogen peroxide‐induced oxidative damage in HEK293 cells with stable expression of human GLP‐1 receptors but not in HEK293T cells without GLP‐1 receptor expression.
We have previously demonstrated that iridoid compounds, including geniposide, geniposidic acid, genipin methyl ether, 1,10‐anhydrogenipin, loganin, catalpol, shanzhiside methylester and 8‐O‐acetyl‐shanzhiside methylester, are agonists of human and rat GLP‐1 receptors and presumably function by binding to the same site as exendin(9‐39) (Gong et al., 2014a; Zhu et al., 2014). This study has added our attempts to the list. Morroniside is an atypical secoiridoid with a broken double bond at C7 and C8 in the five‐membered carbon ring and is replaced by a six‐membered cyclic inner ether fragment. Thus, our current data demonstrated that the atypical secoiridoid can retain its biological activity regardless of the presence of the double bond in the scaffold of cyclopentapyran. In fact, the change in structure may be responsible for the higher potency than typical iridoid glycosides, in activation of GLP‐1 receptors. The data from our current study showed the EC50 (3.6 × 10−7 M) for morroniside to protect cell damage from hydrogen peroxide was at least one order of magnitude lower than those of shanzhiside methylester (9.3 × 10−5 M), 8‐O‐acetyl shanzhiside methylester (1.2 × 10−4 M), geniposide (1.6 × 10−4 M), geniposidic acid (2.4 × 10−4 M), loganin (1.6 × 10−4 M) and catalpol (2.0 × 10−4 M) (Zhu et al., 2014; Gong et al., 2014c). Furthermore, intrathecal injection of morroniside more potently inhibited neuropathic pain (ED50 of 7.1 μg or 1.7 × 10−7 mol) than shanzhiside methylester (ED50 of 40.4 μg or 9 × 10−7 mol) (Fan et al., 2016).
In addition, these iridoid glycoside compounds activated GLP‐1 receptors expressed in rat PC12 cells (Zhu et al., 2014; Gong et al., 2014c; Fan et al., 2015), human HEK293 cells (Zhu et al., 2014; present study) and mouse N9 cells (present study), suggesting that iridoid compounds, including morroniside, like the peptide agonist exenatide and the non‐peptide agonist WB4‐24, do not exhibit apparent species specificity to activate GLP‐1 receptors between human, rat and mouse cells, providing pharmacological evidence for the feasibility of clinical use of morroniside in control of chronic pain. On the other hand, GLP‐1 receptor peptidic agonists (exenatide, liraglutide and lixisenatide) are used to treat type 2 diabetes mellitus mainly by induction of insulin secretion and promotion of insulin sensitivity (Baggio and Drucker, 2007; Koole et al., 2013). Indeed, oral administration of morroniside and loganin decreased blood glucose levels in diabetic mice (He et al., 2016), and C. officinalis extracts attenuated diabetic angiopathy and the early stage of diabetic nephropathy (Xu et al., 2004; Xu et al., 2006). It is worth further investigating their anti‐diabetic effects in relation to activation of GLP‐1 receptors.
Our results demonstrated that morroniside, given both orally and intrathecally, produced long‐lasting mechanical antiallodynia and thermal hyperalgesia in the ipsilateral paws of rats with neuropathy induced by tight ligation of L5 and L6 spinal nerves. The peak effect occurred 1 h or earlier after administration and lasted longer than 4 h. Morroniside exhibited higher efficacy in thermal antihyperalgesia (100% inhibition) than mechanical antiallodynia (approximately 70% inhibition). In contrast, morroniside did not alter normal nociceptive responses to either mechanical or thermal stimuli in the contralateral paws. The mechanical antiallodynic effects of oral and intrathecal administration of morroniside were dose‐dependent, with maximum inhibition of 67% and 75% and ED50 values of 335 mg·kg−1 and 7.1 μg respectively. The GLP‐1 receptors in the spinal dorsal horn have been recently been identified as a potential target in control of chronic pain (Gong et al., 2014c; Fan et al., 2015). Systemic and intrathecal injection of the peptide GLP‐1 receptor agonists GLP‐1(7‐36) and exenatide, the non‐peptide agonist WB4‐2 and iridoid agonists (shanzhiside methylester, geniposide, loganin and catalpol) produced antinociception in a variety of rodent models of pain hypersensitivity, including neuropathic pain, formalin‐induced tonic pain, complete Freund's adjuvant‐induced inflammatory pain (both mechanical and thermal hypersensitivity), bone cancer pain and diabetic neuropathic pain. Their antinociceptive effects on pain hypersensitivity were completely blocked by specific GLP‐1 receptor antagonist exendin(9‐39) or knockdown of GLP‐1 receptor expression using GLP‐1 receptor siRNA (Zhu et al., 2014; Gong et al., 2014c; Fan et al., 2015; Fan et al., 2016). Similarly, the mechanical antiallodynic effects of intrathecal and particularly oral administration of morroniside were completely blocked by pretreatment with intrathecal injection of exendin(9‐39). Thus, we suggest that morroniside produced its antihypersensitivity effects through activation of GLP‐1 receptors, localized in the spinal cord, rather than the brain ventral area or peripheral nociceptors. The results imply that morroniside and C. officinalis may be effective in treating neuropathic pain clinically and further support the notion that spinal GLP‐1 receptors are a potential target for the treatment of chronic pain, including neuropathic pain (Zhu et al., 2014, Gong et al., 2014a, Gong et al., 2014c, Fan et al., 2015, Fan et al., 2016).
The development of tolerance to morphine analgesia is a serious disadvantage for its long‐term use, in control of chronic pain (Collett, 1998; Inturrisi, 2002). We explored whether morroniside would also induce tolerance to antinociception following chronic treatments. Our data showed that intrathecal morroniside injections, repeated over 7 days, did not apparently induce mechanical antiallodynic tolerance, in contrast to the 7 day treatments with morphine which induced complete antinociceptive tolerance in rats with spinal nerve ligation (Gong et al., 2014b; Fan et al., 2016). The results are consistent with our previous study showing that GLP‐1 receptor activation by exenatide did not induce antinociceptive tolerance in neuropathic pain (Gong et al., 2014c).
In summary, morroniside, like exenatide, protected microglial N9 cells and HEK293 cells that express mouse and human GLP‐1 receptors, respectively, from oxidative damage induced by hydrogen peroxide. The protective effects of morroniside and exenatide were also competitively antagonized by exendin(9‐39), suggesting that morroniside is an orthosteric GLP‐1 receptor agonist with a full efficacy, probably acting at the same binding site as exenatide and exendin(9‐39). Oral and intrathecal morroniside administration in a rat model of neuropathic pain produced mechanical antiallodynia and thermal antihyperalgesia, which were completely blocked by pretreatment with intrathecal exendin(9‐39). Furthermore, repeated daily injections of morroniside did not induce antinociceptive tolerance. Our study suggests that morroniside is an orthosteric agonist of GLP‐1 receptors and produces antihypersensitivity activities in neuropathic pain models by activation of spinal GLP‐1 receptors.
Author contributions
Y.X.W. and M.X. conceived and designed the experiments; M.X., H.Y.W., H.L., N.G. and Y.R.W. performed the experiments; Y.X.W. and M.X. analysed the data; and Y.X.W., M.X. and N.G. wrote the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
This study was supported, in part, by grants from the National Natural Science Foundation of China (no. 81374000) and the Shanghai Industrial Translational Project (no. 15401901300). We thank Dr Dongshen Xie at Shanghai Jiao Tong University School of Pharmacy for his constructive discussion of the morroniside structure and bioactivity.
Xu, M. , Wu, H.‐Y. , Liu, H. , Gong, N. , Wang, Y.‐R. , and Wang, Y.‐X. (2017) Morroniside, a secoiridoid glycoside from Cornus officinalis , attenuates neuropathic pain by activation of spinal glucagon‐like peptide‐1 receptors. British Journal of Pharmacology, 174: 580–590. doi: 10.1111/bph.13720.
References
- Ai HX, Wang W, Sun FL, Huang WT, An Y, Li L (2008). Morroniside inhibits H2O2‐induced apoptosis in cultured nerve cells. Zhongguo Zhong Yao Za Zhi 33: 2109–2112. [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015). The Concise Guide to PHARMACOLOGY 2015/16: G Protein‐Coupled Receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avila‐Martin G, Galan‐Arriero I, Gomez‐Soriano J, Taylor J (2011). Treatment of rat spinal cord injury with the neurotrophic factor albumin‐oleic acid: translational application for paralysis, spasticity and pain. PLoS One 6: e26107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baggio LL, Drucker DJ (2007). Biology of incretins: GLP‐1 and GIP. Gastroenterology 132: 2131–2157. [DOI] [PubMed] [Google Scholar]
- Collett BJ (1998). Opioid tolerance: the clinical perspective. Br J Anaesth 81: 58–68. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doxey J, Smith C, Walker JM (1977). Selectivity of blocking agents for pre‐ and postsnaptic‐adrenoceptors. Br J Pharmacol 60: 91–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan H, Gong N, Li TF, Ma AN, Wu XY, Wang MW et al. (2015). The non‐peptide GLP‐1 receptor agonist WB4‐24 blocks inflammatory nociception by stimulating beta‐endorphin release from spinal microglia. Br J Pharmacol 172: 64–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan H, Li T‐F, Gong N, Wang Y‐X (2016). Shanzhiside methylester, the principle effective iridoid glycoside from the analgesic herb Lamiophlomis rotata, reduces neuropathic pain by stimulating spinal microglial β‐endorphin expression. Neuropharmacology 101: 98–109. [DOI] [PubMed] [Google Scholar]
- Gong N, Fan H, Ma AN, Xiao Q, Wang YX (2014a). Geniposide and its iridoid analogs exhibit antinociception by acting at the spinal GLP‐1 receptors. Neuropharmacology 84: 31–45. [DOI] [PubMed] [Google Scholar]
- Gong N, Li XY, Xiao Q, Wang YX (2014b). Identification of a novel spinal dorsal horn astroglial D‐amino acid oxidase‐hydrogen peroxide pathway involved in morphine antinociceptive tolerance. Anesthesiology 120: 962–975. [DOI] [PubMed] [Google Scholar]
- Gong N, Xiao Q, Zhu B, Zhang CY, Wang YC, Fan H et al. (2014c). Activation of spinal glucagon‐like peptide‐1 receptors specifically suppresses pain hypersensitivity. J Neurosci 34: 5322–5334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen HH, Fabricius K, Barkholt P, Niehoff ML, Morley JE, Jelsing J et al. (2015). The GLP‐1 receptor agonist liraglutide improves memory function and increases hippocampal CA1 neuronal numbers in a senescence‐accelerated mouse model of Alzheimer's disease. J Alzheimers Dis 46: 877–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harkavyi A, Whitton PS (2010). Glucagon‐like peptide 1 receptor stimulation as a means of neuroprotection. Br J Pharmacol 159: 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He K, Song S, Zou Z, Feng M, Wang D, Wang Y et al. (2016). The hypoglycemic and synergistic effect of loganin, morroniside, and ursolic acid isolated from the fruits of Cornus officinalis . Phytother Res 30: 283–291. [DOI] [PubMed] [Google Scholar]
- Hickman SE, Allison EK, El Khoury J (2008). Microglial dysfunction and defective beta‐amyloid clearance pathways in aging Alzheimer's disease mice. J Neurosci 28: 8354–8360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holscher C (2012). Potential role of glucagon‐like peptide‐1 (GLP‐1) in neuroprotection. CNS Drugs 26: 871–882. [DOI] [PubMed] [Google Scholar]
- Inturrisi CE (2002). Clinical pharmacology of opioids for pain. Clin J Pain 18: S3–13. [DOI] [PubMed] [Google Scholar]
- Jia Y, Gong N, Li TF, Zhu B, Wang YX (2015). Peptidic exenatide and herbal catalpol mediate neuroprotection via the hippocampal GLP‐1 receptor/beta‐endorphin pathway. Pharmacol Res 102: 276–285. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG, Group NCRRGW (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Chung JM (1992). An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain 50: 355–363. [DOI] [PubMed] [Google Scholar]
- Kim S, Moon M, Park S (2009). Exendin‐4 protects dopaminergic neurons by inhibition of microglial activation and matrix metalloproteinase‐3 expression in an animal model of Parkinson's disease. J Endocrinol 202: 431–439. [DOI] [PubMed] [Google Scholar]
- Koole C, Pabreja K, Savage EE, Wootten D, Furness SG, Miller LJ et al. (2013). Recent advances in understanding GLP‐1R (glucagon‐like peptide‐1 receptor) function. Biochem Soc Trans 41: 172–179. [DOI] [PubMed] [Google Scholar]
- Lee KY, Sung SH, Kim SH, Jang YP, Oh TH, Kim YC (2009). Cognitive‐enhancing activity of loganin isolated from Cornus officinalis in scopolamine‐induced amnesic mice. Arch Pharm Res 32: 677–683. [DOI] [PubMed] [Google Scholar]
- Li CY, Li L, Li YH, Ai HX, Zhang L (2005). Effects of extract from Cornus officinalis on nitric oxide and NF‐jB in cortex of cerebral infarction rat model. China J Chinese Mater Med 30: 1667–1671. [PubMed] [Google Scholar]
- Li X, Huo C, Wang Q, Zhang X, Sheng X, Zhang L (2007). Identification of new metabolites of morroniside produced by rat intestinal bacteria and HPLC‐PDA analysis of metabolites in vivo. J Pharm Biomed Anal 45: 268–274. [DOI] [PubMed] [Google Scholar]
- Li TF, Fan H, Wang YX (2015). Epidural sustained release ropivacaine prolongs anti‐allodynia and anti‐hyperalgesia in developing and established neuropathic pain. PLoS One 10: e0117321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Yin F, Zheng X, Jing J, Hu Y (2007). Geniposide, a novel agonist for GLP‐1 receptor, prevents PC12 cells from oxidative damage via MAP kinase pathway. Neurochem Int 51: 361–369. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oeseburg H, De Boer RA, Buikema H, Van der Harst P, Van Gilst WH, Sillje HH (2010). Glucagon‐like peptide 1 prevents reactive oxygen species‐induced endothelial cell senescence through the activation of protein kinase A. Arterioscler Thromb Vasc Biol 30: 1407–1414. [DOI] [PubMed] [Google Scholar]
- Osswald H (1975). Renal effects of adenosine and their inhibition by theophylline in dogs. Naunyn Schmiedebergs Arch Pharmacol 288: 79–86. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44 (Database Issue): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker WS, Gatewood J, Olivas E, Askew D, Havenith CE (1995). Mouse microglial cell lines differing in constitutive and interferon‐gamma‐inducible antigen‐presenting activities for naive and memory CD4+ and CD8+ T cells. J Neuroimmunol 63: 163–174. [DOI] [PubMed] [Google Scholar]
- Wang YX, Pang CC (1993). Functional integrity of the central and sympathetic nervous systems is a prerequisite for pressor and tachycardic effects of diphenyleneiodonium, a novel inhibitor of nitric oxide synthase. J Pharm Exp Ther 265: 263–272. [PubMed] [Google Scholar]
- Wang YX, Poon CI, Pang CC (1993). Vascular pharmacodynamics of NG‐nitro‐L‐arginine methyl ester in vitro and in vivo. J Pharmacol Exp Ther 267: 1091–1099. [PubMed] [Google Scholar]
- Wang SF, Chen XG, Hu ZD, Ju Y (2003). Analysis of three effective components in Fructus corni and its preparations by micellar electrokinetic capillary chromatography. Biomed Chrom 17: 306–311. [DOI] [PubMed] [Google Scholar]
- Wang W, Huang W, Li L, Ai H, Sun F, Liu C et al. (2008). Morroniside prevents peroxide‐induced apoptosis by induction of endogenous glutathione in human neuroblastoma cells. Cell Mol Neurobiol 28: 293–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Sun F, An Y, Ai H, Zhang L, Huang W et al. (2009a). Morroniside protects human neuroblastoma SH‐SY5Y cells against hydrogen peroxide‐induced cytotoxicity. Eur J Pharmacol 613: 19–23. [DOI] [PubMed] [Google Scholar]
- Wang W, Sun F, An Y, Huang W, Ai H, Zhang L et al. (2009b). Morroniside inhibits hydrogen peroxide‐induced calcium overload and cytotoxicity in SH‐SY5Y neuroblastoma cells. Chin J Rehabil Theory Pract 15: 201–202. [Google Scholar]
- Wang W, Xu J, Li L, Wang P, Ji X, Ai H et al. (2010). Neuroprotective effect of morroniside on focal cerebral ischemia in rats. Brain Res Bull 83: 196–201. [DOI] [PubMed] [Google Scholar]
- Willard FS, Bueno AB, Sloop KW (2012). Small molecule drug discovery at the glucagon‐like peptide‐1 receptor. Exp Diabetes Res 2012: 709893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H‐Q, Hao H‐P, Zhang X, Pan Y (2004). Morroniside protects cultured human umbilical vein endothelial cells from damage by high ambient glucose. Acta Pharmacol Sin 25: 412–415. [PubMed] [Google Scholar]
- Xu H, Shen J, Liu H, Shi Y, Li L, Wei M (2006). Morroniside and loganin extracted from Cornus officinalis have protective effects on rat mesangial cell proliferation exposed to advanced glycation end products by preventing oxidative stress. Can J Physiol Pharmacol 84: 1267–1273. [DOI] [PubMed] [Google Scholar]
- Yao RQ, Zhang L, Wang W, Li L (2009). Cornel iridoid glycoside promotes neurogenesis and angiogenesis and improves neurological function after focal cerebral ischemia in rats. Brain Res Bull 79: 69–76. [DOI] [PubMed] [Google Scholar]
- Zhu B, Gong N, Fan H, Peng CS, Ding XJ, Jiang Y et al. (2014). Lamiophlomis rotata, an orally available Tibetan herbal painkiller, specifically reduces pain hypersensitivity states through the activation of spinal glucagon‐like peptide‐1 receptors. Anesthesiology 121: 835–851. [DOI] [PubMed] [Google Scholar]