

Abstract
Background and Purpose
Ribosomal protein S3 (RPS3) is a 40S ribosomal protein of the S3P family essential for implementing protein translation. RPS3 has recently been found to interact with the p65 subunit of the NF‐κB complex and promote p65 DNA‐binding activity. Persistent activation of the NF‐κB pathway is evident in allergic asthma. We hypothesized that gene silencing of lung RPS3 can ameliorate allergic airway inflammation.
Experimental Approach
The gene silencing efficacy of RPS3 siRNA was screened in three different mouse cell lines by real‐time PCR and immunoblotting. Protective effects of intratracheal RPS3 siRNA in a house dust mite (HDM) mouse asthma model were determined by measuring cell counts in lung lavage fluid and lung sections, lung cytokine profiles and airway hyperresponsiveness (AHR).
Key Results
RPS3 siRNA markedly knocked down RPS3 levels in all mouse cell lines tested, and in mouse lung tissues, blocked TNF‐α‐ or HDM‐induced release of mediators by the cultured cells and reduced eosinophil counts in lung lavage fluid from the HDM mouse asthma model. RPS3 siRNA lessened HDM‐induced airway mucus hypersecretion, cytokine production and serum IgE elevation. Moreover, RPS3 knockdown significantly suppressed methacholine‐induced AHR in experimental asthma. RPS3 siRNA disrupted TNF‐α‐induced NF‐κB activation in a NF‐κB reporter gene assay in vitro and prevented the nuclear accumulation of p65 subunit and p65 transcriptional activation in HDM‐challenged lungs and cells.
Conclusions and Implications
RPS3 gene silencing ameliorates experimental asthma, probably by disrupting NF‐κB activity. RPS3 could be a novel therapeutic target for allergic airway inflammation.
Abbreviations
- AHR
airway hyperresponsiveness
- BAL
bronchoalveolar lavage
- Cdyn
dynamic compliance
- HDM
house dust mite
- IKK
inhibitory κB kinase
- iNOS
inducible NOS
- KH
K homology
- Muc5ac
mucin 5AC
- Rl
lung resistance
- RPS3
ribosomal protein S3
- siRNA
small interfering RNA
- TBP
TATA binding protein
Tables of Links
| LIGANDS | |
|---|---|
| Eotaxin (CCL11) | IL‐5 |
| GM‐CSF | IL‐6 |
| IL‐1β | IL‐13 |
| IL‐4 | KC (CXCL1) |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,bAlexander et al., 2015a,b).
Introduction
Allergic asthma is characterized by chronic airway inflammation and the infiltration of immune cells, especially eosinophils and neutrophils, mucus hypersecretion, airway hyperresponsiveness (AHR) and airway remodelling (Lambrecht and Hammad, 2015). Among many clinically relevant allergens linked to the development of asthma, house dust mite (HDM) is the most commonly encountered aeroallergen (Calderón et al., 2014). Corticosteroids are the first line anti‐inflammatory controllers for asthma, and one of their mechanisms of action is through inhibition of NF‐κB, a master transcription factor for pro‐inflammatory gene expression (Coutinho and Chapman, 2011). Histological evidence has revealed significant activation of NF‐κB in the isolated bronchial epithelium from asthmatics (Trejo Bittar et al., 2015). While corticosteroids are highly effective, their usage comes with severe side effects, and about 5–10% of severe asthmatics are steroid‐refractory (Barnes, 2013). Direct inhibitors of inhibitory κB kinase β (IKKβ) and the NF‐κB complex have been shown to have protective effects in experimental asthma models (Edwards et al., 2009; Ogawa et al., 2011). To further minimize potential adverse effects, a strategy that targets accessory signalling molecules, which positively regulate the NF‐κB pathway, becomes more pharmacologically viable. We have recently reported potent anti‐inflammatory effects of a small interfering RNA (siRNA) targeting receptor interacting protein 2 (Rip2), an inducible transcriptional product of NF‐κB activation and an upstream positive regulator of NF‐κB pathway, in an ovalbumin‐induced mouse model of asthma (Goh et al., 2013).
Ribosomal protein S3 (RPS3) is a 40S ribosomal protein that belongs to the RPS3P family essential for protein translation. RPS3 consists of a nuclear protein K homology (KH) domain and a nuclear localization signal domain (Wan et al., 2007). Recent findings have shown that RPS3 plays an extra‐ribosomal role by being an integral part of the NF‐κB complex (Wan et al., 2007; Wan and Lenardo, 2010). The KH domain facilitates the binding of RPS3 to the p65 subunit of NF‐κB, and the subsequent nuclear accumulation and binding of the NF‐κB complex to the κB sites of a number of target genes. The electrophoretic mobility shift assay (EMSA) has revealed a synergistic effect of RPS3 on the DNA‐binding activity of p65 homodimers and p50‐p65 heterodimers (Wan et al., 2007). While more than 100 other proteins have been identified to be able to interact with the p65 subunit, none can dramatically increase the DNA‐binding affinity of NF‐κB (Wan and Lenardo, 2010). RPS3 has been shown to promote the expression of Ig‐κ light chain gene in B cells, T cell proliferation and IL‐2 and IL‐8 production in a NF‐κB‐dependent manner (Wan et al., 2007).
RNA interference is a natural process produced by turning down specific gene expression via endogenous siRNA (Kole et al., 2012). Gene silencing by siRNA is highly targeted and sequence‐specific, and synthetic siRNA can be given by inhalation to knock down specific protein targets in the lungs as an experimental tool or therapeutic approach (Durcan et al., 2008). The lung has a large absorption surface area and is lined with surfactants, a family of cationic glycolipids that function as a vehicle to facilitate siRNA uptake into lung cells (Durcan et al., 2008). The objective of the present study was to investigate the role of RPS3 in allergic airway inflammation in a house dust mite (HDM) mouse asthma model.
To the best of our knowledge, we have shown for the first time that the level of RPS3 protein in the lung is up‐regulated in experimental asthma. Intratracheal RPS3 siRNA effectively knocked down RPS3 in HDM‐challenged lungs and markedly suppressed pulmonary immune cell infiltration, mucus hypersecretion, cytokine production and AHR. In addition, RPS3 lung gene silencing decreased serum total IgE and HDM‐specific IgE levels in experimental asthma. RPS3 knock down was found to be associated with a significant drop in the nuclear accumulation and transactivation of NF‐κB in the inflamed airways. Our findings demonstrate that RPS3 is a novel therapeutic target for the treatment of allergic asthma.
Methods
In vitro characterization of RPS3 siRNA
A mouse macrophage cell line RAW 264.7 and a mouse fibroblast cell line NIH/3T3 (American Type Culture Collection, Rockville, MD, USA) were maintained in DMEM (Invitrogen, Carlsbad, CA, USA). A mouse lung epithelial cell line LA‐4 was maintained in Ham's F12 medium (Biowest, Rue de la Caille, Nuaille, France) containing 1% MEM non‐essential amino acids (Invitrogen). Cells were seeded at 60–70% confluency in antibiotic‐free media 24 h before transfection. To detect RPS3 gene silencing, cells were transfected with 100 nM RPS3 siRNA or non‐targeting control siRNA for 6 h at 37°C in OptiMEM containing Lipofectamine 2000 (Invitrogen). On‐target plus siRNA and siSTABLE siRNA targeting mouse RPS3 mRNA with the following sequences: S1, 5′‐UCAUGUGAGCAUCGUGGAA‐3′, S2, 5′‐AGUUGAAGUCCGAGUUACA‐3′, S3, 5′‐UCUGGAGGCGAUUUAAUAA‐3′ and S4, 5′‐GCCAGUGCCUACAGCAUAA‐3′, and non‐targeting control siRNA, 5′‐UGGUUUACAUGUCGACUAA‐3′, were purchased from Thermo Scientific (Waltham, MA, USA). Transfected cells were allowed to recover in complete DMEM or Ham's F12 medium for 18, 42 or 66 h before they were analysed for RPS3 mRNA and protein expression. Total mRNA was extracted using TRIzol reagent (Invitrogen). PCR amplifications were performed using primers listed in Table S1, and PCR products were analysed by quantitative real‐time PCR (ABI 7500 Cycler; Applied Biosystems, Carlsbad, CA, USA). The mRNA expression levels were normalized to the level of β‐actin internal control. Total protein lysates (20 μg per lane) were separated by 10% SDS‐PAGE, probed with anti‐RPS3 mAb (Abcam, Cambridge, M, USA) and anti‐β‐actin mAb (Cell Signaling, Beverly, MA, USA) as an internal control. Band intensity was quantified using ImageJ software (National Institutes of Health, Bethesda, MD, USA) and normalized to β‐actin control. To detect potential anti‐inflammatory effects of RPS3 siRNA in vitro, transfected cells were allowed to recover for 18 h followed by stimulation with 50 ng·mL−1 TNF‐α (Abcam) or 100 μg·mL−1 HDM (Dermatophagoides pteronyssinus extracts, Greer Laboratories, Lenior, NC, USA) (Hammad et al., 2009) for another 24 h before analysis of pro‐inflammatory gene expression. To detect the cytotoxicity of RPS3 siRNA, transfected cells were allowed to recover for 18, 42 or 66 h before they were evaluated by use of the MTS assay (Promega, Madison, WI, USA).
NF‐κB luciferase reporter gene assay
NF‐κB luciferase reporter NIH/3T3 stable cells (Signosis, Santa Clara, CA, USA) were cultured in DMEM supplemented with high glucose, sodium pyruvate and L‐glutamine. One day before transfection, cells were seeded in a 96‐well plate at 5 × 104 cells .100 μL−1. Cells were then transfected with siRNAs for 6 h, allowed to recover for 18 h and stimulated with 10 ng·mL−1 TNF‐α for an additional 6 h. Cells were then lysed in luciferase lysis buffer (Promega), and luciferase activity was measured using a luminometer. Luciferase experiments were conducted in duplicate and repeated three times.
Animals
Female BALB/c mice 6 to 8 weeks old were purchased from the InVivos Pte. Ltd. (Singapore). Mice were anaesthetized with isoflurane (Halocarbon Products, River Edge, NJ, USA) and were given 40 μL of 100 μg HDM or saline (negative control) intratracheally on days 0, 7 and 14 to develop experimental asthma (Peh et al., 2015). RPS3 siRNA (1 and 5 nmol) or non‐targeting control siRNA in 30 μL PBS was given once daily via the intratracheal route on days 11–13 (Goh et al., 2013). Mice were killed by an i.p. injection of 300 μL of anaesthetic mixture (7.5 mg·mL−1 ketamine plus 0.1 mg·mL−1 medetomidine, obtained from National University of Singapore Animal Holding Unit) on day 17 (Figure S1A). Animal experiments were performed following the guidelines of the Institutional Animal Care and Use Committee of the National University of Singapore. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
Bronchoalveolar lavage fluid and serum analyses
Bronchoalveolar lavage (BAL) was performed as previously described (Goh et al., 2013), and the total and differential cell counts in the BAL fluid were determined by flow cytometry (Fortessa, BD) (van Rijt et al., 2004). BAL fluid levels of cytokines and chemokines were measured using multiplex elisa (EMD Millipore, Billerica, MA, USA). Blood was collected by cardiac puncture, and serum levels of total IgE and HDM‐specific IgE were measured using elisa (BD Biosciences, San Jose, CA, USA). Peripheral complete blood counts, serum alanine transaminase/aspartate transaminase and creatinine levels of blood samples were also measured for potential toxicity.
Lung histology
Lungs were fixed in 10% neutral formalin, treated with paraffin, cut into 5 μm sections and stained with haematoxylin and eosin for examining cell infiltration and with periodic acid‐fluorescence Schiff for mucus production. Quantitative analyses of cell infiltration and mucus production were performed blinded as described previously (Goh et al., 2013).
Measurements of AHR
Mice were anaesthetized, and tracheotomy and intubation were performed (Peh et al., 2015). The trachea was intubated with a cannula that was connected to the pneumotach, ventilator and nebulizer. Lung resistance (Rl) and dynamic compliance (Cdyn) in response to nebulized methacholine (0.5–8.0 mg·mL−1) were recorded using a whole‐body plethysmography chamber (FinePointe RC System, Buxco Research Systems, Wilmington, NC, USA) and the FinePointe™ data acquisition and analysis software (Buxco). Results are expressed as a percentage of the respective basal values in response to PBS.
Lung tissue analysis
Total protein lysates and nuclear extracts (20 μg per lane) were separated by 10% SDS‐PAGE and probed with anti‐p65 mAb (Cell Signaling) or anti‐RPS3 mAb (Abcam), and anti‐β‐actin mAb (Cell Signaling) or anti‐TATA binding protein mAb (Abcam) as the internal control. Band intensity was quantified using ImageJ software (National Institutes of Health, Bethesda, MD, USA). Nuclear proteins were also analysed for NF‐κB p65 DNA‐binding activity using the TransAM™ NF‐κB p65 transcription factor assay kit (Active Motif, Carlsbad, CA, USA). Total mRNA was extracted using TRIzol reagent (Invitrogen). PCR amplifications were performed using primers listed in Table S1, and PCR products were analysed by quantitative real‐time PCR (ABI 7500 Cycler). The mRNA expression levels were normalized to the level of β‐actin internal control.
Immunofluorescence staining
Raw 264.7 and LA‐4 cells were seeded onto chamber slides and transfected with 100 nM RPS3 siRNA or non‐targeting control siRNA. Transfected cells were then allowed to recover for 18 h followed by stimulation with 100 μg·mL−1 HDM for 6 h at 37°C. Cells were fixed, permeabilized and probed with antibodies targeted at NF‐κB p65 (Cell Signaling), followed by 4’‐6‐diamidino‐2‐phenylindole (DAPI; Sigma‐Aldrich, St. Louis, Missouri, United States) staining. Images were captured with an Olympus FluoView™ FV1000 confocal microscope (Olympus, Shinjuku, Tokyo, Japan) and quantified with ImageJ software (National Institutes of Health). Experiments were repeated for four times and the % of p65‐positive cells were quantified.
Statistical analysis
Data are presented as mean ± SEM. Normal distribution was confirmed by the Kolmogorov–Smirnov‐test. One‐way ANOVA of raw data without prior normalization followed by Dunnett's test was used to determine the significant differences between treatment groups. Significant levels were set at P < 0.05 as compared with non‐targeting control siRNA. The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015).
Results
In vitro characterization of RPS3 siRNA
RAW 264.7 (Figure 1A) and NIH/3T3 (Figure 1B) cells are used routinely in this laboratory for screening the relevant siRNA sequences as previously reported (Goh et al., 2013). The effects of gene silencing of four RPS3 siRNA sequences (S1–S4) targeted at different sites of the coding region of mouse RPS3 mRNA were examined. S1, S2, S3 and S4 markedly silenced RPS3 mRNA expression in both cell lines 24 h after transfection by about 70% as compared with the control siRNA. The four sequences produced significant knockdown of RPS3 protein expression by at least 60–80% in both RAW 264.7 cells and NIH/3T3 cells, especially at 24 and 48 h. Among the four sequences, S1 consistently knocked down RPS3 mRNA and protein with the highest silencing efficiency and the least variability and was chosen as the lead siRNA for subsequent in vitro and in vivo experiments. S1 was then tested in a relevant respiratory epithelial cell line, LA‐4, to confirm the down‐regulation of RPS3 mRNA expression by 60–70% and protein expression by 60–80% (Figure 1C).
Figure 1.
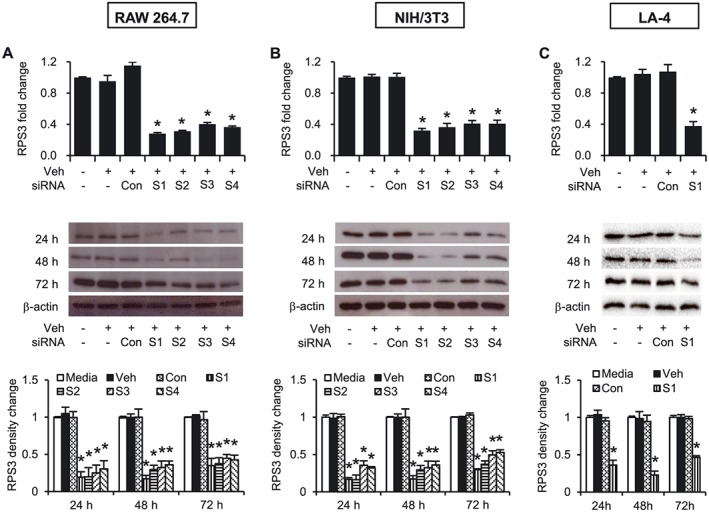
Characterization of RPS3 siRNAs. mRNA and protein levels of RPS3 after transfection of non‐targeting control siRNA or ON‐TARGETplus RPS3 siRNAs in (A) RAW 264.7, (B) NIH/3T3 and (C) LA‐4 cells. Four sequences of ON‐TARGETplus RPS3 siRNAs (100 nM) designated as S1, S2, S3 or S4 were transfected and characterized individually in the cells as indicated. Total mRNAs were extracted 18 h later (n = 3), and total protein lysates were prepared 18, 42 and 66 h later (n = 3 per time point) post‐transfection. The immunoblots were probed with anti‐RPS3 mAb, and band intensities were analysed using ImageJ software and normalized to β‐actin as an internal control. Veh, Lipofectamine 2000; Con, control siRNA. Values are shown as means ± SEM. *Significant difference from control siRNA, P < 0.05.
RPS3 knockdown by S1 siRNA markedly prevented TNF‐α‐induced gene expression of IL‐1β, IL‐5, KC (CXCL1) and inducible NOS (iNOS) in RAW 264.7 cells (Figure 2A); IL‐1β, IL‐5, mucin 5AC (Muc5ac) and MMP9 in NIH/3T3 cells (Figure 2B); and IL‐1β, IL‐5 and IL‐6, MMP9 in LA‐4 cells (Figure 2C). In addition, RPS3 gene silencing significantly blocked direct aeroallergen HDM‐stimulated gene expression of IL‐1β, IL‐6, KC and granulocyte‐macrophage colony‐stimulating factor (GM‐CSF) in RAW 264.7 cells (Figure 3A); IL‐6, Muc5ac, TNF‐α and MMP9 in NIH/3T3 cells (Figure 3B); and IL‐1β, IL‐6 and KC (CXCL1), MMP9 in LA‐4 cells (Figure 3C).
Figure 2.
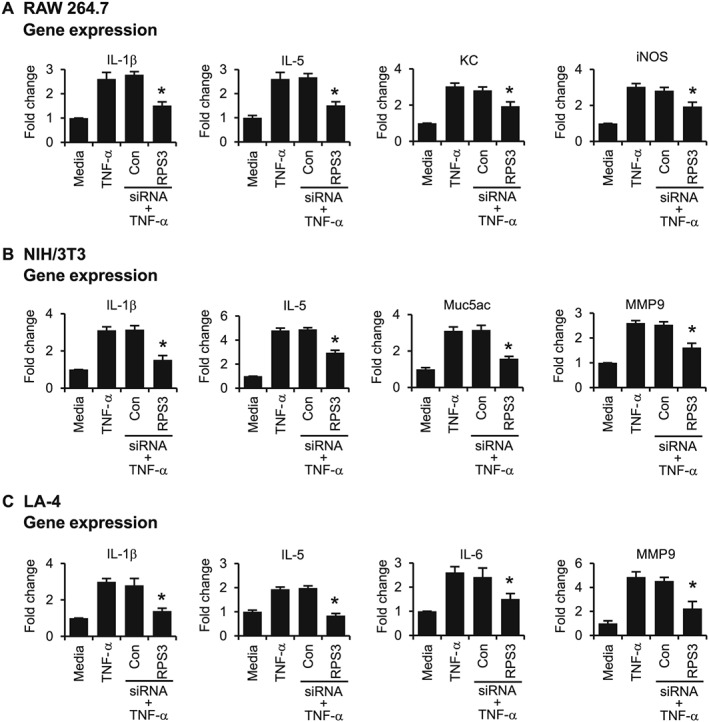
Effects of RPS3 siRNA on TNF‐α‐induced expression of inflammatory and remodelling mediators in vitro. mRNA expression of TNF‐α‐induced mediators after transfection with RPS3 siRNA in (A) RAW 264.7, (B) NIH/3T3 and (C) LA‐4 cells (n = 6). Values are shown as means ± SEM. *Significant difference from control siRNA, P < 0.05.
Figure 3.
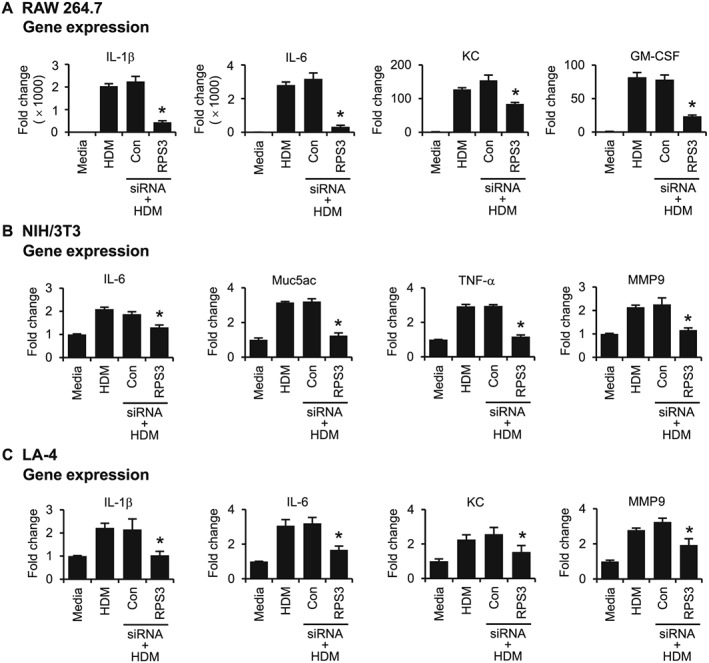
Effects of RPS3 siRNA on HDM‐induced expression of inflammatory and remodelling mediators in vitro. mRNA expression of HDM‐induced mediators after transfection with RPS3 siRNA in (A) RAW 264.7, (B) NIH/3T3 and (C) LA‐4 cells (n = 6). Values are shown as means ± SEM. *Significant difference from control siRNA, P < 0.05.
RPS3 gene silencing attenuates HDM‐induced allergic airway inflammation
Daily intratracheal administration of 5 nmol S1 siRNA to naïve BALB/c mice for three consecutive days was able to knock down the RPS3 protein lung level for up to 72 h after the last siRNA dose (Figure 4A). In the HDM‐induced experimental asthma model, we observed for the first time that the RPS3 protein level in lung tissues was moderately, but significantly, elevated (Figure 4B). However, three consecutive daily doses of S1 siRNA were sufficient to down‐regulate RPS3 protein level in lung tissues from HDM‐challenged mice (Figure 4C).
Figure 4.
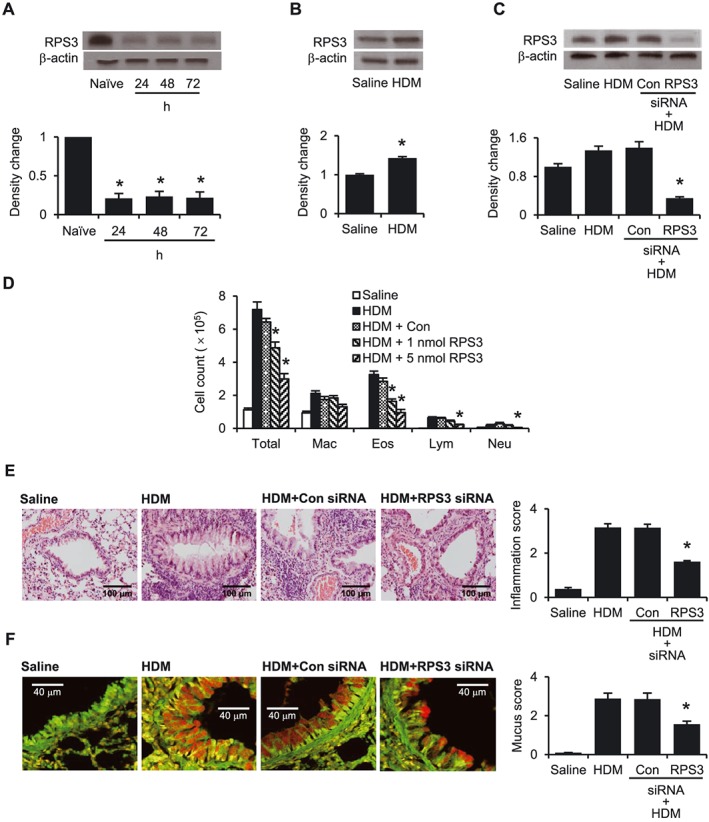
Effects of RPS3 gene silencing in an HDM‐induced mouse asthma model. (A) RPS3 protein level in mouse lung tissues. Naïve mice were given 5 nmol RPS3 siRNA for 3 days consecutively by intratracheal administration. RPS3 protein levels were measured 24, 48 and 72 h after the last RPS3 siRNA administration (n = 3). (B) RPS3 protein levels in saline and HDM‐challenged mouse lungs (n = 3). (C) RPS3 protein levels in HDM‐challenged mouse lungs after intratracheal administration of RPS3 siRNA (n = 3). Data are shown as representative of three independent experiments. (D) Inflammatory cell counts in BAL fluid 72 h after the last challenge [Saline, n = 8; HDM, n = 7; control siRNA (Con), n = 7; 1 nmol RPS3 siRNA, n = 9; 5 nmol RPS3 siRNA, n = 9]. Differential cell counts were performed on a minimum of 500 cells to identify eosinophils (Eos), macrophages (Mac), neutrophils (Neu) and lymphocytes (Lym). Representative images of (E) haematoxylin and eosin‐stained lung sections (200× magnification) and (F) periodic acid‐fluorescence Schiff‐stained lung sections (900× magnification) with corresponding quantitative analyses of inflammation score and mucus score respectively (n = 4). Values are shown as means ± SEM. *Significant difference from control siRNA, P < 0.05.
HDM challenge markedly increased total cell, eosinophil and macrophage counts and slightly but significantly increased lymphocyte and neutrophil counts, as compared with the saline control (Figure 4D). Intratracheal S1 siRNA (1 and 5 nmol) dose‐dependently decreased (P < 0.05) total cell and eosinophil counts in BAL fluid as compared with control siRNA. At a dose of 5 nmol, S1 siRNA was able to significantly lower BAL fluid neutrophil and lymphocyte counts as well (Figure 4D). In addition, RPS3 gene silencing markedly reduced HDM‐induced leukocyte infiltration and mucus hypersecretion (Figure 4E–F).
RPS3 gene silencing attenuates the HDM‐induced increase in inflammatory mediators
HDM challenge evidently increased BAL fluid levels of Th2 cytokines, including IL‐4, IL‐5, IL‐13, eotaxin (CCL11), IL‐1β and KC (CXCL1) (Figure 5A), and lung gene expression of pro‐inflammatory mediators, including Muc5ac, TNF‐α, IL‐1β, IL‐6, KC, iNOS, E‐selectin and GM‐CSF (Figure 5B), as compared with saline control. RPS3 siRNA dose‐dependently suppressed BAL fluid (Figure 5A) and lung tissue (Figure 5B) levels of these pro‐inflammatory mediators (Figure 5A), as compared with the non‐targeting control siRNA. An elevation of serum IgE levels is a hallmark of the Th2 immune response. RPS3 gene silencing markedly lowered HDM‐induced increases in total and HDM‐specific IgE levels in a dose‐dependent manner (Figure 5C).
Figure 5.
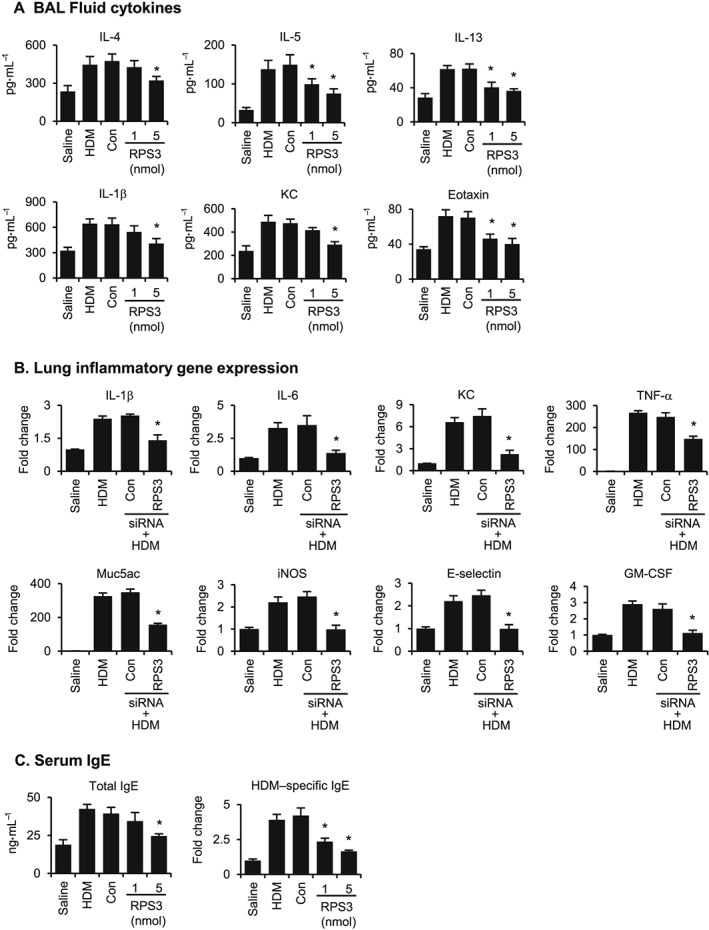
Effects of RPS3 gene silencing on BAL fluid cytokine levels, lung tissue inflammatory gene expressions and serum IgE levels in a HDM‐induced mouse asthma model. (A) BAL fluid cytokine levels of IL‐4, IL‐5, IL‐13, IL‐1β, KC and eotaxin (n = 7–9). (B) Lung tissue inflammatory gene expressions. Lung tissues were collected 24 h after the last challenge. The relative quantity of target gene expression was automatically normalized to that of β‐actin as an internal control, and values shown are ratios of various treatments to saline group (n = 7–9). (C) Mouse serum lgE levels. Mouse sera were collected 24 h after the last challenge. The levels of total IgE and ovalbumin‐specific IgE were analysed using elisa (n = 7–9). Values are shown as means ± SEM. *Significant difference from control siRNA, P < 0.05.
RPS3 gene silencing reduces HDM‐induced AHR
To detect the effects of RPS3 gene silencing on HDM‐induced AHR, we measured the lung resistance (Rl) and dynamic compliance (Cdyn) in mechanically ventilated mice in response to increasing concentrations of nebulized methacholine. Rl is defined as the pressure driving respiration divided by flow. Cdyn is defined as the change in volume of the lung produced by a change in pressure across the lung. HDM‐challenged mice developed AHR, which is typically reflected by an enhanced Rl (Figure 6A) and declining Cdyn (Figure 6B). RPS3 siRNA (5 nmol) drastically reduced Rl (Figure 6A) and restored Cdyn (Figure 6B) to the saline control levels, suggesting that airway pathology was largely modified by RPS3 gene silencing.
Figure 6.
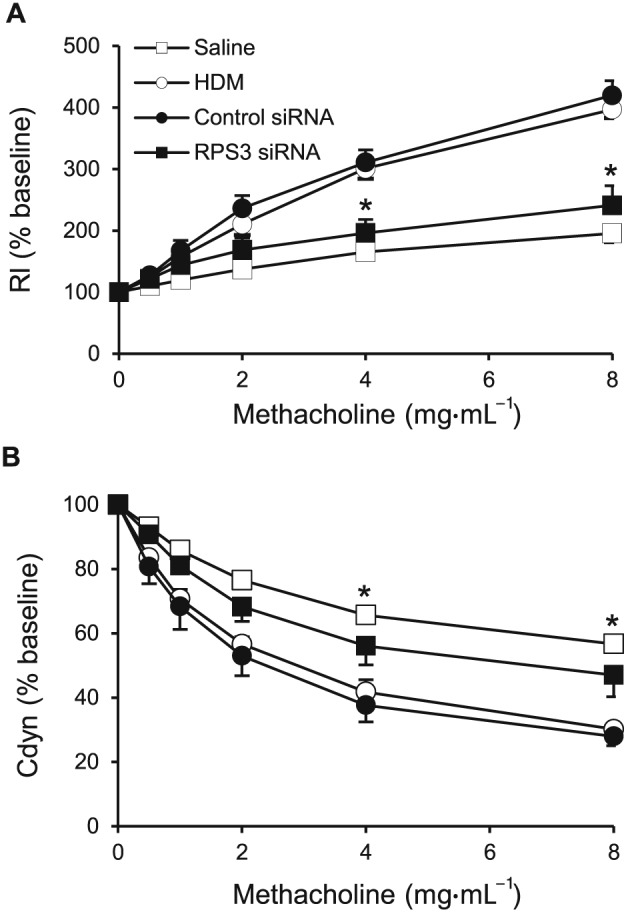
Effects of RPS3 gene silencing on HDM‐induced AHR. AHR in response to increasing concentrations of aerosolized methacholine was measured 24 h after the last challenge with (pretreatment) 5 nmol control or RPS3 siRNA. AHR is expressed as percentage change compared with the baseline level of (A) lung resistance (Rl; n = 6) and (B) dynamic compliance (Cdyn; n = 6). Values are shown as means ± SEM. *Significant difference from control siRNA, P < 0.05.
RPS3 gene silencing disrupts NF‐κB signalling
Nuclear translocation of NF‐κB was captured using immunofluorescence staining in RAW 264.7 (Figure 7A) and LA‐4 (Figure 7B) cells. HDM treatment of macrophages and epithelial cell lines promoted p65 accumulation in the nucleus, and the p65 nuclear translocation was markedly inhibited by RPS3 siRNA. HDM challenge promoted NF‐κB p65 subunit and RPS3 nuclear accumulation in mouse lung tissues. Intratracheal administration of RPS3 siRNA (5 nmol) significantly reduced nuclear p65 and RPS3 levels as compared with non‐targeting control siRNA (Figure 7C). In addition, RPS3 gene silencing lessened the HDM‐induced increase in p65 DNA‐binding activity (Figure 7D). Using an NF‐κB luciferase reporter assay to further confirm the disruption of the NF‐κB pathway caused by RPS3 knockdown, we observed a strong TNF‐α‐mediated NF‐κB‐dependent increase in luciferase activity in NIH/3T3 cells, but RPS3 siRNA markedly suppressed luciferase activity as compared with the control siRNA (Figure 7E).
Figure 7.
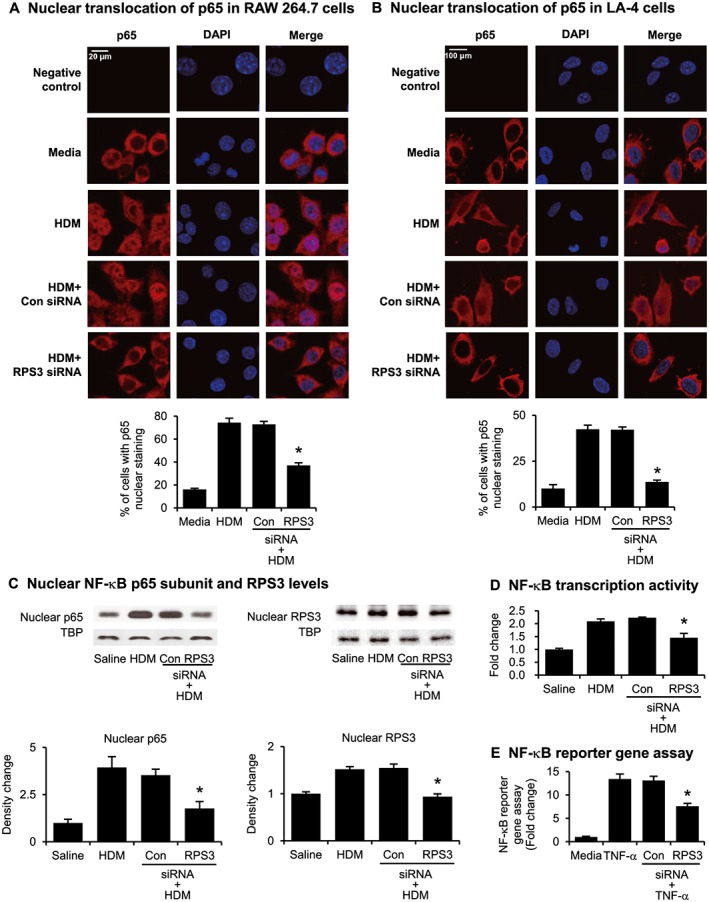
Effects of RPS3 gene silencing on NF‐κB translocation and activity. Nuclear translocation of p65 induced by HDM was captured using immunofluorescence staining in (A) RAW 264.7 and (B) LA‐4 cells. The percentage of cells with p65 nuclear staining was quantified. Negative control panels were probed with the secondary antibody only to indicate potential background or non‐specific staining. (C) Immunoblot of nuclear NF‐κB subunit p65 and RPS3 accumulation. Mouse lung nuclear proteins were separated by 10% SDS‐PAGE and probed with anti‐p65, anti‐RPS3 or anti‐TATA binding protein (TBP) mAb. TBP were used as a nuclear protein loading control (n = 7–9). (D) Nuclear p65 DNA‐binding activity was determined using a TransAM™ p65 transcription factor elisa kit (n = 7–9). (E) NF‐κB reporter gene assay in NF‐κB luciferase reporter NIH/3T3 stable cell line pretreated with RPS3 siRNA and then stimulated with TNF‐α. Results are expressed as fold change relative to media control. The luciferase assay was conducted in duplicate wells for each sample, with three independent experiments. Values are shown as means ± SEM. *Significant difference from control siRNA, P < 0.05.
Discussion
Persistent activation of the NF‐κB pathway has been observed in allergic asthma (Edwards et al., 2009; Trejo Bittar et al., 2015). RPS3 has recently been found to bind to the p65 subunit of the NF‐κB complex via its KH domain, facilitate nuclear translocation of NF‐κB complex and enhance the binding affinity of p65 subunit to downstream gene targets (Wan and Lenardo, 2010). In an RPS3‐deficient condition, it has been shown that overexpression of p65 failed to enhance NF‐κB activity, implicating an obligatory role of RPS3 in p65 function (Wan et al., 2007). Noticeably, activation of IKKβ, upstream in the NF‐κB pathway can lead to phosphorylation of RPS3 at Ser209, which is crucial for subsequent RPS3 nuclear translocation (Wan et al., 2011). In addition, RPS3 knockdown disrupted the RPS3‐p65 interaction and significantly attenuates cytokine production, T cell proliferation and immunoglobulin κ light chain gene expression in B cells (Wier et al., 2012). In this study, we revealed that the lung RPS3 protein level was elevated in HDM‐induced mouse asthma, and intratracheally administered RPS3 siRNA markedly attenuated allergic airway inflammation. We also demonstrated that p65 and RPS3 nuclear translocation were elevated in HDM‐challenged mice, and RPS3 gene silencing significantly inhibited their nuclear accumulation. In addition, RPS3 siRNA markedly suppressed the nuclear translocation of NF‐κB p65 induced by TNF‐α in the NF‐κB luciferase reporter NIH/3T3 stable cells or by HDM in RAW 264.7 and LA‐4 cells.
Among the four siRNA sequences (S1–S4) tested, S1 was selected as the most potent RPS3‐specific siRNA based on its consistently efficient knockdown effects of at least 60–80% on RPS3 gene and protein levels in the RAW 264.7 or NIH/3T3 cell line for up to 72 h. The knockdown of RPS3 by S1 was confirmed in LA‐4 mouse lung epithelial cells. The cells were stimulated with TNF‐α or HDM, and the expression of pro‐inflammatory mediators was measured. Different cell types responded differently to TNF‐α or HDM, resulting in differential inflammatory gene expressions. The inhibitory effects of S1 on direct TNF‐α‐ or HDM‐induced production of cell type‐specific inflammatory mediators were then measured and shown in Figures 2 and 3. To investigate the role of RPS3 in asthma, three consecutive daily doses of S1, given via the intratracheal route, were found to be sufficient to knockdown RPS3 protein in the lungs for up to 72 h in a HDM experimental mouse asthma model.
In addition, potential toxic side effects of RPS3 siRNA were measured in vitro and in vivo. The MTS assay revealed that RPS3 siRNA had no negative influence on the proliferation of all three cell lines tested, as compared with control siRNA (Figure S1B). Complete blood count and chemical analysis of serum kidney and liver markers confirmed that intratracheal RPS3 siRNA had no adverse effects on complete blood count and serum kidney and liver markers in the HDM experimental mouse asthma model (Table S1).
Experimental asthma in mice with IKKβ conditional knockout or transgenic IκBα mutant expression in the airway epithelium produced significantly less IL‐4, IL‐5, IL‐13 and eotaxin (CCL11) in the airways (Broide et al., 2005). Th2 cytokines including IL‐4, IL‐5, IL‐13 and eotaxin (CCL11), together with TNF‐α, IL‐1β, IL‐6, IL‐8 and GM‐CSF, have been shown to play an essential role in the pathogenesis of asthma (Gandhi et al., 2016; Chung, 2015). We observed a marked drop in BAL fluid levels of IL‐4, IL‐5, IL‐13, IL‐1β, KC (CXCL1) and eotaxin (CCL11), and in lung gene expression of TNF‐α, IL‐6 and GM‐CSF, in RPS3 siRNA‐treated HDM‐challenged mice. There is increasing evidence supporting the role of IL‐1β, which is expressed in a NF‐κB‐dependent manner and activated by NLRP3 inflammasome in allergic airways, in inducing IL‐5 and IL‐13 production in asthma (Kim et al., 2014). Taken together, these results indicate that RPS3 gene silencing can suppress a wide spectrum of pro‐inflammatory cytokines in experimental mouse asthma and this effect is probably mediated by disruption of the NF‐κB pathway.
RPS3 knockdown markedly reduced total cell and eosinophil counts and, to a lesser extent, lymphocyte and neutrophil counts, in BAL fluid, and induced tissue eosinophilia in lung sections. Eosinophil transmigration into the airways is promoted by Th2 cytokines IL‐5 and IL‐13 as well as TNF‐α and facilitated by chemokines such as eotaxin (CCL11) in combination with adhesion molecules such as E‐selectin (Hogan et al., 2008). IL‐13 has been shown to induce eotaxin production from airway epithelial cells (Pope et al., 2001), while TNF‐α induces E‐selectin expression in human endothelial cells (Ceolotto et al., 2014). GM‐CSF can prolong the survival of eosinophils, and IL‐8 promotes neutrophil chemotaxis and activation (Pelaia et al., 2015). Intratracheal RPS3 siRNA strongly suppressed eotaxin (CCL11), TNF‐α, KC (CXCL1) and E‐selectin levels in HDM‐challenged lungs. These results are likely to be due to interruption of NF‐κB transcriptional activity by RPS3 knockdown, as the genes for eotaxin (CCL11), TNF‐α, GM‐CSF, IL‐6, KC (CXCL1), IL‐13 and E‐selectin contain the κB site for NF‐κB within their promoters (Kumar et al., 2004).
Cumulative evidence has revealed that goblet cell hyperplasia and Muc5ac expression can be induced by IL‐4, IL‐5, IL‐8 or IL‐13 (Zhang et al., 2015; Bautista et al., 2009). Muc5ac gene expression is dependent on the transcriptional activity of NF‐κB (Hewson et al., 2010). Selective ablation of NF‐κB function in airway epithelium has been shown to reduce allergen‐induced mucus production in mice (Edwards et al., 2009; Ogawa et al., 2011). Recent studies have revealed the important role of IL‐1β and TNF‐α in mucus production in experimental asthma and in airway epithelial cells (Lora et al., 2005; Fujisawa et al., 2009). Therefore, the marked reduction in mucus production and Muc5ac expression in the lungs of RPS3 siRNA‐treated mice is probably a consequence of the inhibiton of pro‐inflammatory cytokine production and a direct disruption of the NF‐κB pathway in airway epithelium.
Elevated serum IgE levels are a hallmark of the Th2 immune response. NF‐κB plays an essential role in B cell proliferation and development (Gerondakis and Siebenlist, 2010). IL‐4 and IL‐13 are important in driving B cell growth, differentiation and secretion of IgE (Lambrecht and Hammad, 2015; Gandhi et al., 2016). The biological activities of IgE are mediated through its interaction with the FcεRI on mast cells and basophils. Cross‐linking of FcεRI initiates multiple signalling cascades leading to NF‐κB activation and the production of lipid mediators, cytokines and chemokines (Galli et al., 2008). Our results show that RPS3 siRNA substantially reduced serum levels of total IgE and HDM‐specific IgE. The observed reduction is likely to be mediated by its disruptive action on NF‐κB activation in B cells and on IL‐4‐ and IL‐13‐mediated class switching to IgE.
Increased exhaled NO is positively correlated with increased iNOS expression in the lung epithelium in asthma (Jiang et al., 2009). IL‐13, IL‐1β and TNF‐α have been shown to induce iNOS expression in human bronchial epithelial cells leading to an elevatation in NO production (Suresh et al., 2007; Jiang et al., 2009). Furthermore, iNOS gene expression is controlled by the NF‐κB pathway (Bogdan, 2001). Our results show that RPS3 gene silencing markedly suppressed the HDM‐induced iNOS expression in the lungs, which may be due to the direct interruption of NF‐κB signalling and the reduced levels of IL‐13, IL‐1β and TNF‐α in the allergic airways.
IL‐4, IL‐5, IL‐6, IL‐13 and TNF‐α have been implicated in AHR by mobilizing and activating esoinophils, leading to the release of pro‐inflammatory mediators including major basic protein and cysteinyl‐leukotrienes, which are closely linked to AHR development (Leckie et al., 2000; Brightling et al., 2008; Oh et al., 2010; Tanaka et al., 2012). In addition, inhibition of the NF‐κB pathway has also been shown to attenuate AHR in experimental asthma (Tully et al., 2013). We observed that RPS3 gene silencing strongly suppressed methacholine‐induced AHR, which may be the result of the marked drop in pro‐inflammatory cytokines and tissue eosinophilia induced by RPS3 siRNA treatment.
We demonstrated that the lung RPS3 level was substantially elevated in experimental asthma, and RPS3 gene silencing markedly attenuated HDM‐induced airway inflammation and AHR, probably by disruption of the NF‐κB signalling pathway. Our findings strongly indicate that gene silencing of RPS3 using siRNA can be a novel therapeutic strategy for asthma.
Author contributions
J.D., W.L. and W.S.F.W. conceived and designed the study. J.D., H.Y.P., T.K.C., W.S.D.T., L.L. and A.Y. participated in the acquisition of samples and data. J.D., W.L., H.Y.P. and T.K.C. analysed and interpreted the data. J.D., W.L., H.Y.P., T.K.C. and W.S.F.W. wrote and revised the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 (A) HDM‐induced mouse asthma model. (B) Effects of RPS3 siRNA on cell proliferation by MTS assay. Percentage of cell viability compared with media control after transfection with control or RPS3 siRNA in RAW 264.7, NIH/3 T3 and LA‐4 cells (n = 6).
Table S1 Primer sequences for real‐time PCR.
Table S2 Complete blood count and chemical analysis of mice serum.
Acknowledgements
This work was supported by a grant NMRC/CBRG/0027/2012 from the National Medical Research Council of Singapore (W.S.F.W.), a grant R‐184‐000‐269‐592 from the National Research Foundation of Singapore (W.S.F.W) and a bridging grant R‐184‐000‐267‐733 from NUHS (W.S.F.W.).
Dong, J. , Liao, W. , Peh, H. Y. , Chan, T. K. , Tan, W. S. D. , Li, L. , Yong, A. , and Wong, W. S. F. (2017) Ribosomal protein S3 gene silencing protects against experimental allergic asthma. British Journal of Pharmacology, 174: 540–552. doi: 10.1111/bph.13717.
References
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015a). The concise guide to PHARMACOLOGY 2015/16: Overview. Br J Pharmacol 172: 5729–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes PJ (2013). Corticosteroid resistance in patients with asthma and chronic obstructive pulmonary disease. J Allergy Clin Immunol 131: 636–645. [DOI] [PubMed] [Google Scholar]
- Bautista MV, Chen Y, Ivanova VS, Rahimi MK, Watson AM, Rose MC (2009). IL‐8 regulates mucin gene expression at the posttranscriptional level in lung epithelial cells. J Immunol 183: 2159–2166. [DOI] [PubMed] [Google Scholar]
- Bogdan C (2001). Nitric oxide and the immune response. Nat Immunol 2: 907–916. [DOI] [PubMed] [Google Scholar]
- Brightling C, Berry M, Amrani Y (2008). Targeting TNF‐α: a novel therapeutic approach for asthma. J Allergy Clin Immunol 121: 5–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broide DH, Lawrence T, Doherty T, Cho JY, Miller M, McElwain K et al. (2005). Allergen‐induced peribronchial fibrosis and mucus production mediated by IκB kinase β‐dependent genes in airway epithelium. Proc Natl Acad Sci U S A 102: 17723–17728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderón MA, Linneberg A, Kleine‐Tebbe J, De Blay F, de Rojas DHF, Virchow JC et al. (2014). Respiratory allergy caused by house dust mites: what do we really know? J Allergy Clin Immunol 136: 38–48. [DOI] [PubMed] [Google Scholar]
- Ceolotto G, De Kreutzenberg SV, Cattelan A, Fabricio AS, Squarcina E, Gion M et al. (2014). Sirtuin 1 stabilization by HuR represses TNF‐α‐ and glucose‐induced E‐selectin release and endothelial cell adhesiveness in vitro: relevance to human metabolic syndrome. Clin Sci 127: 449–461. [DOI] [PubMed] [Google Scholar]
- Chung KF (2015). Targeting the interleukin pathway in the treatment of asthma. Lancet 386: 1086–1096. [DOI] [PubMed] [Google Scholar]
- Coutinho AE, Chapman KE (2011). The anti‐inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol Cell Endocrinol 335: 2–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SPA, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durcan N, Murphy C, Cryan SA (2008). Inhalable siRNA: potential as a therapeutic agent in the lungs. Mol Pharm 5: 559–566. [DOI] [PubMed] [Google Scholar]
- Edwards MR, Bartlett NW, Clarke D, Birrell M, Belvisi M, Johnston SL (2009). Targeting the NF‐κB pathway in asthma and chronic obstructive pulmonary disease. Pharmacol Ther 121: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujisawa T, Velichko S, Thai P, Hung LY, Huang F, Wu R (2009). Regulation of airway MUC5AC expression by IL‐1β and IL‐17A; the NF‐κB paradigm. J Immunol 183: 6236–6243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galli SJ, Tsai M, Piliponsky AM (2008). The development of allergic inflammation. Nature 454: 445–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi NA, Bennett BL, Graham NM, Pirozzi G, Stahl N, Yancopoulos GD (2016). Targeting key proximal drivers of type 2 inflammation in disease. Nat Rev Drug Discov 15: 35–50. [DOI] [PubMed] [Google Scholar]
- Gerondakis S, Siebenlist U (2010). Roles of the NF‐κB pathway in lymphocyte development and function. Cold Spring Harb Perspect Biol 2: a000182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh FY, Cook KL, Upton N, Tao L, Lah LC, Leung BP et al. (2013). Receptor‐interacting protein 2 gene silencing attenuates allergic airway inflammation. J Immunol 191: 2691–2699. [DOI] [PubMed] [Google Scholar]
- Hammad H, Chieppa M, Perros F, Willart MA, Germain RN, Lambrecht BN (2009). House dust mite allergen induces asthma via Toll‐like receptor 4 triggering of airway structural cells. Nat Med 15: 410–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewson CA, Haas JJ, Bartlett NW, Laza‐Stanca V, Kebadze T, Caramori G et al. (2010). Rhinovirus induces MUC5AC in a human infection model and in vitro via NF‐κB and EGFR pathways. Eur Respir J 36: 1425–1435. [DOI] [PubMed] [Google Scholar]
- Hogan SP, Rosenberg HF, Moqbel R, Phipps S, Foster PS, Lacy P et al. (2008). Eosinophils: biological properties and role in health and disease. Clin Exp Allergy 38: 709–750. [DOI] [PubMed] [Google Scholar]
- Jiang J, Malavia N, Suresh V, George SC (2009). Nitric oxide gas phase release in human small airway epithelial cells. Respir Res 10: 10–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SR, Kim DI, Kim SH, Lee H, Lee KS, Cho SH et al. (2014). NLRP3 inflammasome activation by mitochondrial ROS in bronchial epithelial cells is required for allergic inflammation. Cell Death Dis 5: e1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kole R, Krainer AR, Altman S (2012). RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nat Rev Drug Discov 11: 125–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Takada Y, Boriek AM, Aggarwal BB (2004). Nuclear factor‐κB: its role in health and disease. J Mol Med 82: 434–448. [DOI] [PubMed] [Google Scholar]
- Lambrecht BN, Hammad H (2015). The immunology of asthma. Nat Immunol 16: 45–56. [DOI] [PubMed] [Google Scholar]
- Leckie MJ, ten Brinke A, Khan J, Diamant Z, O'Connor BJ, Walls CM et al. (2000). Effects of an interleukin‐5 blocking monoclonal antibody on eosinophils, airway hyper‐responsiveness, and the late asthmatic response. Lancet 356: 2144–2148. [DOI] [PubMed] [Google Scholar]
- Lora JM, Zhang DM, Liao SM, Burwell T, King AM, Barker PA et al. (2005). Tumor necrosis factor‐α triggers mucus production in airway epithelium through an IκB kinase β‐dependent mechanism. J Biol Chem 280: 36510–36517. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa H, Azuma M, Muto S, Nishioka Y, Honjo A, Tezuka T et al. (2011). IκB kinase β inhibitor IMD‐0354 suppresses airway remodelling in a Dermatophagoides pteronyssinus‐sensitized mouse model of chronic asthma. Clin Exp Allergy 41: 104–115. [DOI] [PubMed] [Google Scholar]
- Oh CK, Geba GP, Molfino N (2010). Investigational therapeutics targeting the IL‐4/IL‐13/STAT‐6 pathway for the treatment of asthma. Eur Respir Rev 19: 46–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peh HY, Ho WE, Cheng C, Chan TK, Seow ACG, Lim AY et al. (2015). Vitamin E isoform γ‐Tocotrienol downregulates house dust mite‐induced asthma. J Immunol 195: 437–444. [DOI] [PubMed] [Google Scholar]
- Pelaia G, Vatrella A, Busceti MT, Gallelli L, Calabrese C, Terracciano R et al. (2015). Cellular mechanisms underlying eosinophilic and neutrophilic airway inflammation in asthma. Mediators Inflamm. doi:10.1155/2015/879783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope SM, Brandt EB, Mishra A, Hogan SP, Zimmermann N, Matthaei KI et al. (2001). IL‐13 induces eosinophil recruitment into the lung by an IL‐5‐ and eotaxin‐dependent mechanism. J Allergy Clin Immunol 108: 594–601. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44 (D1): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suresh V, Mih JD, George SC (2007). Measurement of IL‐13‐induced iNOS‐derived gas phase nitric oxide in human bronchial epithelial cells. Am J Respir Cell Mol Biol 37: 97–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Narazaki M, Kishimoto T (2012). Therapeutic targeting of the interleukin‐6 receptor. Annu Rev Pharmacol Toxicol 52: 199–219. [DOI] [PubMed] [Google Scholar]
- Trejo Bittar HE, Yousem SA, Wenzel SE (2015). Pathobiology of severe asthma. Annu Rev Pathol 10: 511–545. [DOI] [PubMed] [Google Scholar]
- Tully JE, Hoffman SM, Lahue KG, Nolin JD, Anathy V, Lundblad LK et al. (2013). Epithelial NF‐κB orchestrates house dust mite‐induced airway inflammation, hyperresponsiveness, and fibrotic remodeling. J Immunol 191: 5811–5821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rijt LS, Kuipers H, Vos N, Hijdra D, Hoogsteden HC, Lambrecht BN (2004). A rapid flow cytometric method for determining the cellular composition of bronchoalveolar lavage fluid cells in mouse models of asthma. J Immunol Methods 288: 111–121. [DOI] [PubMed] [Google Scholar]
- Wan F, Lenardo MJ (2010). The nuclear signaling of NF‐κB: current knowledge, new insights, and future perspectives. Cell Res 20: 24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan F, Anderson DE, Barnitz RA, Snow A, Bidere N, Zheng L et al. (2007). Ribosomal protein S3: a KH domain subunit in NF‐κB complexes that mediates selective gene regulation. Cell 131: 927–939. [DOI] [PubMed] [Google Scholar]
- Wan F, Weaver A, Gao X, Bern M, Hardwidge PR, Lenardo MJ (2011). IKKβ phosphorylation regulates RPS3 nuclear translocation and NF‐κB function during Escherichia coli O157:H7 infection. Nat Immunol 12: 335–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wier EM, Neighoff J, Sun X, Fu K, Wan F (2012). Identification of an N‐terminal truncation of the NF‐κB p65 subunit that specifically modulates ribosomal protein S3‐dependent NF‐κB gene expression. J Biol Chem 287: 43019–43029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Derycke L, Zhang L, Holtappels G, Wang X, Zhang N et al. (2015). Th2 cytokines orchestrate the secretion of MUC5AC and MUC5B in chronic rhinosinusitis with nasal polyps. J Allergy Clin Immunol 135: AB53. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 (A) HDM‐induced mouse asthma model. (B) Effects of RPS3 siRNA on cell proliferation by MTS assay. Percentage of cell viability compared with media control after transfection with control or RPS3 siRNA in RAW 264.7, NIH/3 T3 and LA‐4 cells (n = 6).
Table S1 Primer sequences for real‐time PCR.
Table S2 Complete blood count and chemical analysis of mice serum.