

Abstract
X‐linked juvenile retinoschisis (XLRS) is a hereditary retinal dystrophy in young males, caused by mutations in the RS1 gene. The function of the encoded protein, termed retinoschisin, and the molecular mechanisms underlying XLRS pathogenesis are still unresolved, although a direct interaction partner of the secreted retinoschisin, the retinal Na/K‐ATPase, was recently identified. Earlier gene expression studies in retinoschisin‐deficient (Rs1h −/Y) mice provided a first indication of pathological up‐regulation of mitogen‐activated protein (MAP) kinase signalling in disease pathogenesis. To further investigate the role for retinoschisin in MAP kinase regulation, we exposed Y‐79 cells and murine Rs1h −/Y retinae to recombinant retinoschisin and the XLRS‐associated mutant RS1‐C59S. Although normal retinoschisin stably bound to retinal cells, RS1‐C59S exhibited a strongly reduced binding affinity. Simultaneously, exposure to normal retinoschisin significantly reduced phosphorylation of C‐RAF and MAP kinases ERK1/2 in Y‐79 cells and murine Rs1h −/Y retinae. Expression of MAP kinase target genes C‐FOS and EGR1 was also down‐regulated in both model systems. Finally, retinoschisin treatment decreased pro‐apoptotic BAX‐2 transcript levels in Y‐79 cells and Rs1h −/Y retinae. Upon retinoschisin treatment, these cells showed increased resistance against apoptosis, reflected by decreased caspase‐3 activity (in Y‐79 cells) and increased photoreceptor survival (in Rs1h −/Y retinal explants). RS1‐C59S did not influence C‐RAF or ERK1/2 activation, C‐FOS or EGR1 expression, or apoptosis. Our data imply that retinoschisin is a novel regulator of MAP kinase signalling and exerts an anti‐apoptotic effect on retinal cells. We therefore discuss that disturbances of MAP kinase signalling by retinoschisin deficiency could be an initial step in XLRS pathogenesis.
Keywords: X‐linked juvenile retinoschisis, retinoschisin, RS1, Na/K‐ATPase, MAP kinase signalling, apoptosis
Introduction
Pathogenic alterations affecting the RS1 gene on chromosome Xp22.1 have been shown to cause XLRS (OMIM #312700) 1, a macular degeneration disorder in young males with a prevalence of approximately 1:5000 to 1:20,000 2. Disorganization of retinal layers and distinct abnormalities in the electroretinogram (ERG) are hallmarks of the disease. Specifically, a characteristic splitting of retinal layers, presenting as a bilateral foveal schisis, is found at an early stage of the disease and results in cystic degeneration of the central retina 3, 4, 5, 6. Additionally, defects in signal transmission from photoreceptor to bipolar cells as visualized by ERG recordings are observed and reveal a characteristic reduction in the b‐wave amplitude, whereas the a‐wave remains almost unaffected 4, 7. Comparable pathological features are also evident in XLRS mice, generated via a targeted disruption of the murine orthologue of RS1, the Rs1h gene 8, 9, 10. Due to the close resemblance of the retinal phenotype in Rs1h knockout mice and XLRS patients, the retinoschisin‐deficient mouse represents an excellent disease model widely used in experimental studies addressing the mechanisms of XLRS pathology but also novel treatment approaches 11, 12, 13, 14, 15, 16.
The RS1 gene is organized into six exons and encodes a 224‐amino acid (aa) precursor protein 1. It is specifically expressed in the retina by photoreceptor and bipolar cells, as well as in pinealocytes of the pineal gland 1, 17, 18. During protein synthesis, a 23‐aa signal sequence is cleaved to produce a 201‐aa mature polypeptide which is secreted from photoreceptors and bipolar cells as a homooctamer held together by intermolecular disulphide bonds between aa 223 and aa 59 19, 20, 21, 22. So far, over 190 unique XLRS‐associated sequence variants in RS1 have been reported (Leiden Open Variation Database, http://grenada.lumc.nl/LOVD2/eye/home.php?select_db=RS1, accessed May 2016). Functional assessment of a subset of these variants demonstrated that the vast majority of mutations result in a complete loss of the functional protein 4.
Despite intensive research, the precise molecular function of retinoschisin remains unresolved. Searching for retinoschisin interaction partners, Molday et al. 14 identified the retina‐specific Na/K‐ATPase composed of the two subunits ATP1A3 (α3) and ATP1B2 (β2). Subsequently, our group confirmed the Na/K‐ATPase to be required for anchoring retinoschisin to plasma membranes 23. The Na/K‐ATPase is a plasma membrane spanning ion pump, responsible for maintaining the cellular membrane potential by transporting Na+ and K+ ions across the plasma membrane against their electrochemical gradient 24, 25. Despite this essential task, Na/K‐ATPases also mediate intercellular adhesion 26, 27, 28 and induce activation of intracellular signalling pathways upon binding of glycoside hormones such as ouabain 25, 29, 30, 31, 32, 33, 34. Members of the FXYD family, a class of Na/K‐ATPase‐binding proteins 35, 36, were reported to be important regulators of the Na/K‐ATPase, modulating its pump activity and mediation of intercellular adhesion 37, 38, 39. Similar to FXYD proteins, one could consider retinoschisin to exert a role as a modulator of Na/K‐ATPase activity.
A genomewide expression analysis of the Rs1h‐deficient (Rs1h −/Y) murine retina first indicated an increased activation of the ERK pathway in early XLRS pathogenesis, prior to apoptotic photoreceptor degeneration 40. The ERK pathway is one of the four major MAP kinase pathways 41, 42 known to play a crucial role in fundamental developmental and physiological processes such as apoptosis, neuroprotection, neuronal development and adhesion 41, 43, 44, 45, 46, 47, 48, 49, 50. It is tempting to speculate that misregulation of MAP kinase signalling caused by retinoschisin deficiency could be an initial step in XLRS pathogenesis. However, aberrant MAP kinase activation could also be a secondary event, caused by alterations of the cellular/retinal homeostasis in the XLRS disease process.
In this study, we examined whether retinoschisin binding to retinal membranes directly modulates MAP kinase signalling. Our findings in cultured Y‐79 cells and in retinal explants of Rs1h −/Y mice demonstrate that the addition of recombinant retinoschisin, but not recombinant mutant retinoschisin, significantly down‐regulates MAP kinase signalling, as well as protects against apoptosis. We conclude that retinoschisin deficiency could be a trigger for disease pathogenesis by a defective control of MAP kinase signalling and apoptosis in the retina.
Materials and methods
Animal models
The Rs1h −/Y mouse was generated as described earlier 9 and kept on a C57BL/6 background. Mice were housed under specific pathogen‐free barrier conditions at the Central Animal Facility of the University of Regensburg and maintained under conditions established by the institution for their use, in strict compliance with NIH guidelines. Mice were sacrificed 10 or 18 days after birth by decapitation or cervical dislocation after inhalation of carbon dioxide, respectively.
Cell culture
Y‐79 and Weri‐Rb1 (ATCC, Manassas, VA, USA) cells were cultivated in RPMI medium with 10% FCS as well as 100 U/ml penicillin/streptomycin. ARPE‐19 cells (ATCC) were maintained in DMEM/Ham's F12 medium containing 10% FCS and 100 U/ml penicillin/streptomycin. BV‐2 cells were grown in RPMI‐1640 with 5% FCS, 100 U/ml penicillin/streptomycin and 195 nM β‐mercaptoethanol. Hek293 cells (Invitrogen, Carlsbad, CA, USA) were maintained in DMEM high glucose medium containing 10% FCS, 100 U/ml penicillin/streptomycin and 500 μg/ml G418. All media and cell culture supplies were purchased from Life Technologies (Carlsbad, CA, USA). Cell lines were grown in a 37°C incubator with a 5% CO2 environment and subcultured when they reached 90% confluency for Hek293, BV‐2 and ARPE‐19 or a concentration of 4–5 × 105 cells/ml for Y‐79 and Weri‐Rb1. Only Y‐79 cells passaged less than 10 times were applied in signalling or apoptosis assays.
RNA analysis
RNA was isolated from cell lines using the Qiagen RNeasy Mini Kit (Qiagen, Venlo, the Netherlands). RNA from murine retinae and cultured retinal explants was isolated using the PureLink™ RNA Micro Kit (Invitrogen), according to the manufacturers’ protocols. One microgram of total RNA was transcribed into cDNA using RevertAid M‐MuLV Reverse Transcriptase (Fermentas, St Leon‐Rot, Germany) and poly(dT) primers according to the manufacturer's instructions. Semiquantitative RT‐PCR was performed as described by 23, with primers given in Table S1. Quantitative real‐time RT‐PCR was performed and analysed as published 51 with primers given in Table S1.
Western blot analysis
Proteins were separated after application of Laemmli buffer 52 on 12.5% gels or gradient gels 4–20% Mini‐PROTEAN® TGX™ Precast Protein Gels (Bio‐Rad Laboratories, Hercules, CA, USA; for analysis of retinoschisin octamers). For Western blotting, proteins were transferred to polyvinylidene difluoride (PVDF) membranes (Immobilon; Millipore, Schwalbach, Germany) Antibodies were used as follows: Antibodies against Myc tag, phospho‐c‐Raf (Ser338), c‐Raf and phospho‐44/42‐MAPK (Erk1/2; Thr202/Tyr204) were obtained from Cell Signaling Technologies (Danvers, MA, USA). Antibodies against ACTB and ERK1/2 were from Sigma‐Aldrich (St. Louis, MO, USA). Secondary anti‐rabbit or antimouse IgG horseradish peroxidase (HRP)‐linked antibodies were from Calbiochem (Merck Chemicals GmbH, Schwalbach, Germany). Antibody dilutions were applied according to the manufacturer's recommendations. RS1 primary antibody (diluted 1:10,000) was kindly provided by Prof. Robert Molday, University of British Columbia, Vancouver, Canada. Clarity™ Western ECL Substrate (Bio‐Rad Laboratories) and an Odyssey FC imager (LI‐COR, Lincoln, NE, USA) were used to visualize Western blots. Densitometric evaluation of Western blots was carried out using ImageJ (imagej.nih.gov).
Immunolabelling of retinal cryosections
Retinal explants were washed in PBS (2.7 mM KCl, 140 mM NaCl, 10 mM phosphate, pH 7.4) once. Subsequently, they were submerged in 4% (w/v) paraformaldehyde and incubated for 1 hr at room temperature. Retinae were washed in PBS twice before they were put in 30% (w/v) sucrose overnight. Single retinae were then embedded in Richard‐Allan Scientific™ Neg‐50™ Frozen Section Medium (Thermo Fisher Scientific, Waltham, MA, USA) and fast frozen in liquid nitrogen. About 10 μm cryosections were cut. Immunolabelling with anti‐ATP1B2 and antiretinoschisin antibodies was performed as described by 23. Cone visualization was performed with Alexa 488‐conjugated peanut agglutinin (1:250, PNA; Invitrogen). Rhodopsin staining was performed with Rho‐1D4 antibody (1:1000), kindly provided by Prof. Robert Molday, University of British Columbia, Vancouver, Canada. The sections were counterstained with 4′,6‐diamidino‐2‐phenylindol (DAPI, 1:1000; Molecular Probes, Leiden, the Netherlands). Images were taken with custom‐made VisiScope CSU‐X1 Confocal System (Visitron Systems, Puchheim, Germany) equipped with high‐resolution sCMOS camera (PCO AG, Kehlheim, Germany).
Expression cloning
The coding sequence of non‐mutant retinoschisin (NM_000330.3) was amplified from cDNA of retinal tissue using oligonucleotide primers containing a EcoRI restriction site at the 5′ end and a XhoI restriction site at the 3′ end of the RS1 coding sequence (for primer sequences, see Table S1). The coding sequence of the XLRS‐associated RS1 mutant RS1‐C59S (NM_000330.3 (RS1):c.175T>A [p.Cys59Ser] http://grenada.lumc.nl/LOVD2/eye/home.php?select_db=RS1) was generated by site‐directed mutagenesis on the retinoschisin coding sequence (primer sequences shown in Table S1). For purification, the two RS1 variants were each tagged with an N‐terminal Myc tag, following the leader sequence (after aa 23). This peptide insertion into the full‐length RS1 coding sequence was performed by fusing the N‐terminal part of both RS1 coding sequences from positions 1 to 69 (aa 1–23) to the N‐terminal half of the Myc tag sequence (for primers, see Table S1). The C‐terminal part of both RS1 coding sequences (aa 70 to stop codon) was fused to the C‐terminal half of the Myc tag sequence (for primers, see Table S1). C‐ and N‐terminal RS1 fragments were ligated via a BclI restriction site which was introduced into the Myc tag, and inserted into the pCDNA3.1™ expression vector (Thermo Fisher Scientific).
Expression and purification of recombinant RS1 variants
Expression constructs were transfected into Hek293 cells using calcium phosphate transfection as described by 53. About 7 hrs after transfection, the culture medium was replaced by Opti‐MEM® containing 100 U/ml penicillin/streptomycin (Life Technologies) and cells were cultured for additional 48 hrs.
Myc‐tagged retinoschisin and RS1‐C59S were isolated from cultivation media by immunoprecipitation using Pierce™ Anti‐c‐Myc Agarose (Thermo Fisher Scientific) according to the manufacturer's instructions. Concentrations of purified proteins were determined using the Bio‐Rad DC™ Protein Assay Kit (Bio‐Rad Laboratories).
For use as a treatment control, Hek293 cells were transfected with empty pCDNA3.1™ expression vector, and cultivation medium of these cells was subjected to purification procedure exactly like medium from cells transfected with RS1 variants.
Purity of purified Myc‐tagged RS1 proteins and control eluate was verified via silver staining, Coomassie Blue staining and Western blot analysis using antibodies against the Myc tag and against retinoschisin (Fig. S1).
Binding of RS1 protein variants to membranes
Retinoschisin binding to adherent cell lines (BV‐2, ARPE and Hek293) as well as to murine retinal membranes (P10) was assessed as described by 23, but with a prolonged incubation time of 1 hr.
Retinoschisin binding to suspension cell lines Y‐79 and Weri‐Rb1 was analysed by incubating 4 × 106 cells in 5 ml RS1 containing medium (from supernatant of stably transfected Hek293 cells 23) for 1 hr, with subsequent washing steps as described 23.
For comparing binding affinity of retinoschisin and RS1‐C59S to Y‐79 cells and Rs1h −/Y murine retinal membranes, 6 μg of purified retinoschisin or RS1‐C59S was added to 5 ml cultivation medium and incubated for 10, 30 and 60 min. Subsequent steps were performed as above.
For localization of bound recombinant RS1 variants on Rs1h −/Y murine retinae, Rs1h −/Y murine retinal explants were incubated with 1 μg RS1, RS1‐C59S or control eluate, as described in signalling experiments. After 30 min. of incubation, the retinal tissue was washed with PBS once before immunolabelling of retinal cryosections was performed.
Analysis of signalling pathways in retinal explants and Y‐79 cells
Y‐79 cells were grown to a concentration of 4–5 × 105 cells/ml in 10 ml medium. The experiment was started by adding 1 μg purified retinoschisin or RS1‐C59S, or equal volume of control eluate. After 10 or 30 min. of incubation at 37°C, cells were harvested by centrifugation. For Western blotting, cells were resuspended in 200 μl of pre‐cooled PBS with PhosSTOP™ phosphatase inhibitor (Sigma‐Aldrich) and lysed by sonication (10 sec., 40% intensity). For RNA isolation, cells were washed once with pre‐cooled PBS before they were subjected to RNA isolation.
Eyes from Rs1h −/Y mice at post‐natal day 10 were enucleated and retinal explants were dissected as described by 54. Retinae were incubated in 800 μl DMEM/Ham's F12 containing 10% FCS, 100 U/ml penicillin/streptomycin, 2 mM l‐glutamine and 2 μg/ml insulin (Thermo Fisher Scientific). One microgram purified retinoschisin or RS1‐C59S or equal volumes of control eluate were added. After 10 or 30 min. of incubation at 37°C, retinal explants were removed from medium and transferred to 200 μl of pre‐cooled PBS containing PhosSTOP™ phosphatase inhibitor. For Western blot analysis, retinal explants were sonicated for 10 sec. at 40% intensity. For RNA isolation, retinal explants were immediately transferred into lysis buffer (PureLink™ RNA Micro Kit; Invitrogen).
Analysis of caspase‐3 activity in Y‐79 cells
About 2 × 106 cells/well were seeded onto poly‐l‐lysine‐coated 24‐well plates. Cells were allowed to adhere overnight before 0.1 μg of purified retinoschisin or RS1‐C59S, or equal volumes of control eluate were added to 1 ml medium per well. After 1 hr, the culture medium was changed to 1 ml RPMI (containing RS1 variants or control eluate as before) and 0.2 mM H2O2 to induce apoptosis or 0 mM H2O2 as control. After 2 hrs, the medium was replaced by 1 ml RPMI containing RS1 variants or control eluate as before. Cells were allowed to recover for 18 hrs before they were subjected to a caspase‐3 activity test using the EnzChek® Caspase‐3 Assay Kit #2 by Thermo Fisher Scientific according to the manufacturer's directions.
Analysis of photoreceptor degeneration in retinal explants
Eyes were enucleated from mice at post‐natal day 18 and retinae were dissected as described before 55. Five retinae each were subjected to treatment with retinoschisin, RS1‐C59S and control protein. Retinal explants were transferred into pre‐warmed medium (DMEM/Ham's F12 containing 10% FCS, 100 U/ml antibiotic–antimytotic, 2 mM l‐glutamine and 2 μg/ml insulin, all from Life Technologies) immediately after preparation, rinsed once in pre‐warmed medium and then transferred onto Track Etch Membrane Filters (Whatman plc, Maidstone, UK) in 35‐mm tissue culture dishes containing 3 ml of medium to which 1 μg of purified retinoschisin, RS1‐C59S or control eluate had been added. The retinal explants on the filters were covered in a small droplet of medium. Cultivation was carried out under sterile conditions in a 37°C incubator with a 5% CO2 environment. Medium was replaced every 36 hrs and total cultivation time was 1 week. Subsequently, the retinal explants were fixed, cryopreserved and cut into 10‐μm sections for subsequent histological analyses as described before. After PNA staining, cones were counted in each two different sections of the same retina, in 200‐nm‐wide regions to the left and the right side of the optic nerve. Rods were stained with anti‐Rho‐1D4 antibody. Staining signals of each two different sections of the same retina, in 200‐nm‐wide regions to the left and the right side of the optic nerve, were quantified using ImageJ.
Results
Increased MAP kinase signalling in murine Rs1h −/Y retinae
A study by Gehrig et al. 40 previously indicated up‐regulated MAP kinase activity in early retinal development of Rs1h −/Y mice. The authors found in the murine Rs1h −/Y retinae increased phosphorylation of extracellular‐signal‐regulated kinases 1 and 2 (Erk1/2), as well as up‐regulated expression of Egr1 (early growth response protein 1), a prominent target gene of activated MAP kinases 56.
To first verify these results, we analysed phosphorylation of Erk1/2 as well as Egr1 expression in retinae of wild‐type and Rs1h −/Y mice, 7, 10 and 14 days after birth (P7, P10 and P14). Furthermore, we investigated phosphorylation of c‐Raf, a central constituent of the ERK pathway, the activation of which precedes and is required for Erk1/2 phosphorylation 57, in P7, P10 and P14 retinae. As an additional MAP kinase target gene, we assessed expression of the FBJ murine osteosarcoma viral oncogene homologue gene (c‐Fos), which is expressed in response to transient and sustained ERK signalling 56, 58, 59, 60, 61.
Western blot analyses showed an increase in c‐Raf and Erk1/2 phosphorylation in Rs1h −/Y retinae compared with wild‐type retina (Fig. 1A). C‐Raf phosphorylation levels in Rs1h −/Y retinae increased to around 150–200%, whereas Erk1/2 phosphorylation levels rose to around 250–300% above normal (Fig. 1B). These differences were obtained in all stages (P7, P10 and P14), and each were statistically significant (P < 0.05, except for c‐Raf at P10; Fig. 1A and B) Retinoschisin deficiency had no influence on levels of total c‐Raf and Erk1/2, independent of the post‐natal stages.
Figure 1.
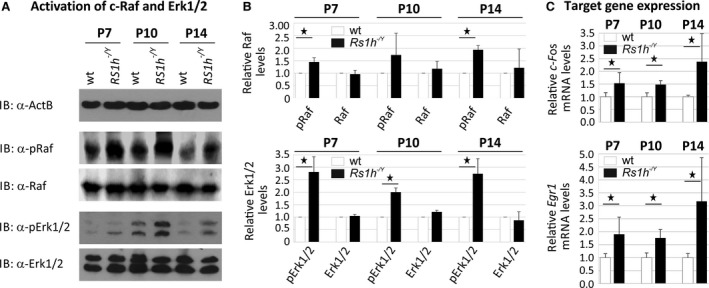
Influence of retinoschisin deficiency on MAP kinase signalling in the murine retina. C‐Raf and Erk1/2 phosphorylation in murine wild‐type and Rs1h −/Y retinae, harvested at post‐natal days 7, 10 and 14 (P7, P10 and P14). Retinal lysates were subjected to Western blot analyses with antibodies against phosphorylated c‐Raf (pRaf), total c‐Raf (Raf), phosphorylated Erk1 and Erk2 (pErk1/2), total Erk1 and Erk2 (Erk1/2), as well as ActB as a control (A). Densitometric quantification (B) was performed with immunoblots from three independent sample sets. Signals for pErk1/2, Erk1/2, pRaf and Raf were normalized against ActB and calibrated against signals for wild‐type retinae. Data represent the mean + S.D. (C) C‐Fos and Egr1 expression in murine wild‐type and Rs1h −/Y retinae harvested at post‐natal days 7, 10 and 14 (P7, P10 and P14). mRNA expression of C‐Fos and Egr1 was determined via quantitative real‐time RT‐PCR. Five independent sample sets were analysed. Results were normalized to Hprt transcript levels and calibrated with the control. The mean + S.D. for the three (immunoblot analyses) or five (quantitative RT‐PCR) independent sample sets is given. Asterisks mark statistically significant (*P < 0.05) and highly significant (**P < 0.01) differences.
Expression of MAP kinase target genes C‐Fos and Egr1 was increased in Rs1h −/Y retinae, at all developmental stages; c‐Fos expression levels in Rs1h −/Y retinae were between 150% and 250%, and Egr1 expression levels between 175% and 300% compared with wild‐type retinae. Differences in expression were statistically significant between wild‐type and Rs1h −/Y retinae (P < 0.05).
Binding of retinoschisin to different retinal cell types
Searching for an in vitro model system applicable for analysing the influence of retinoschisin on MAP kinase signalling, we tested different retinal cells including murine microglial cell line BV‐2, human RPE‐derived cell line ARPE‐19, the human retinoblastoma cell lines Y‐79 and Weri‐Rb1 for their capacity to bind retinoschisin (Fig. 2A). In these cells, we also investigated endogenous expression of the retinal Na/K‐ATPase subunits ATP1A3 and ATP1B2, required for anchoring retinoschisin to retinal plasma membranes 23 (Fig. 2B and C).
Figure 2.
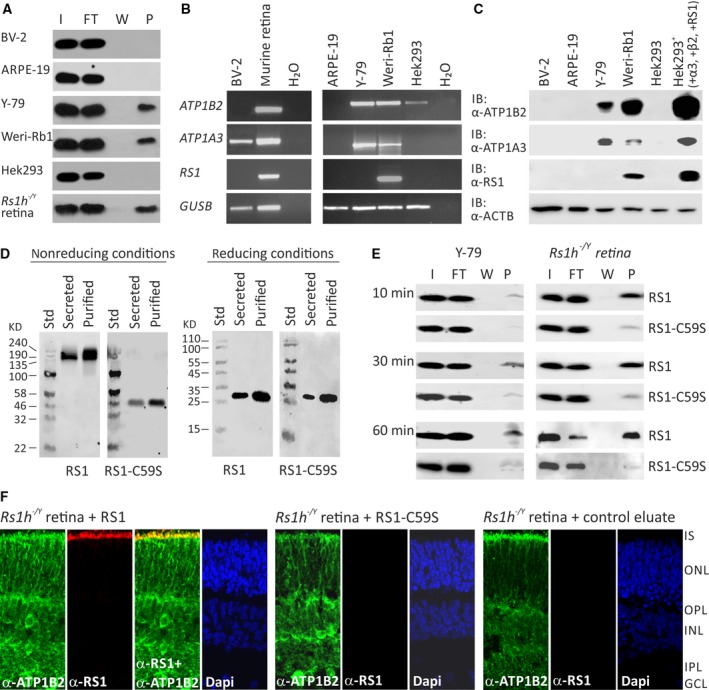
Binding of RS1 variants to retinal cells (A) Binding of retinoschisin to cultured retinal cell lines ARPE‐19, Y‐79, Weri‐Rb1 and BV‐2: Cells were incubated for 60 min. with retinoschisin containing supernatant (I, input) of cells stably transfected with a retinoschisin expression vector. Subsequently, cells were centrifuged and supernatant (FT, flowthrough) was discarded. After further washing steps (last supernatant, W), cells were pelleted (pellet, P). Fractions were subjected to Western blot analyses using an antiretinoschisin antibody. Retinoschisin binding to Hek293 cells and murine Rs1h −/Y retinal membranes served as negative and positive controls, respectively. (B) RT‐PCR analysis of ATP1B2, ATP1A3 and RS1 gene expression in cell lines derived from murine microglia (BV‐2), human retinal pigment epithelium (ARPE‐19), human retinoblastoma (Y‐79 and Weri‐Rb1) and human embryonic kidney (Hek293). GUSB gene expression was assessed as control for RNA integrity. (C) Cell lysates from BV‐2, ARPE‐19, Y‐79, Weri‐Rb1 and Hek293 were subjected to Western blot analyses using antibodies against ATP1B2, ATP1A3 and retinoschisin. Hek293+ cells served as positive control. The ACTB immunoblot was performed as loading control. (D) Oligomerization of RS1 variants (non‐mutant retinoschisin and RS1‐C59S) before and after purification. About 48 hrs after transfection of Hek293 cells with expression constructs for N‐terminally Myc‐tagged RS1 variants, the cell culture medium (supernatant) was harvested and Myc‐tagged proteins were purified from the supernatant. Aliquots of supernatant and purified RS1 fractions were subjected to SDS‐PAGE under non‐reducing and reducing conditions, followed by Western blot analyses using an antiretinoschisin antibody. (E) Binding of RS1 variants to retinal cells. Y‐79 cells and murine Rs1h −/Y retinal explants were incubated with purified RS1 variants (I, input) for 10, 30 and 60 min. Cells were centrifuged and supernatant (FT, flowthrough) was discarded. After several washing steps (last supernatant, W), cells were pelleted (pellet, P). Fractions were subjected to Western blot analyses using an antiretinoschisin antibody. (F) Localization of recombinant RS1 variants on retinal membranes. Rs1h −/Y retinal explants (P10) were incubated for 30 min. with retinoschisin, RS1‐C59S or control protein, the latter purified from supernatant of empty expression vector‐transfected cells. After washing and embedding, cryosections of these explants were subjected to immunohistochemical analyses using antibodies against ATP1B2 and retinoschisin. 4′,6‐Diamidino‐2‐phenylindol (DAPI) staining shows the nuclei of the different retinal layers. IS, inner segments; ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer.
Retinoschisin binding assays were performed on intact cells as described by 23. Retinoschisin binding to Hek293 cells and crude membranes of Rs1h −/Y retinae served as negative and positive controls, respectively 23. Efficient retinoschisin binding was found with Y‐79, Weri‐Rb1 and membranes of Rs1h −/Y retinae, but not with ARPE‐19, BV‐2 and Hek293.
Semiquantitative RT‐PCR (Fig. 2B) and Western blot analyses (Fig. 2C) revealed endogenous expression of ATP1A3 and ATP1B2 only in Y‐79 and Weri‐Rb1. Weri‐Rb1 cells also weakly expressed retinoschisin (Fig. 2B and C). In all further analyses, Y‐79 cells were used to assess effects of externally added retinoschisin on intracellular signalling.
Different binding affinities of normal retinoschisin and the XLRS‐associated mutant protein RS1‐C59S to retinal membranes
To analyse functional properties of retinoschisin, we heterologously expressed RS1 (NM_000330.3) and the XLRS‐associated mutant protein (NM_000330.3(RS1):c.175T>A [p.Cys59Ser], termed RS1‐C59S, in Hek293 cells. The mutation c.175T>A [p.Cys59Ser] is one of the rare XLRS variants which are not subjected to co‐ or post‐translational degradation, but instead are translated and secreted from cells, although not as a stable octamer but instead as a dimer 21, 22. For purification, retinoschisin (normal and mutant) was fused to an N‐terminal Myc tag, which did not influence secretion, oligomerization or binding capacities of the resulting protein (Fig. 2D and E).
Y‐79 cells and Rs1h −/Y retinal explants exposed to recombinant retinoschisin for 10, 30 and 60 min. stably bound the externally added retinoschisin, even after only 10 min. of incubation (Fig. 2E). Notably, RS1‐C59S exhibited a strongly reduced binding affinity to Y‐79 cells and Rs1h −/Y retinal explants (Fig. 2E).
Immunohistochemical stainings of murine Rs1h −/Y retinal explants after treatment with retinoschisin for 30 min. (Fig. 2F) confirmed the binding of externally added recombinant retinoschisin. Recombinant retinoschisin colocalized with the endogenously expressed retinal Na/K‐ATPase of the murine retina at the inner segments of photoreceptor cells. This is in agreement with the localization of retinoschisin in wild‐type retinae 23. In contrast to the known retinoschisin localization, no recombinant retinoschisin was detected in the plexiform layers of the retinal explants, which could possibly be explained by limited diffusion of the externally added retinoschisin through the retinal layers. No retinoschisin staining was observed in immunohistochemical analyses of Rs1h −/Y retinal explants treated with RS1‐C59S or control protein (Fig. 2F).
Extracellular retinoschisin modulates ERK 1/2 signalling in Y‐79 cells and Rs1h −/Y murine retinal explants
To assess the capacity of retinoschisin to directly modulate intracellular ERK1/2 signalling, we investigated an influence of extracellularly added recombinant retinoschisin (normal and mutant) on phosphorylation of C‐RAF and ERK1/2 in Y‐79 cells (Fig. 3) and Rs1h −/Y murine retinal explants (Fig. 4). As control, we applied protein purified from supernatant of mock vector‐transfected cells.
Figure 3.
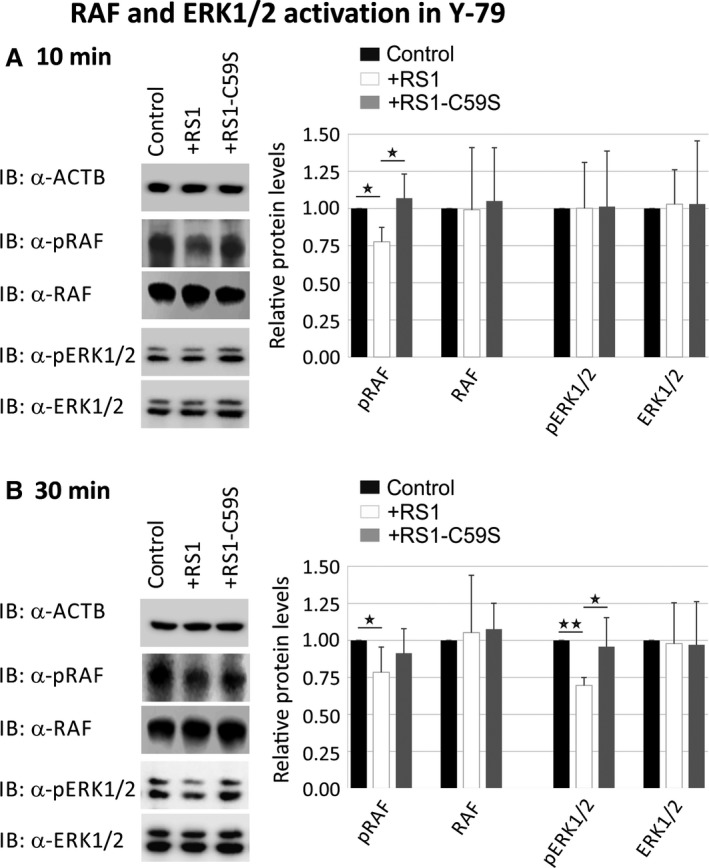
Influence of retinoschisin on the ERK pathway in Y‐79 cells. Retinoschisin‐dependent C‐RAF and ERK1/2 phosphorylation in Y‐79 cells. Y‐79 cells were treated for 10 min. (A) or 30 min. (B) with retinoschisin, RS1‐C59S or control protein (purified from supernatant of empty expression vector‐transfected cells). Subsequently, the cells were subjected to Western blot analyses with antibodies against phosphorylated C‐RAF (pRAF), total C‐RAF (RAF), phosphorylated ERK1 and ERK2 (pERK1/2), total ERK1 and ERK2 (ERK1/2), as well as ACTB as a control. (A and B) Densitometric quantification was performed with immunoblots from five independent experiments. Signals for pRAF RAF, pERK1/2 and ERK1/2 were normalized against ACTB and calibrated against the control. Data represent the mean + S.D. Asterisks mark statistically significant (*P < 0.05) and highly significant (**P < 0.01) differences.
Figure 4.
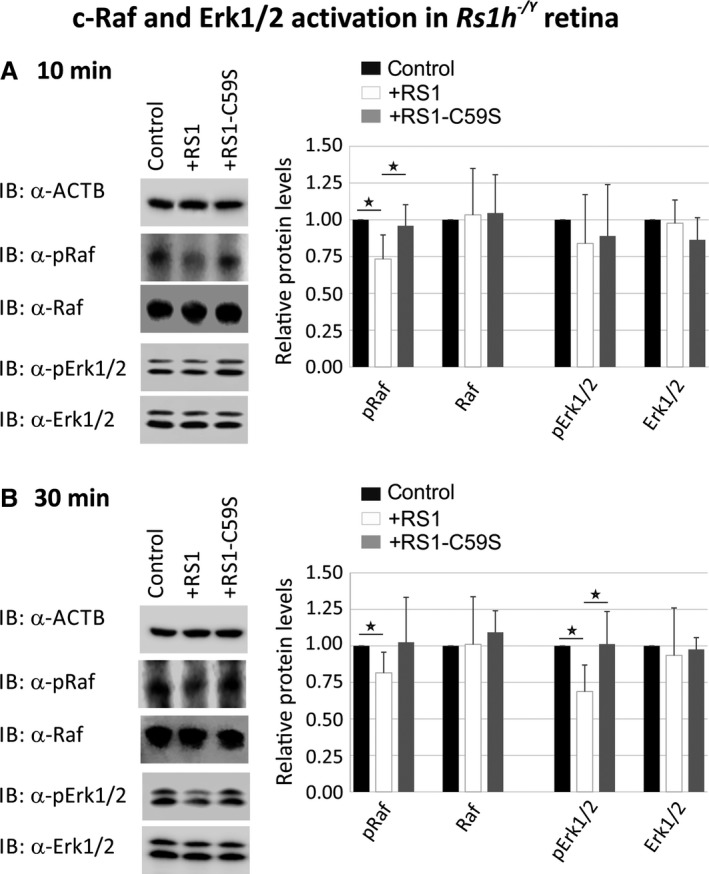
Influence of retinoschisin on the ERK pathway in murine Rs1h −/Y retinal explants. Retinoschisin‐dependent c‐Raf and Erk1/2 phosphorylation in murine Rs1h −/Y retinal explants. Retinal explants harvested 10 days after birth (P10) were treated for 10 min. (A) or 30 min. (B) with retinoschisin, RS1‐C59S or control protein (purified from supernatant of empty expression vector‐transfected cells). Subsequently, the retinal lysates were subjected to Western blot analyses with antibodies against phosphorylated c‐Raf (pRaf), total c‐Raf (Raf), phosphorylated Erk1 and Erk2 (pErk1/2), total Erk1 and Erk2 (Erk1/2), as well as ActB as a control. (A and B) Densitometric quantification was performed with immunoblots from five independent experiments. Signals for pRaf Raf, pErk1/2 and Erk1/2 were normalized against ActB and calibrated against the control. Data represent the mean + S.D. Asterisks mark statistically significant differences (*P < 0.05).
In Y‐79 cells, we observed a down‐regulation of phosphorylated C‐RAF (77.6 ± 9.7%) after 10 min. of treatment with recombinant retinoschisin (Fig. 3A). In contrast, RS1‐C59S failed to inhibit C‐RAF phosphorylation (Fig. 3A). The differences in phosphorylated C‐RAF levels between retinoschisin treatment and control or RS1‐C59S treatment were statistically significant (P < 0.05). The effect of retinoschisin on C‐RAF phosphorylation was still evident after 30 min. of treatment with retinoschisin, where C‐RAF phosphorylation was reduced to 78.4 ± 17.0% (Fig. 3B).
In contrast to its effect on C‐RAF in Y‐79 cells, retinoschisin treatment failed to show a significant decrease in ERK1/2 phosphorylation after 10 min. of incubation (Fig. 3A). Thirty minutes of retinoschisin treatment (Fig. 3B), however, reduced ERK activation to around 69.6 ± 5.3% in Y‐79 cells (P < 0.05 compared with control protein or RS1‐C59S). No alterations in total C‐RAF and total ERK1/2 protein levels were detected, excluding an effect of retinoschisin on expression or stability of the two proteins (Fig. 3).
MAP kinase signalling in murine Rs1h −/Y retinal explants was similarly affected by retinoschisin treatment (Fig. 4). After 10 min. of incubation with retinoschisin, phosphorylated c‐Raf levels were decreased to 73.4 ± 16.3% (Fig. 4A). RS1‐C59S treatment had no effect on c‐Raf phosphorylation. The differences in phosphorylated c‐Raf levels were statistically highly significant (P < 0.01) between control and retinoschisin treatment and statistically significant (P < 0.05) between retinoschisin and RS1‐C59S treatment. After 30 min. of incubation, the reduction in c‐Raf phosphorylation by retinoschisin treatment was still observable (Fig. 4B): Retinoschisin decreased c‐Raf phosphorylation to 81.4 ± 14.1%, compared with the control.
Erk1/2 phosphorylation was not affected after 10 min. (Fig. 4A), but only after 30 min. of treatment with recombinant retinoschisin (Fig. 4B). In contrast to control or RS1‐C59S treatment, incubation with retinoschisin resulted in a clear reduction in phosphorylated Erk1/2 (68.7 ± 18.2% compared with control, Fig. 4B). Differences in phosphorylated Erk1/2 levels were statistically highly significant (P < 0.01) when compared between control and retinoschisin treatment and statistically significant (P < 0.05) between retinoschisin and RS1‐C59S treatment. The different treatments caused no changes in total c‐Raf and total Erk1/2 levels in the retinal explants (Fig. 4).
Extracellular retinoschisin modulates ERK1/2 target gene expression in Y‐79 cells and Rs1h −/Y murine retinal explants
Upon treatment with recombinant retinoschisin, a statistically significant down‐regulation of C‐FOS mRNA expression was observed in Y‐79 cells (84.6 ± 7.7%) and in Rs1h −/Y murine retinal explants (68.1 ± 18.9%) by quantitative RT‐PCR when compared to control treatment (P < 0.05, Fig. 5A). In contrast, no prominent decrease in C‐FOS transcripts was found after treatment with RS1‐C59S (98.4 ± 7.3% for Y‐79; 97.7 ± 17.9% for retinal explants). The differences between the retinoschisin and the control or RS1‐C59S treatment were statistically significant (P < 0.05; Fig. 5A).
Figure 5.
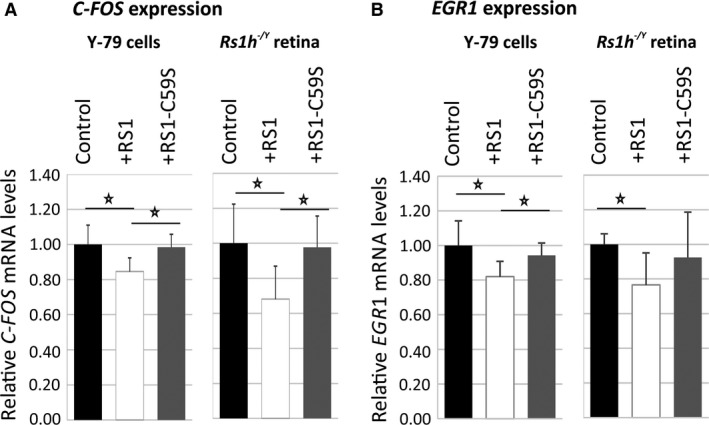
Influence of retinoschisin on the expression of ERK1/2 pathway target genes. Retinoschisin‐dependent C‐FOS (A) and EGR1 (B) expression in Y‐79 cells and murine Rs1h −/Y retinal explants. Cells/retinal explants were treated as described in Figures 3 and 4 for 2 hrs (Y‐79 cells) and 30 min. (murine retinal explants). C‐FOS and EGR1 mRNA expression was determined via quantitative real‐time RT‐PCR. Five independent experiments were performed. Results were normalized to HPRT transcript levels and calibrated with the control. The mean + S.D. for the five independent experiments is given. Asterisks mark statistically significant differences (*P < 0.05).
Similarly, retinoschisin treatment caused a statistically significant decrease in EGR1 expression (Fig. 5B): EGR1 transcript levels were reduced to 82.0 ± 8.8% in Y‐79 cells and to 76.6 ± 18.8% in Rs1h −/Y murine retinal explants (P < 0.05) when compared to control treatment. No prominent decrease in EGR1 mRNA levels was found after treatment with RS1‐C59S (94.3 ± 7.1% for Y‐79 cells, P < 0.05 compared with retinoschisin treatment, and 92.4 ± 26.2% for retinal explants).
Retinoschisin is protective for apoptotic events
Mitogen‐activated protein kinase signalling is an important mediator and regulator of several physiological processes implicated in XLRS pathogenesis 41, 43, 44, 45, 46, 47, 48, 49, 50, 62. As one characteristic feature of Rs1h −/Y mice is an early photoreceptor degeneration due to apoptotic cell death 63, we analysed the influence of retinoschisin on the expression of apoptosis marker BAX in Y‐79 cells and murine Rs1h −/Y retinal explants. Additionally, we assessed retinoschisin‐dependent caspase‐3 activation as a prominent marker for apoptosis induction 64, 65, 66 in Y‐79 cells, as well as the influence of recombinant retinoschisin on cone degeneration in murine Rs1h −/Y retinal explants (Fig. 6).
Figure 6.
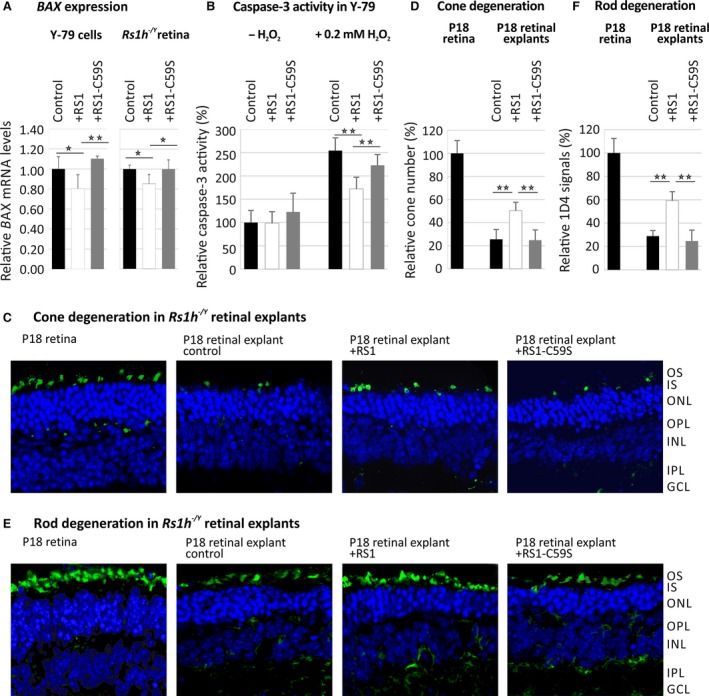
Influence of retinoschisin on apoptosis. (A) Retinoschisin‐dependent BAX expression in Y‐79 cells and murine Rs1h −/Y retinal explants. Y‐79 cells or retinal explants were treated with retinoschisin, RS1‐C59S or control protein for 20 hrs or 30 min., respectively. BAX mRNA expression was determined via quantitative real‐time RT‐PCR. Five independent experiments were performed. Results were normalized to HPRT transcript levels and calibrated with the control. The mean + S.D. for the five independent experiments is given. Asterisks mark statistically significant (*P < 0.05) and highly significant (**P < 0.01) differences. (B) Retinoschisin‐dependent activation of caspase‐3 in Y‐79 cells subjected to oxidative stress. Y‐79 cells, exposed to retinoschisin, RS1‐C59S or control protein were treated with 0.2 mM H2O2 for 2 hrs. About 18 hrs later, apoptosis was assayed by following caspase‐3‐specific proteolytic activity. Data represent the mean + S.D. of six independent experiments. Asterisks mark statistically highly significant differences (**P < 0.01). (C–F) Retinoschisin‐dependent photoreceptor degeneration in murine Rs1h −/Y retinal explants. Retinal explants harvested 18 days after birth (P18) were cultured for 1 week in medium containing retinoschisin, RS1‐C59S or control protein (purified from supernatant of empty expression vector‐transfected cells). After washing and embedding, cryosections of these explants were subjected to staining for nuclei, cones and rods. OS, outer segments; IS, inner segments; ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer. DAPI staining shows the nuclei of the different retinal layers. (C) Alexa488‐conjugated peanut agglutinin (PNA) staining was applied to visualize cones. (D) The total number of cones per analyzed section was counted after staining with PNA. (E) Anti‐Rho‐1D4 antibody staining was applied to visualize rod specific Rhodopsin. (F) Rhodopsin signals per analyzed section were measured using ImageJ (imagej.nih.gov). Data represent the mean + SD. Asterisks mark statistically highly significant differences (**P < 0.01).
Short time exposure to retinoschisin caused no change in BAX transcript levels in Y‐79 cells (data not shown). After 20 hrs of incubation, however, BAX transcript levels were significantly decreased (77.6 ± 14.0%) compared with control (P < 0.05) or RS1‐C59S treatment (P < 0.01, Fig. 6A). In Rs1h −/Y retinal explants, 30 min. of incubation with retinoschisin reduced BAX transcript levels to 85.4 ± 9.3% (P < 0.01 compared with control and RS1‐C59S‐treated retinae, Fig. 6A). In contrast, incubation with RS1‐C59S had no significant effect on BAX expression in Y‐79 cells (109.5 ± 2.6%) or retinal explants (103.3 ± 11.8%, Fig. 6A).
Caspases were shown to play a prominent role in photoreceptor cell death in the retinoschisin‐deficient mouse 63. We thus followed caspase‐3 activation in Y‐79 cells upon stress‐induced apoptosis after treatment with 0.2 mM hydrogen peroxide [H2O2] 64, 65, 66. H2O2 treatment caused an about 2.5‐fold increase in caspase‐3 activity compared with unstimulated cells (Fig. 6B). Notably, in cells stimulated with H2O2, retinoschisin strongly decreased caspase‐3 activation (67.6 ± 9.9%) compared with control protein or RS1‐C59S (P < 0.01). RS1‐C59S led to a slight, albeit statistically not significant, reduction in caspase‐3 activity (87.6 ± 9.1%, P = 0.59) in H2O2‐treated cells.
Finally, we addressed retinoschisin‐dependent photoreceptor survival by following cone and rod degeneration in murine Rs1h −/Y retinal explants 63. Photoreceptor cell death in Rs1h −/Y mice is triggered by apoptotic events initiated around 14 days after birth (P14, 63). We isolated Rs1h −/Y retinae 18 days after birth (P18) and incubated them in medium containing retinoschisin, RS1‐C59S or control protein. One week of cultivation resulted in a strong degeneration of retinal explants, shown by a markedly decreased thickness of the central retina and a significant reduction in photoreceptor cells (Fig. 6C–F). More specifically, compared with untreated retinae, 1 week of cultivation reduced the number of cones to around 25% in control and RS1‐C59S‐treated explants (25.6 ± 8.5% for control and 24.8 ± 8.9% for RS1‐C59S treatment (Fig. 6C and D). Notably, in explants treated with retinoschisin, the cone number was decreased to only about 50% (50.3 ± 7.3% compared with untreated retinae), with a statistically highly significant difference to control and RS1‐C59S‐treated explants (P < 0.01). Investigations on rod degeneration revealed similar results (Fig. 6E and F). After 1 week of cultivation, rod signals were reduced to around 24.8 ± 9.3% for control treated and to 28.9 ± 4.7% for RS1‐C59S‐treated explants. Treatment with retinoschisin lead to a rod signal decrease of only 59.3 ± 7.6% compared with untreated retinae, with statistically highly significant differences to control and RS1‐C59S‐treated explants (P < 0.01).
Discussion
In this study, we investigated the role of retinoschisin in the regulation of intracellular MAP kinase signalling. Firstly, our experiments confirmed strongly increased MAP kinase signalling in early retinal development of Rs1h −/Y mice. Secondly, we demonstrated that retinoschisin binding directly decreased phosphorylation of C‐RAF and MAP kinases ERK1 and ERK2, as well as expression of the MAP kinase target genes C‐FOS and EGR1 in a retinal (Y‐79) cell line and in murine Rs1h −/Y retinal explants. Thirdly, our data suggest a protective effect of retinoschisin against apoptotic cell death in Y‐79 cells and Rs1h −/Y retinal explants. As a stringent control, the XLRS mutant RS1‐C59S was deficient in binding to retinal membranes, and failed to reveal regulation on MAP kinase signalling or effects on apoptosis. Together, our results demonstrate that retinoschisin is a novel regulator of intracellular signalling and protects retinal cells from apoptosis. We suggest that aberrant MAP kinase signalling due to retinoschisin deficiency could be an initial trigger in XLRS pathogenesis.
In recent years, the importance of MAP kinase signalling in retinal development and homeostasis has attracted increasing attention 67, 68, 69. Not surprisingly, several retinal dystrophies such as age‐related macular degeneration 70, 71, 72, diabetic retinopathy 73 or retinitis pigmentosa 74, 75 were linked to malfunctioning MAP kinase pathways. Aberrant MAP kinase signalling was also observed during early retinal development in the XLRS mouse model 40. Our study verified the earlier observations from Gehrig et al. 40 by showing increased activation of central constituents of the ERK pathway, c‐Raf and Erk1/2 57, in Rs1h −/Y retinae of 7‐, 10‐ and 14‐day‐old mice. Furthermore, we showed up‐regulation of prominent target genes of MAP kinase signalling, namely c‐Fos and Egr1 56, indicating an early and sustained alteration in MAP kinase signalling in disease development of Rs1h −/Y mice.
To assess whether retinoschisin has the capacity to directly modulate MAP kinase signalling, we investigated the effect of recombinant retinoschisin on activation of the ERK pathway in two retinal model systems; the human retinoblastoma cell line Y‐79 and Rs1h −/Y murine retinal explants, both capable to bind extracellularly added retinoschisin due to an endogenous expression of the NA/K‐ATPase subunits α3 and β2. The addition of recombinant retinoschisin had an immediate and significant influence on MAP kinase signalling in these two model systems, reflected by decreased C‐RAF and ERK1/2 phosphorylation. C‐RAF activation (10 min.) occurred before ERK1/2 phosphorylation (30 min. after addition of retinoschisin), in agreement with the established sequence of C‐RAF and ERK1/2 activation in the ERK signalling cascade 57, 76. Subsequently, retinoschisin treatment also induced down‐regulation of C‐FOS and EGR1 expression in Y79 cells and Rs1h −/Y murine retinal explants. These results establish retinoschisin as an important regulator of the MAP kinase pathway in retinal cells.
The contribution of MAP kinase signalling to various disease processes can be explained by its key role in the regulation of complex physiological processes such as apoptosis, adhesion, proliferation, differentiation or development 41, 44, 49. For instance, several studies showed a pro‐apoptotic effect of ERK activation specifically connected to neuronal cells, for example in neurodegenerative disease processes 77, 78, 79, 80. Of note, a characteristic increase in ERK1/2 activation with an effect size similar to our results has been described for early disease stages of Alzheimer's disease with 25% less ERK1/2 activation in temporal cortex of healthy individuals compared with patients 81, or of ocular ischaemic syndrome where 29% less ERK1 and 21% less ERK2 activation in murine retinae of control mice were found when compared to a mouse model of ocular ischaemic syndrome 82. Additionally, comparably small alterations in MAP kinase signalling, related to cellular survival, were found in natural killer cells of chronic fatigue syndrome 83 and in lymphocytes of patients with Alzheimer's and Parkinson's disease 84.
Consistently, we demonstrate a protective influence of retinoschisin against apoptosis: Transcript levels of the pro‐apoptotic BAX protein 85, 86 were down‐regulated in Y‐79 cells and Rs1h −/Y retinae exposed to recombinant retinoschisin. Furthermore, in Y‐79 cells subjected to oxidative stress, caspase‐3 activity, a marker for the induction of apoptosis 65, was significantly decreased by retinoschisin. Similarly, apoptosis‐induced cone and rod degeneration 63 in murine Rs1h −/Y retinal explants was strongly reduced in the presence of recombinant retinoschisin. Further studies are required to verify the direct contribution of increased MAP kinase activation to photoreceptor apoptosis in XLRS pathogenesis. Nevertheless, considering the current state of knowledge on the pathological role of MAP kinase activation in neurodegeneration 77, 78, 79, 80, 81, 82, we speculate that increased MAP kinase signalling due to retinoschisin deficiency can induce or contribute to XLRS‐associated neurodegenerative processes in humans, and apoptotic photoreceptor degeneration in the XLRS mouse model 40.
Our investigations included studies on the functionality of the XLRS‐associated retinoschisin mutant, RS1‐C59S. Unlike most RS1 mutants, RS1‐C59S is translated and secreted from cells, but with defective oligomerization 21, 22. The functional consequences of this structural alteration have not been elucidated, so far. Here, we show that RS1‐C59S cannot bind to retinal membranes and can thus not fulfil its function as a regulator of intracellular signalling.
The present data do not allow elucidation of how extracellular retinoschisin binding affects intracellular MAP kinase signalling. Previous analysis identified the retinal Na/K‐ATPase as the specific binding partner for retinoschisin on retinal membranes 14, 23. Several groups reported that in addition to their function as an ion pump 87, 88, Na/K‐ATPases are important regulators of intracellular MAP kinase signalling 31, 89, 90, 91, although the exact mechanism of signal transduction from Na/K‐ATPases to the MAP cascade is under debate 30, 92, 93, 94, 95, 96. It would thus be conceivable that retinoschisin modulates the capacity of the Na/K‐ATPase to regulate intracellular signalling. A disruption of this retinoschisin‐Na/K‐ATPase signalosome complex by retinoschisin deficiency could therefore result in defective MAP kinase regulation by the Na/K‐ATPase.
Taken together, we provide evidence that retinoschisin is a novel regulator of MAP kinase signalling in the retina with the capacity to protect cells against apoptotic cell death. We suggest that disturbances of intracellular MAP kinase signalling by retinoschisin deficiency might be one of the initial steps in XLRS pathology. Thus, our data could provide a novel basis for considerations to therapeutic treatments for this progressive and currently untreatable disease.
Conflict of interest
None declared.
Supporting information
Figure S1 Purity of Myc‐tagged RS1 proteins.
Table S1 Primers used in RNA analyses and expression cloning.
Acknowledgements
This work was supported in parts by a grant from the Deutsche Forschungsgemeinschaft (DFG) (FR 3377/1‐1 to U.F.). We thank T.L. (Laboratory for Experimental Immunology of the Eye, Department of Ophthalmology, University of Cologne, Germany) for providing BV‐2 cells. We thank L.P., M.R. and D.S. (Institute of Human Genetics, University of Regensburg, Germany) for excellent technical assistance.
[Correction added on 06 February 2017, after first online publication: The last lane (positive control) of the Western blot panel in figure 2C has incorrect label and this has been amended in this version.]
References
- 1. Sauer CG, Gehrig A, Warneke‐Wittstock R, et al Positional cloning of the gene associated with X‐linked juvenile retinoschisis. Nat Genet. 1997; 17: 164–70. [DOI] [PubMed] [Google Scholar]
- 2. George ND, Yates JR, Bradshaw K, et al Infantile presentation of X linked retinoschisis. Br J Ophtalmol. 1995; 79: 653–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kellner U, Brummer S, Foerster MH, et al X‐linked congenital retinoschisis. Graefes Arch Clin Exp Ophthalmol. 1990; 228: 432–7. [DOI] [PubMed] [Google Scholar]
- 4. Molday RS, Kellner U, Weber BH. X‐linked juvenile retinoschisis: clinical diagnosis, genetic analysis, and molecular mechanisms. Prog Retin Eye Res. 2012; 31: 195–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pimenides D, George ND, Yates JR, et al X‐linked retinoschisis: clinical phenotype and RS1 genotype in 86 UK patients. J Med Genet. 2005; 42: e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yu J, Ni Y, Keane PA, et al Foveomacular schisis in juvenile X‐linked retinoschisis: an optical coherence tomography study. Am J Ophthalmol. 2010; 149: 973–8 e2. [DOI] [PubMed] [Google Scholar]
- 7. Renner AB, Kellner U, Fiebig B, et al ERG variability in X‐linked congenital retinoschisis patients with mutations in the RS1 gene and the diagnostic importance of fundus autofluorescence and OCT. Doc Ophthalmol. 2008; 116: 97–109. [DOI] [PubMed] [Google Scholar]
- 8. Takada Y, Vijayasarathy C, Zeng Y, et al Synaptic pathology in retinoschisis knockout (Rs1‐/y) mouse retina and modification by rAAV‐Rs1 gene delivery. Invest Ophthalmol Vis Sci. 2008; 49: 3677–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Weber BH, Schrewe H, Molday LL, et al Inactivation of the murine X‐linked juvenile retinoschisis gene, Rs1h, suggests a role of retinoschisin in retinal cell layer organization and synaptic structure. Proc Natl Acad Sci USA. 2002; 99: 6222–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zeng Y, Takada Y, Kjellstrom S, et al RS‐1 gene delivery to an adult Rs1h knockout mouse model restores ERG b‐wave with reversal of the electronegative waveform of X‐linked retinoschisis. Invest Ophthalmol Vis Sci. 2004; 45: 3279–85. [DOI] [PubMed] [Google Scholar]
- 11. Bush RA, Wei LL, Sieving PA. Convergence of human genetics and animal studies: gene therapy for X‐linked retinoschisis. Cold Spring Harb Perspect Med. 2015; 5: a017368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Byrne LC, Ozturk BE, Lee T, et al Retinoschisin gene therapy in photoreceptors, Muller glia or all retinal cells in the Rs1h‐/‐ mouse. Gene Ther. 2014; 21: 585–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ou J, Vijayasarathy C, Ziccardi L, et al Synaptic pathology and therapeutic repair in adult retinoschisis mouse by AAV‐RS1 transfer. J Clin Invest. 2015; 125: 2891–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Molday LL, Wu WW, Molday RS. Retinoschisin (RS1), the protein encoded by the X‐linked retinoschisis gene, is anchored to the surface of retinal photoreceptor and bipolar cells through its interactions with a Na/K ATPase‐SARM1 complex. J Biol Chem. 2007; 282: 32792–801. [DOI] [PubMed] [Google Scholar]
- 15. Park TK, Wu Z, Kjellstrom S, et al Intravitreal delivery of AAV8 retinoschisin results in cell type‐specific gene expression and retinal rescue in the Rs1‐KO mouse. Gene Ther. 2009; 16: 916–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sikkink SK, Biswas S, Parry NR, et al X‐linked retinoschisis: an update. J Med Genet. 2007; 44: 225–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Molday LL, Hicks D, Sauer CG, et al Expression of X‐linked retinoschisis protein RS1 in photoreceptor and bipolar cells. Invest Ophthalmol Vis Sci. 2001; 42: 816–25. [PubMed] [Google Scholar]
- 18. Takada Y, Fariss RN, Muller M, et al Retinoschisin expression and localization in rodent and human pineal and consequences of mouse RS1 gene knockout. Mol Vis. 2006; 12: 1108–16. [PubMed] [Google Scholar]
- 19. Bush M, Setiaputra D, Yip CK, et al Cog‐wheel octameric structure of RS1, the discoidin domain containing retinal protein associated with X‐linked retinoschisis. PLoS One. 2016; 11: e0147653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tolun G, Vijayasarathy C, Huang R, et al Paired octamer rings of retinoschisin suggest a junctional model for cell‐cell adhesion in the retina. Proc Natl Acad Sci USA. 2016; 113: 5287–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wu WW, Molday RS. Defective discoidin domain structure, subunit assembly, and endoplasmic reticulum processing of retinoschisin are primary mechanisms responsible for X‐linked retinoschisis. J Biol Chem. 2003; 278: 28139–46. [DOI] [PubMed] [Google Scholar]
- 22. Wu WW, Wong JP, Kast J, et al RS1, a discoidin domain‐containing retinal cell adhesion protein associated with X‐linked retinoschisis, exists as a novel disulfide‐linked octamer. J Biol Chem. 2005; 280: 10721–30. [DOI] [PubMed] [Google Scholar]
- 23. Friedrich U, Stohr H, Hilfinger D, et al The Na/K‐ATPase is obligatory for membrane anchorage of retinoschisin, the protein involved in the pathogenesis of X‐linked juvenile retinoschisis. Hum Mol Genet. 2011; 20: 1132–42. [DOI] [PubMed] [Google Scholar]
- 24. Blanco G, Mercer RW. Isozymes of the Na‐K‐ATPase: heterogeneity in structure, diversity in function. Am J Physiol. 1998; 275: F633–50. [DOI] [PubMed] [Google Scholar]
- 25. Skou JC, Esmann M. The Na, K‐ATPase. J Bioenerg Biomembr. 1992; 24: 249–61. [DOI] [PubMed] [Google Scholar]
- 26. Antonicek H, Persohn E, Schachner M. Biochemical and functional characterization of a novel neuron‐glia adhesion molecule that is involved in neuronal migration. J Cell Biol. 1987; 104: 1587–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gloor S, Antonicek H, Sweadner KJ, et al The adhesion molecule on glia (AMOG) is a homologue of the beta subunit of the Na,K‐ATPase. J Cell Biol. 1990; 110: 165–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vagin O, Dada LA, Tokhtaeva E, et al The Na‐K‐ATPase alpha(1)beta(1) heterodimer as a cell adhesion molecule in epithelia. Am J Physiol Cell Physiol. 2012; 302: C1271–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kaplan JH. Biochemistry of Na,K‐ATPase. Annu Rev Biochem. 2002; 71: 511–35. [DOI] [PubMed] [Google Scholar]
- 30. Li Z, Xie Z. The Na/K‐ATPase/Src complex and cardiotonic steroid‐activated protein kinase cascades. Pflugers Arch. 2009; 457: 635–44. [DOI] [PubMed] [Google Scholar]
- 31. Xie Z, Askari A. Na(+)/K(+)‐ATPase as a signal transducer. Eur J Biochem. 2002; 269: 2434–9. [DOI] [PubMed] [Google Scholar]
- 32. Xie Z, Cai T. Na+‐K+–ATPase‐mediated signal transduction: from protein interaction to cellular function. Mol Interv. 2003; 3: 157–68. [DOI] [PubMed] [Google Scholar]
- 33. Aperia AC, Akkuratov EE, Fontana JM, et al Na+, K + ‐ATPase, a new class of plasma membrane receptors. Am J Physiol Cell Physiol. 2016; 1: C491–5. [DOI] [PubMed] [Google Scholar]
- 34. Schoner W, Scheiner‐Bobis G. Endogenous and exogenous cardiac glycosides and their mechanisms of action. Am J Cardiovasc Drugs. 2007; 7: 173–89. [DOI] [PubMed] [Google Scholar]
- 35. Geering K. FXYD proteins: new regulators of Na‐K‐ATPase. Am J Physiol Renal Physiol. 2006; 290: F241–50. [DOI] [PubMed] [Google Scholar]
- 36. Sweadner KJ, Rael E. The FXYD gene family of small ion transport regulators or channels: cDNA sequence, protein signature sequence, and expression. Genomics. 2000; 68: 41–56. [DOI] [PubMed] [Google Scholar]
- 37. Jones DH, Li TY, Arystarkhova E, et al Na, K‐ATPase from mice lacking the gamma subunit (FXYD2) exhibits altered Na+ affinity and decreased thermal stability. J Biol Chem. 2005; 280: 19003–11. [DOI] [PubMed] [Google Scholar]
- 38. Lubarski I, Pihakaski‐Maunsbach K, Karlish SJ, et al Interaction with the Na, K‐ATPase and tissue distribution of FXYD5 (related to ion channel). J Biol Chem. 2005; 280: 37717–24. [DOI] [PubMed] [Google Scholar]
- 39. Tokhtaeva E, Sun H, Deiss‐Yehiely N, et al FXYD5 O‐glycosylated ectodomain impairs adhesion by disrupting cell‐cell trans‐dimerization of Na,K‐ATPase beta1 subunits. J Cell Sci. 2016; 129: 2394–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gehrig A, Langmann T, Horling F, et al Genome‐wide expression profiling of the retinoschisin‐deficient retina in early postnatal mouse development. Invest Ophthalmol Vis Sci. 2007; 48: 891–900. [DOI] [PubMed] [Google Scholar]
- 41. Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001; 410: 37–40. [DOI] [PubMed] [Google Scholar]
- 42. Lewis TS, Shapiro PS, Ahn NG. Signal transduction through MAP kinase cascades. Adv Cancer Res. 1998; 74: 49–139. [DOI] [PubMed] [Google Scholar]
- 43. Karmarkar SW, Bottum KM, Krager SL, et al ERK/MAPK is essential for endogenous neuroprotection in SCN2.2 cells. PLoS One. 2011; 6: e23493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kolkova K, Novitskaya V, Pedersen N, et al Neural cell adhesion molecule‐stimulated neurite outgrowth depends on activation of protein kinase C and the Ras‐mitogen‐activated protein kinase pathway. J Neurosci. 2000; 20: 2238–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wruck CJ, Gotz ME, Herdegen T, et al Kavalactones protect neural cells against amyloid beta peptide‐induced neurotoxicity via extracellular signal‐regulated kinase 1/2‐dependent nuclear factor erythroid 2‐related factor 2 activation. Mol Pharmacol. 2008; 73: 1785–95. [DOI] [PubMed] [Google Scholar]
- 46. Johnson GL, Lapadat R. Mitogen‐activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002; 298: 1911–2. [DOI] [PubMed] [Google Scholar]
- 47. Sun Y, Liu WZ, Liu T, et al Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J Recept Signal Transduct Res. 2015; 35: 600–4. [DOI] [PubMed] [Google Scholar]
- 48. Xian J, Shao H, Chen X, et al Nucleophosmin mutants promote adhesion, migration and invasion of human leukemia THP‐1 cells through MMPs up‐regulation via Ras/ERK MAPK signaling. Int J Biol Sci. 2016; 12: 144–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhang W, Liu HT. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002; 12: 9–18. [DOI] [PubMed] [Google Scholar]
- 50. Widmann C, Gibson S, Jarpe MB, et al Mitogen‐activated protein kinase: conservation of a three‐kinase module from yeast to human. Physiol Rev. 1999; 79: 143–80. [DOI] [PubMed] [Google Scholar]
- 51. Friedrich U, Myers CA, Fritsche LG, et al Risk‐ and non‐risk‐associated variants at the 10q26 AMD locus influence ARMS2 mRNA expression but exclude pathogenic effects due to protein deficiency. Hum Mol Genet. 2011; 20: 1387–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970; 227: 680–5. [DOI] [PubMed] [Google Scholar]
- 53. Sambrook J, Russell DW. Calcium‐phosphate‐mediated transfection of eukaryotic cells with plasmid DNAs. CSH Protoc. 2006; 2006: doi: 10.1101/pdb.prot3871. [DOI] [PubMed] [Google Scholar]
- 54. Hsiau TH, Diaconu C, Myers CA, et al The cis‐regulatory logic of the mammalian photoreceptor transcriptional network. PLoS One. 2007; 2: e643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lee J, Myers CA, Williams N, et al Quantitative fine‐tuning of photoreceptor cis‐regulatory elements through affinity modulation of transcription factor binding sites. Gene Ther. 2010; 17: 1390–9. [DOI] [PubMed] [Google Scholar]
- 56. Whitmarsh AJ. Regulation of gene transcription by mitogen‐activated protein kinase signaling pathways. Biochim Biophys Acta. 2007; 1773: 1285–98. [DOI] [PubMed] [Google Scholar]
- 57. Roskoski R Jr. ERK1/2 MAP kinases: structure, function, and regulation. Pharmacol Res. 2012; 66: 105–43. [DOI] [PubMed] [Google Scholar]
- 58. Hazzalin CA, Mahadevan LC. MAPK‐regulated transcription: a continuously variable gene switch? Nat Rev Mol Cell Biol. 2002; 3: 30–40. [DOI] [PubMed] [Google Scholar]
- 59. Hess J, Angel P, Schorpp‐Kistner M. AP‐1 subunits: quarrel and harmony among siblings. J Cell Sci. 2004; 117: 5965–73. [DOI] [PubMed] [Google Scholar]
- 60. Murphy LO, Blenis J. MAPK signal specificity: the right place at the right time. Trends Biochem Sci. 2006; 31: 268–75. [DOI] [PubMed] [Google Scholar]
- 61. Whitmarsh AJ, Davis RJ. Transcription factor AP‐1 regulation by mitogen‐activated protein kinase signal transduction pathways. J Mol Med. 1996; 74: 589–607. [DOI] [PubMed] [Google Scholar]
- 62. Wada T, Penninger JM. Mitogen‐activated protein kinases in apoptosis regulation. Oncogene. 2004; 23: 2838–49. [DOI] [PubMed] [Google Scholar]
- 63. Gehrig A, Janssen A, Horling F, et al The role of caspases in photoreceptor cell death of the retinoschisin‐deficient mouse. Cytogenet Genome Res. 2006; 115: 35–44. [DOI] [PubMed] [Google Scholar]
- 64. Demelash A, Karlsson JO, Nilsson M, et al Selenium has a protective role in caspase‐3‐dependent apoptosis induced by H2O2 in primary cultured pig thyrocytes. Eur J Endocrinol. 2004; 150: 841–9. [DOI] [PubMed] [Google Scholar]
- 65. Li P, Nijhawan D, Budihardjo I, et al Cytochrome c and dATP‐dependent formation of Apaf‐1/caspase‐9 complex initiates an apoptotic protease cascade. Cell. 1997; 91: 479–89. [DOI] [PubMed] [Google Scholar]
- 66. Supanji, Shimomachi M, Hasan MZ, et al HtrA1 is induced by oxidative stress and enhances cell senescence through p38 MAPK pathway. Exp Eye Res. 2013; 112: 79–92. [DOI] [PubMed] [Google Scholar]
- 67. Darling NJ, Cook SJ. The role of MAPK signalling pathways in the response to endoplasmic reticulum stress. Biochim Biophys Acta. 2014; 1843: 2150–63. [DOI] [PubMed] [Google Scholar]
- 68. Donovan M, Doonan F, Cotter TG. Differential roles of ERK1/2 and JNK in retinal development and degeneration. J Neurochem. 2011; 116: 33–42. [DOI] [PubMed] [Google Scholar]
- 69. Mongan M, Wang J, Liu H, et al Loss of MAP3K1 enhances proliferation and apoptosis during retinal development. Development. 2011; 138: 4001–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Dridi S, Hirano Y, Tarallo V, et al ERK1/2 activation is a therapeutic target in age‐related macular degeneration. Proc Natl Acad Sci USA. 2012; 109: 13781–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. SanGiovanni JP, Lee PH. AMD‐associated genes encoding stress‐activated MAPK pathway constituents are identified by interval‐based enrichment analysis. PLoS One. 2013; 8: e71239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Yating Q, Yuan Y, Wei Z, et al Oxidized LDL induces apoptosis of human retinal pigment epithelium through activation of ERK‐Bax/Bcl‐2 signaling pathways. Curr Eye Res. 2015; 40: 415–22. [DOI] [PubMed] [Google Scholar]
- 73. Dong N, Chang L, Wang B, et al Retinal neuronal MCP‐1 induced by AGEs stimulates TNF‐alpha expression in rat microglia via p38, ERK, and NF‐kappaB pathways. Mol Vis. 2014; 20: 616–28. [PMC free article] [PubMed] [Google Scholar]
- 74. Kang MJ, Chung J, Ryoo HD. CDK5 and MEKK1 mediate pro‐apoptotic signalling following endoplasmic reticulum stress in an autosomal dominant retinitis pigmentosa model. Nat Cell Biol. 2012; 14: 409–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Sekimukai D, Honda S, Negi A. RNA interference for apoptosis signal‐regulating kinase‐1 (ASK‐1) rescues photoreceptor death in the rd1 mouse. Mol Vis. 2009; 15: 1764–73. [PMC free article] [PubMed] [Google Scholar]
- 76. Qi M, Elion EA. MAP kinase pathways. J Cell Sci. 2005; 118: 3569–72. [DOI] [PubMed] [Google Scholar]
- 77. Cheung EC, Slack RS. Emerging role for ERK as a key regulator of neuronal apoptosis. Sci STKE. 2004; 2004: PE45. [DOI] [PubMed] [Google Scholar]
- 78. Kulich SM, Chu CT. Sustained extracellular signal‐regulated kinase activation by 6‐hydroxydopamine: implications for Parkinson's disease. J Neurochem. 2001; 77: 1058–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Lu Z, Xu S. ERK1/2 MAP kinases in cell survival and apoptosis. IUBMB Life. 2006; 58: 621–31. [DOI] [PubMed] [Google Scholar]
- 80. Stanciu M, Wang Y, Kentor R, et al Persistent activation of ERK contributes to glutamate‐induced oxidative toxicity in a neuronal cell line and primary cortical neuron cultures. J Biol Chem. 2000; 275: 12200–6. [DOI] [PubMed] [Google Scholar]
- 81. Arendt T, Holzer M, Grossmann A, et al Increased expression and subcellular translocation of the mitogen activated protein kinase kinase and mitogen‐activated protein kinase in Alzheimer's disease. Neuroscience. 1995; 68: 5–18. [DOI] [PubMed] [Google Scholar]
- 82. Du R, Wang JL, Wang YL. Role of RhoA/MERK1/ERK1/2/iNOS signaling in ocular ischemic syndrome. Graefes Arch Clin Exp Ophthalmol. 2016; doi: 10.1007/s00417‐016‐3456‐1. [DOI] [PubMed] [Google Scholar]
- 83. Huth TK, Staines D, Marshall‐Gradisnik S. ERK1/2, MEK1/2 and p38 downstream signalling molecules impaired in CD56 dim CD16 + and CD56 bright CD16 dim/‐ natural killer cells in chronic fatigue syndrome/myalgic encephalomyelitis patients. J Transl Med. 2016; doi: 10.1186/s12967‐016‐0859‐z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Wang S, Zhang C, Sheng X, et al Peripheral expression of MAPK pathways in Alzheimer's and Parkinson's diseases. J Clin Neurosci. 2014; 21: 810–4. [DOI] [PubMed] [Google Scholar]
- 85. Miyashita T, Krajewski S, Krajewska M, et al Tumor suppressor p53 is a regulator of bcl‐2 and bax gene expression in vitro and in vivo . Oncogene. 1994; 9: 1799–805. [PubMed] [Google Scholar]
- 86. Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell. 1995; 80: 293–9. [DOI] [PubMed] [Google Scholar]
- 87. Geering K. Na, K‐ATPase. Curr Opin Nephrol Hypertens. 1997; 6: 434–9. [DOI] [PubMed] [Google Scholar]
- 88. Therien AG, Blostein R. Mechanisms of sodium pump regulation. Am J Physiol Cell Physiol. 2000; 279: C541–66. [DOI] [PubMed] [Google Scholar]
- 89. Desfrere L, Karlsson M, Hiyoshi H, et al Na, K‐ATPase signal transduction triggers CREB activation and dendritic growth. Proc Natl Acad Sci USA. 2009; 106: 2212–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Haas M, Wang H, Tian J, et al Src‐mediated inter‐receptor cross‐talk between the Na+/K + ‐ATPase and the epidermal growth factor receptor relays the signal from ouabain to mitogen‐activated protein kinases. J Biol Chem. 2002; 277: 18694–702. [DOI] [PubMed] [Google Scholar]
- 91. Tian J, Cai T, Yuan Z, et al Binding of Src to Na+/K + ‐ATPase forms a functional signaling complex. Mol Biol Cell. 2006; 17: 317–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Banerjee M, Duan Q, Xie Z. SH2 ligand‐like effects of second cytosolic domain of Na/K‐ATPase alpha1 subunit on Src kinase. PLoS One. 2015; 10: e0142119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Gable ME, Abdallah SL, Najjar SM, et al Digitalis‐induced cell signaling by the sodium pump: on the relation of Src to Na(+)/K(+)‐ATPase. Biochem Biophys Res Commun. 2014; 446: 1151–4. [DOI] [PubMed] [Google Scholar]
- 94. Weigand KM, Swarts HG, Fedosova NU, et al Na, K‐ATPase activity modulates Src activation: a role for ATP/ADP ratio. Biochim Biophys Acta. 2012; 1818: 1269–73. [DOI] [PubMed] [Google Scholar]
- 95. Ye Q, Li Z, Tian J, et al Identification of a potential receptor that couples ion transport to protein kinase activity. J Biol Chem. 2011; 286: 6225–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Yosef E, Katz A, Peleg Y, et al Do Src kinase and caveolin interact directly with Na,K‐ATPase? J Biol Chem. 2016; 291: 11736–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Purity of Myc‐tagged RS1 proteins.
Table S1 Primers used in RNA analyses and expression cloning.