

Abstract
Thioalkalivibrio is a genus of obligate chemolithoautotrophic haloalkaliphilic sulfur-oxidizing bacteria. Their habitat are soda lakes which are dual extreme environments with a pH range from 9.5 to 11 and salt concentrations up to saturation. More than 100 strains of this genus have been isolated from various soda lakes all over the world, but only ten species have been effectively described yet. Therefore, the assignment of the remaining strains to either existing or novel species is important and will further elucidate their genomic diversity as well as give a better general understanding of this genus. Recently, the genomes of 76 Thioalkalivibrio strains were sequenced. On these, we applied different methods including (i) 16S rRNA gene sequence analysis, (ii) Multilocus Sequence Analysis (MLSA) based on eight housekeeping genes, (iii) Average Nucleotide Identity based on BLAST (ANIb) and MUMmer (ANIm), (iv) Tetranucleotide frequency correlation coefficients (TETRA), (v) digital DNA:DNA hybridization (dDDH) as well as (vi) nucleotide- and amino acid-based Genome BLAST Distance Phylogeny (GBDP) analyses. We detected a high genomic diversity by revealing 15 new “genomic” species and 16 new “genomic” subspecies in addition to the ten already described species. Phylogenetic and phylogenomic analyses showed that the genus is not monophyletic, because four strains were clearly separated from the other Thioalkalivibrio by type strains from other genera. Therefore, it is recommended to classify the latter group as a novel genus. The biogeographic distribution of Thioalkalivibrio suggested that the different “genomic” species can be classified as candidate disjunct or candidate endemic species. This study is a detailed genome-based classification and identification of members within the genus Thioalkalivibrio. However, future phenotypical and chemotaxonomical studies will be needed for a full species description of this genus.
Introduction
Members of the genus Thioalkalivibrio are sulfur-oxidizing bacteria that thrive under the dual extreme conditions of soda lakes [1,2]. These lakes are characterized by extremely high sodium carbonate concentrations, creating buffered haloalkaline conditions with a pH of around 10 [3,4]. Despite these extreme conditions, the primary production [5–7] and the microbial diversity [8–11] in these soda lakes is high, and they also contain microbial communities that are actively involved in the cycling of the chemical elements, such as carbon, nitrogen and sulfur [12,13]. Until now, ten species have been validly described within the genus Thioalkalivibrio [14–20] and more than 100 strains have been isolated and assigned to this genus [20,21]. The genus Thioalkalivibrio is grouped within the gammaproteobacterial family Ectothiorhodospiraceae [14]. In addition to their haloalkaliphilic and chemolithoautotrophic nature, the members of this genus are also characterized by a versatile energy metabolism as they are able to use different electron donors and acceptors. All strains can use reduced sulfur compounds, such as sulfide, polysulfide, thiosulfate, polythionates and elemental sulfur as an energy source [14–20]. In addition, the type strains Tv. paradoxus ARh1T [15], Tv. thiocyanoxidans ARh2T [15] and Tv. thiocyanodenitrificans ARhD1T [19] are able to use thiocyanate as their energy, sulfur and nitrogen source [22]. Other type strains, such as Tv. denitrificans ALJDT [23], Tv. nitratireducens ALEN2T [17] and Tv. thiocyanodenitrificans ARhD1T [19] can perform sulfur-dependent denitrification under anaerobic conditions. Moreover, some of the strains can grow over a broad range of salt concentrations (from 0.2 to 5 M Na+), and others can even grow with 3.6 M K+ [14–20].
By definition, a bacterial species is described as a collection of strains whose DNA:DNA hybridization (DDH) percentage is at least 70% and whose DNA melting temperature (Tm) lies within 5°C [24]. Apart from these characteristics, a taxonomic species should also reflect a phenotypic coherence [24]. At a higher taxonomic level, a genus is characterized by uniting the assigned strains in a monophyletic branch of a phylogenetic tree, such as 16S rRNA gene sequence analysis or Multilocus Sequence Analysis (MLSA) [25]. In the “All-Species Living Tree Project”, numerous bacterial genera were revealed to be paraphyletic or polyphyletic, which shows that by far not all bacteria are correctly classified at their genus level [26,27]. Whether or not taxa, and in particular genera, are classified in a coherent way, should be assessed, for instance, using modern, genome-based tools as recently shown for the phylum Bacteroidetes [28].
Nowadays, in the genomic era, in silico-based methods are becoming more and more common [29]. All new genome sequence-based approaches for species delineation have to be however evaluated according to their correspondence to the traditional DDH [30], which ensures consistency in prokaryotic species delineation across hitherto and novel methods. The Average Nucleotide Identity (ANI) was proposed as an in silico replacement for the traditional DDH, because it was shown to correlate well with it [31,32] by delineating species from each other using a threshold value of 94–96% [32]. In addition to the ANI calculation, the program JSpecies [32] also provides the tetranucleotide signature correlation index (TETRA) which is a non-alignment based parameter. Another replacement method, the Genome-to-Genome Distance Calculator (GGDC) [33], infers digital DDH (dDDH) estimates from intergenomic distances [33,34] and was shown to provide the highest correlation [33] to conventional DDH without mimicking its pitfalls [35] The dDDH values are predicted on the established DDH scale, along with confidence intervals (CI) that allow conservative taxonomic decisions [33,34] as well as the delineation of bacterial subspecies [36]. The latest GGDC version 2.1 is based on the optimized Genome BLAST Distance Phylogeny (GBDP) method which was originally devised for the inference of highly resolved whole-genome phylogenetic trees using either nucleotide or amino acid data and including branch support [37]. A routine method for the taxonomic classification of bacteria is the analysis of the 16S rRNA gene sequences [30,38] which is however known to have only limited to even no discriminatory power in many bacterial groups [39]. The MLSA approach, which is based on ubiquitous and single-copy housekeeping genes whose proteins have essential and conserved functions, has also been shown to yield highly resolved phylogenetic trees [40, 41]. However, the exclusive application of single-phased and genome-based approaches does still not replace a full and effective taxonomic species description which includes phenotypical, genotypical and chemotaxonomic analysis [42, 43].
Here we describe the genome-based taxonomic classification and identification of strains within the genus Thioalkalivibrio in order to assess its genomic diversity. We applied six different approaches on a dataset of 76 Thioalkalivibrio genome sequences, such as (i) 16S rRNA gene sequence analysis, (ii) MLSA on eight housekeeping genes (atpD, clpA, dnaJ, gyrB, rpoD, rpoH, rpoS and secF), (iii) ANI based on BLAST (ANIb) and MUMmer (ANIm), (iv) tetranucleotide frequency correlation coefficients (TETRA), (v) dDDH and (vi) nucleotide- and amino acid-based GBDP analyses. We revealed 15 new “genomic” species next to the ten already described species, as well as 16 new “genomic” subspecies. We use the term “genomic” species here as the definition of a group of strains which clustered into the same species based on ANIb, ANIm, TETRA and dDDH analysis. Furthermore, phylogenetic and -genomic analyses showed that the genus is not monophyletic. Finally, species within the genus Thioalkalivibrio revealed to have either a candidate disjunct or a candidate endemic biogeographical distribution. This means that they are suggested as a genomic species that harbors strains which are geographically widely separated from each other or that they are only found in a specific area, respectively [44].
Materials and methods
Genomes and gene sequences
Sequences of Thioalkalivibrio
We analyzed the genomic diversity of 76 Thioalkalivibrio strains including ten described type strains (S1 Table). The genome sequences of 73 strains were sequenced and annotated within the Community Science Program of the DOE Joint Genome Institute. In addition to these, we sequenced the genomes of Tv. versutus AL2T, Tv. denitrificans ALJDT and Tv. halophilus HL17T in order to include all described type strains of Thioalkalivibrio in this study.
To obtain these three additional genome sequences, DNA extraction was performed on pure cultures using the PowerSoil DNA Isolation Kit (MoBio Laboratories Inc. (Carlsbad, USA)) following the standard conditions given by the supplier. Paired-end sequencing using Illumina HiSeq 1000 (Illumina; BaseClear B.V. (Leiden, The Netherlands)) was applied. The library was previously prepared by Illumina genomic Nextera XT library. The Illumina reads size was 50 bp and the yield of all three samples was higher than 600 Mb. Quality trimming and genome assembly was done with the CLC Genomics Workbench de novo assembler (version 6.0, CLC bio, Aarhus, Denmark) using default settings. The genome sequences were annotated using the Integrated Microbial Genomes Expert Review (IMG-ER) pipeline [45] and deposited in the IMG database under the project ID’s of 62364 (AL2T), 62363 (ALJDT) and 62362 (HL17T) as well as in the NCBI database under the accession of MVAR00000000 (AL2T), MVBK00000000 (ALJDT) and MUZR00000000 (HL17T).
The genome and gene (clpA, atpD, gyrB, rpoH, secF, dnaJ, rpoD and rpoS) sequences of Thioalkalivibrio sp. K90mix and Tv. sulfidiphilus HL-EbGr7T were obtained from the NCBI RefSeq database and the 16S rRNA gene sequences of the Thioalkalivibrio strains AKL11, AL2T, ALEN2T, ALJ12T, ALJ17, ALJ24, ALJDT, ALM2T, ALSr1, ARhD1T, ARh1T, ARh2T, ARh4, HL17T, HL-EbGr7T and K90mix were extracted from the SILVA database [46]. The other Thioalkalivibrio genome and gene (clpA, atpD, gyrB, rpoH, secF, dnaJ, rpoD, rpoS and 16S rRNA) sequences were taken from JGI IMG database [45].
Sequences of related species
To study the monophyly of Thioalkalivibrio in the phylogenetic and -genomic trees, we selected the closely related Thiorhodospira sibirica A12T (photoautotrophic purple sulfur bacterium), Ectothiorhodospira haloalkaliphila ATCC 51935T (photoautotrophic purple sulfur bacterium), Halorhodospira halophila SL1T (purple sulfur bacterium), Alkalilimnicola ehrlichii MLHE-1T (facultatively autotrophic sulfide-oxidizer) and Thiohalospira halophila HL3T (extremely halophilic lithoautotrophic sulfur-oxidizer) (S2 Table).
Their 16S rRNA gene sequences were obtained from the SILVA database and the gene sequences for SL1T (with exception of rpoH) and MLHE-1T (with exception of dnaJ) came from the NCBI RefSeq database. The genome and the gene sequences (clpA, atpD, gyrB, rpoH, secF, dnaJ, rpoD and rpoS) of A12T, ATCC 51935T and HL3T as well as rpoH of SL1T and dnaJ of MLHE-1T were acquired from the JGI IMG database.
16S rRNA gene sequence analysis
Alignment of 16S rRNA gene sequences of the 76 Thioalkalivibrio strains and the members of the five related genera was done by the online SINA alignment service [47]. Subsequently, the aligned sequences were imported into ARB [48] by which an identity matrix was calculated. The tree was built in the software program MEGA (version 6.06; [49]) by manually trimming the aligned sequences, and by using the maximum likelihood algorithm as tree inference with 1000 bootstrap replicates, the Tamura-Nei substitution model and gamma distributed with invariant sites (+G+I) as rates among sites. The phylogenetic tree was rooted using A. ehrlichii MLHE-1T and H. halophila SL1T. In order to calculate the pairwise and overall mean genetic distances with the Kimura 2-parameter model as well as the number of polymorphic sites, the 16S rRNA gene sequences of Thioalkalivibrio were aligned with aligner option MUSCLE [50] within MEGA and the ends were trimmed manually to obtain the same length for all sequences.
Multilocus sequence analysis
The sequences of the individual housekeeping genes of the 76 Thioalkalivibrio strains as well as those of the five strains from other genera were aligned with the software program MUSCLE [50] within MEGA (version 6.06; [49]) and trimmed manually. Subsequently, the alignments of the eight genes were concatenated in the following order: clpA, atpD, gyrB, rpoH, secF, dnaJ, rpoD and rpoS. Phylogenetic trees of individual genes and of the concatenated sequences were calculated in MEGA using the same parameters and the same rooting as for the 16S rRNA gene sequence analysis. The identity matrix of the concatenated housekeeping genes was calculated in MEGA using a pairwise distance matrix made with the “number of difference” model in which also gaps are included as differences. Both, pairwise and overall mean genetic distance as well as the number of polymorphic sites were calculated in analogy to the 16S rRNA gene sequence analysis.
Average nucleotide identity and TETRA
ANIb, ANIm and TETRA values were calculated based on the 76 Thioalkalivibrio genome sequences via the JSpeciesWS online service using the default parameters [51].
The resulting matrices obtained for ANIb and ANIm were converted into dendrograms by the DendroUPGMA webservice ([52]; http://genomes.urv.cat/UPGMA/index.php) using an average-linkage clustering [53]. The dendrograms were drawn with the software program Dendroscope 3 [54].
Whole-genome sequence-based phylogenomic analysis
For all pairwise combinations among the genome sequences of Thioalkalivibrio (76) and the members of the other genera (5), intergenomic distances were calculated using the latest version of the GBDP approach [33,55], the software on which the Genome-to-Genome Distance Calculator web service is based (GGDC 2.1; freely available at http://ggdc.dsmz.de) [33]. The inference of pairwise distances included the calculation of 100 replicate distances, each to assess pseudo-bootstrap support [37]. All distance calculations were conducted under the settings recommended for the comparison of nucleotide data [33]. The GBDP trimming algorithm and the formula d5 were chosen because of their benefits regarding phylogenetic reconstruction [37]. Finally, to evaluate potentially less resolved groupings in the nucleotide-based tree, a second GBDP analysis was conducted based on the more conserved amino acid data and under recommended settings [37], i.e., also using the trimming algorithm and formula d5. Afterwards, both phylogenomic trees were inferred from intergenomic GBDP distance matrices using FastME v2.07 with enabled tree bisection and reconnection (TBR) postprocessing [56] (“initial building method”: balanced; “branch lengths assigned to the topology”: balanced; “type of tree swapping (NNI)”: none) and rooted with A. ehrlichii MLHE-1T and H. halophila SL1T.
Digital DDH
Using the GGDC 2.1 web service, intergenomic distances were calculated using GBDP [33, 55], followed by the prediction of dDDH values and their CI, for all pairwise comparisons between the genome sequences of the 76 Thioalkalivibrio and the 5 type strains of other genera [33].
Obtaining novel species and subspecies
Since the affiliation of all 76 strains to known type strains is the only relevant taxonomic criterion to assess the actual number of novel species, a previously introduced type-based clustering approach was used to assess the affiliation of strains to known species [57]. The reasoning is that strains within a, for instance, 70% dDDH radius around a known type strain can be safely attributed to the underlying known species or be considered as a novel species else.
In a first step, the different species delineation thresholds were taken from literature and applied to the corresponding dataset in order to identify the strains belonging to a described type species. Therefore, a 70% dDDH radius (including 67% and 73% dDDH that represent its lower and upper CI boundaries) was used for the dDDH dataset, whereas a 94%, 95% and 96% radius for the ANIb and ANIm datasets was used. The TETRA dataset was analysed in the same manner under the published 0.989% and 0.999% thresholds. Since clustering programs frequently require distance data the ANIb, ANIm and TETRA similarity matrices were trivially converted to distances (i.e., subtracting the value from 100% and subsequently dividing it by 100). However, the GGDC's intergenomic distances (on which the dDDH is based) could be directly used as input.
In a second step, the strains that were not found to be affiliated to known species (i.e., representing putative novel species) were de novo-clustered under the aforementioned thresholds for species delineation. Here, the clustering optimization program OPTSIL was applied in version 1.5 [58] on the dDDH, ANIb, ANIm and TETRA matrices to identify these novel species clusters. The OPTSIL program is a tool for the optimization of threshold-based linkage clustering runs [59]. It is primarily driven by two parameters: T and F. Strains are considered to be “linked” if the pairwise distance is smaller or equal than the chosen threshold T. The F parameter defines the fraction of links required among a set of strains before merging them into the same cluster. For example, one can either request that it is already sufficient if at least one distance to a cluster member is a link (single linkage; F = 0.0) or that all distances are links (complete linkage; F = 1.0) [58]. Here, all OPTSIL clustering runs were done with a linkage fraction value F set to 0.5, as previously recommended [36].
In a last step, each strain within each putative novel species cluster was consecutively treated as a new putative type strain and the previously described type-based clustering (step 1) was repeated, respectively. In case two or more newly assigned type strains fell into the same species radius, these were counted as “ambiguities”.
Regarding GGDC's capability to delineate microbial subspecies, a respective distance cutoff of 79% dDDH as described in [36] was used.
Results
16S rRNA gene sequence analysis and MLSA
Phylogenetic trees based on 16S rRNA gene sequences (Fig 1A) and MLSA with eight housekeeping genes (atpD, clpA, dnaJ, gyrB, rpoD, rpoH, rpoS and secF) (Fig 1B) were constructed for the Thioalkalivibrio strains and their close relatives to assess the monophyletic status of the genus.
Fig 1.

Phylogenetic tree constructed from 16S rRNA gene sequence analysis (A) and from MLSA (B). Bootstrap values over 60% were shown at each node. The orange box indicates the outlying Thioalkalivibrio strains, contesting the monophyly of the genus.
16S rRNA gene sequence analysis (Fig 1A) and MLSA (Fig 1B) trees showed a separation between the large group of strains around the type species Tv. versutus AL2T (including the type strains ALM2T, ALJ12T, ARh2T, HL17T, ALEN2T and ARh1T) and four other Thioalkalivibrio strains (ALJDT, ARhD1T, HL-EbGr7T and ALJ17). This separation was however not well supported in the 16S rRNA tree (bootstrap value of 52%). Two bacteria of different genera, Trs. sibirica and E. haloalkaliphila, were situated between the separated groups of the Thioalkalivibrio genus (Fig 1).
The alignment of the 16S rRNA gene sequences of the Thioalkalivibrio strains has a genetic distance ranging from 0 to 0.0824 (mean 0.0216) which corresponds to a sequence identity from 100 to 92.95% as calculated in ARB (Table 1). These identity results show that the 16S rRNA gene sequence conservation among the different strains of this genus is moderate to high. Especially strains which are closely related, and also some which are classified as different species, possess a relatively high 16S rRNA gene sequence identity value. Furthermore, some nodes in the phylogenetic tree have bootstrap values of less than 60% (Fig 1A).
Table 1. Characteristics of 16S rRNA, single housekeeping and concatenated housekeeping genes (MLSA).
| Gene | Length (bp) | % of polymorphic sites | Average G+C content (%) | Minimum genetic distance | Maximum genetic distance | Mean genetic distance |
|---|---|---|---|---|---|---|
| 16S rRNA | 1360 | 17.94 | 55.90 | 0 | 0.0824 | 0.0216 |
| atpD | 1380 | 34.71 | 64.32 | 0 | 0.1882 | 0.0895 |
| clpA | 2299 | 47.72 | 64.55 | 0 | 0.3221 | 0.1362 |
| dnaJ | 1162 | 51.64 | 66.92 | 0 | 0.3539 | 0.1521 |
| gyrB | 2457 | 54.50 | 63.11 | 0 | 0.4145 | 0.2000 |
| rpoD | 1960 | 46.99 | 64.24 | 0 | 0.2807 | 0.0934 |
| rpoH | 897 | 57.53 | 66.19 | 0 | 0.3988 | 0.1834 |
| rpoS | 1156 | 63.75 | 64.88 | 0 | 0.4402 | 0.1995 |
| seqF | 972 | 55.14 | 62.16 | 0 | 0.4124 | 0.1987 |
| MLSA | 12,283 | 50.68 | 64.37 | 0 | 0.3179 | 0.1504 |
The genetic distance of the MLSA alignment was calculated and ranged from 0 to 0.3179 (mean 0.1504) (Table 1) which corresponds to an MLSA sequence identity from 100 to 75.63% (S4 Table).
The individual single gene trees (S1 File) show only minor differences between each other as well as compared to the MLSA tree (Fig 1B). However, more divergences were found between the MLSA (Fig 1B) and the 16S rRNA gene tree (Fig 1A). On average, MLSA is better resolved and presents longer branches. In the 16S rRNA analysis, the type strain Tv jannaschii ALM2T was located on the same branch as the Tv. versutus AL2T (unsupported though), whereas these type strains were separated on two branches in the MLSA.
ANIb, ANIm, TETRA, dDDH and GBDP analyses
ANIb, ANIm, TETRA and dDDH are based on the complete genomic information, enabling the delineation of species among closely-related strains [32,33,35,51]. The ANIb dendrogram is shown in Fig 2. Since dDDH is based on intergenomic GBDP distances, these were used to infer a phylogenomic tree (Fig 3) [37].
Fig 2. Dendrogram constructed from the ANIb analysis.
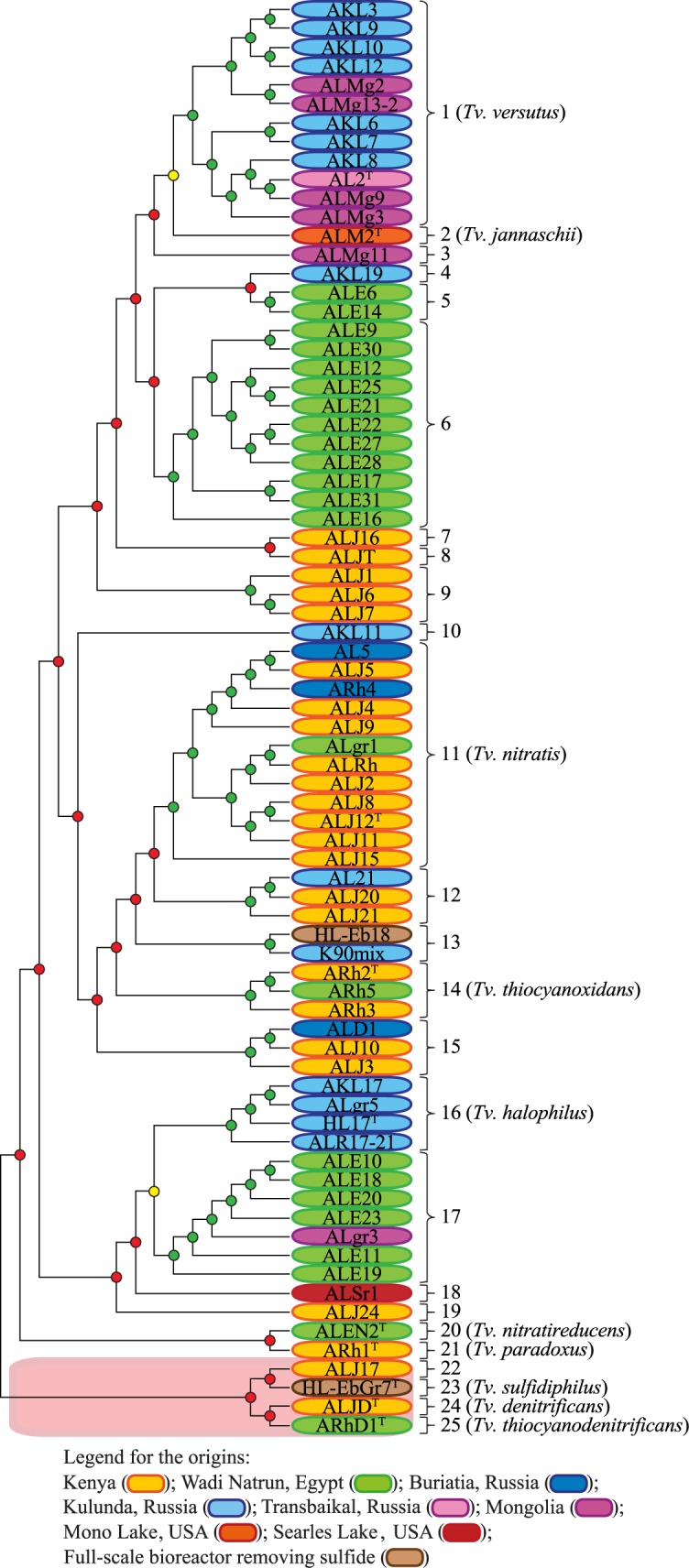
De novo species clusters obtained without consideration of type strains. Clusters are indicated by dots (green: ANI > 96% (strains belong to the same genomic species); yellow: 94% < ANI < 96% (strains might belong to the same genomic species); red: ANI < 94% (strains do not belong to the same genomic species). The genomic species groups are marked by numbers. The orange box indicates the outlying Thioalkalivibrio strains, contesting the monophyly of the genus.
Fig 3. Whole-genome GBDP phylogeny (based on the nucleotide data).

Bootstrap values over 60% are shown at each node. An assignment to genomic species was based on the distance threshold equivalent to 70% dDDH (dDDH ≥ 70% indicates same genomic species) and dDDH < 70% (indicates distinct genomic species). Genomic species groups are marked by numbers whereas genomic subspecies groups are denoted by letters. The orange box indicates the outlying Thioalkalivibrio strains, contesting the monophyly of the genus.
The pairwise similarity/distance values for all different measures were calculated and are listed in S5 Table (ANIb, ANIm, TETRA) and S6 Table (dDDH). The described clustering procedure was applied on all datasets and the resulting clusters are found in S7 Table.
The results of the dDDH dataset (S7 Table) revealed in total 25 non-conflicting (i.e. no ambiguities) genomic species groups under the 70% species delineation threshold, each containing between one and twelve strains per group. From these 25 genomic species groups, 15 new genomic species were identified supplementary to the ten already described species in Thioalkalivibrio. The same non-conflicting clusters were also found using the lower CI boundary (67% dDDH). However, the strains AKL3, AKL9 and AKL12 clustered into a group of their own, separated from the other Tv. versutus strains, under the upper CI boundary (73% dDDH).
Under the 94% delineation threshold, the ANIb dataset (S7 Table) yielded 24 strains that were assigned to multiple type strains (i.e. genomic species groups) at the same time (AL2T/ALM2T and HL17T/ALE10PT) (PT—putative new type strain; chosen to represent its underlying species cluster), whereas, under the 95% threshold delineation threshold, only four of these conflicts were found (AL2T/ALM2T). At the 96% delineation threshold, the ANIb cluster assignments matched the ones found for the dDDH dataset at the 70% threshold.
The ANIm clustering (S7 Table) revealed 42 strains that fell into multiple species groups under the 94% delineation threshold (AL2T/ALM2T, ALJ12T/HL-Eb18PT/AL21PT, ALE10PT/HL17T, ALJ17PT/HL-EbGr7T and ALJ12T/AL21PT), whereas, under the 95% threshold delineation threshold, still 15 strains were ambiguously assigned to multiple genomic species groups (AL2T/ALM2T and HL17T/ALE10PT). At the 96% delineation threshold, the ANIm clustering matched those of the dDDH dataset at the 70% threshold.
TETRA (S7 Table) showed under the 0.989 delineation threshold that almost all strains were ambiguously assigned to multiple genomic species groups at the same time, whereas only 15 strains were affected in that way under the 0.999 delineation threshold (AL2T/ALM2T/ALMg11PT, HL-Eb18PT/ALJ12T and ALE10PT/HL17T).
According to the OPTSIL-based subspecies delineation, using the established dDDH threshold [34], four distinct genomic subspecies were found within the groups 1 (Tv. versutus) and 17, and two subspecies were identified within the groups 6, 9, 13 and 16 (Fig 3). Trivial subspecies (i.e., a single strain in a given species cluster) were not counted.
Except for the genomic species groups 12 and 15, the nucleotide-based phylogenomic tree (Fig 3) demonstrated that all described type strains could be separated from each other as different genomic species by well supported branches. As expected, on the amino acid-level, the respective phylogenomic tree (Fig 4) revealed even more branch support, including maximum support for the genomic species groups 12 and 15.
Fig 4. Whole-genome GDBP phylogeny (based on the amino acid data).

Bootstrap values over 60% were shown at each node. The orange box indicates the outlying Thioalkalivibrio strains, contesting the monophyly of the genus.
Both, the nucleotide- (Fig 3) and the amino acid-based GBDP trees (Fig 4), were inferred to assess the potential monophyly of the genus Thioalkalivibrio, which, in fact, turned out to be paraphyletic. In the nucleotide-based tree, in addition to the strains ARhD1T, ALJDT, HL-EbGr7T and ALJ17, the strains ARh1T and ALEN2T were also separated from the other Thioalkalivibrio by Trs. sibirica and Ths. halophila. However, neither the relevant subtree of the four strains (ARhD1T, ALJDT, HL-EbGr7T and ALJ17) nor of ARh1T and ALEN2T was sufficiently supported by this analysis. In the amino acid-based tree, the strains ARhD1T, ALJDT, HL-EbGr7T and ALJ17 were only separated from the other Thioalkalivibrio by Trs. sibirica and E, haloalkaliphila, and all relevant nodes yielded high bootstrap values throughout. On average, the nucleotide-based GBDP tree (Fig 3) yielded a bootstrap value of 53.7%, whereas the amino acid-based tree (Fig 4) was generally better resolved with an average support of 81.5%, as expected [37].
Discussion
Species classification and identification in Thioalkalivibrio
The 76 Thioalkalivibrio strains could not be uniformly classified into different sets of species groups by ANIb, ANIm, TETRA and dDDH. In the dDDH dataset, all strains were non-ambiguously assigned either to one of the known species or they represented new ones (Fig 3 and S7 Table). The clustering based on ANIb and ANIm revealed conflicts at the 94% and 95% thresholds, however gave the same non-ambiguous genomic species clusters at the 96% threshold as the dDDH at 70% (Fig 2, S1 Fig and S7 Table). The TETRA results showed a high number of conflicts under the 0.989 threshold and a few with 0.999 threshold. A possible reason for the non-conflicting results of dDDH might be due to its better correlations to conventional DDH [33], the main optimality criterion for all such in silico methods. Even though, clustering inconsistencies of ANIb data were previously observed [60], performance parameters, such as cluster consistency, isolation and cohesion indices [34,36], would need to be investigated for a large, representative dataset of bacteria and archaea, as successfully done earlier for dDDH data [34]. Consequently, it seems to be premature to infer any conclusions regarding the (un-)reliability of the other methods, just based on this study.
Among the 25 genomic species clusters, ten were within the radius of an existing type strain and could thus be successfully linked to a described species. Consequently, the 15 remaining groups did not contain a described type strain and therefore, novel species are proposed to be effectively described within the genus Thioalkalivibrio in accordance with the taxonomic rules. These genomic species need to be evaluated by a polyphasic approach in which they need to have a sufficient level of phenotypic and physiological differences with already described species [24,42,43]. The aforementioned clustering conflicts should be carefully investigated in the course of these effective species descriptions, because they might reflect a phenotypic coherence [24].
Furthermore, multiple subspecies groups were found within the genomic species groups 1 (Tv. versutus), 6, 9, 13, 16 and 17 (Fig 3) using the GBDP nucleotide-based analysis [36]. Even though an assignment to subspecies is usually only done for medically relevant strains, we used this approach to gain a better understanding about the diversity within the genus Thioalkalivibrio.
A high genomic diversity is reflected in Thioalkalivibrio through the large number of discovered genomic species and subspecies affiliated to Thioalkalivibrio. Branching patterns of rep-PCR profiles of Thioalkalivibrio strains might indicate that the diversity in Thioalkalivibrio originates from recombination [61]. It is already known that recombination plays an important role in the evolution and diversification of bacterial species [62–64], even more so than mutations [65,66]. Multiple transposases have already been found in the genome of Thioalkalivibrio sp. K90mix [67] and pathogenicity islands as well as prophages in Tv. versutus D301 [68]. Further studies will aid in the clarification of the nature and proportions of the evolutional forces responsible for the diversification within the genus Thioalkalivibrio.
In this study, we found that various Thioalkalivibrio strains have previously been misidentified (S8 Table) [14,20]. Furthermore, the previous studies [69,70,71] consider the strain ALJ15 to represent Tv. versutus, which we identified as a member of the species Tv. nitratis.
16S rRNA gene sequence analysis yielded high identity values among closely related strains and species, and the phylogeny was not well supported. For this reason, this analysis can only distinguish between different Thioalkalivibrio species at a low resolution, which was previously observed for other bacteria [72,39], such as Hyphomonas [73], Thalassospira [74], Acinetobacter [75], Nocardia [76] and Bifidobacterium [77]. Therefore, species affiliation cannot be based on 16S rRNA gene sequence analysis alone due to the fact that different taxa might have different diversification rates of their 16S rRNA gene sequences [78]. Additionally, incorrect assignments can be made using only a single housekeeping gene such as the 16S rRNA gene sequence, because horizontal gene transfer might even occur (though unlikely) for the 16S rRNA gene sequence [79–81]. Indeed, different studies demonstrated that a higher taxonomic resolution and consistency in accepted classification is achieved using a set of at least five housekeeping genes in MLSA [29,36,82,83] or in supertree analysis with single-copy orthologous core genes [75]. It was even demonstrated that the taxonomy of whole phyla can be extensively and reliably revised based on the principles of phylogenetic classification and trees inferred from genome-scale data [28]. In this study, the GBDP (Figs 3 and 4) and MLSA (Fig 1B) showed on average a better resolution, higher bootstrap values and more clusters than the 16S rRNA gene sequence analysis (Fig 1A), supporting the expected higher distinguishing power of these methods.
Comparing the identity results of the MLSA to those of the ANIb and the values of the dDDH, a threshold value for the genomic species delimitation based on the sequence identity given by MLSA could be proposed (S4 Table). With the set of strains and gene sequences used in this study, it was found that strains with a sequence identity higher than 98.13% belong to the same genomic species, whereas identity values below 97.77% indicated that they were not associated to the same genomic species. In between these two values, a grey area exists. However, these values might change if new strains are added in the future to the current set of strains. With this knowledge, we propose that MLSA can be used as a fast and preliminary assessment of the species relatedness for new isolates in Thioalkalivibrio. This method has the advantage that the whole genome sequence is not needed (at this point) and it provides more phylogenetic resolution at species level than the 16S rRNA gene sequence analysis for Thioalkalivibrio. However, the 16S rRNA gene sequence still has the advantage of having a large database linked to it. If genome sequences are available, respective whole-genome sequence-based approaches should be preferred and chosen regarding their clustering performance assessed in this comprehensive study.
Thioalkalivibrio’s phyletic structure at genus level
The genus Thioalkalivibrio is not monophyletic according to the phylogenetic and phylogenomic analyses (Figs 1, 3 and 4), because type strains from other genera disconnect a group of strains including Tv. sulfidiphilus HL-EbGr7T, ALJ17, Tv. denitrificans ALJDT and Tv. thiocyanodenitrificans ARhD1T from the major group of Thioalkalivibrio that includes their type species Tv. versutus. The amino acid-based GBDP analysis supported the MLSA in this respect and, furthermore, yielded higher bootstrap values for all relevant nodes. This is explained by the more conserved nature of the amino acid sequences as well as that GBDP is bootstrapping entire genes [37] which was previously suggested to reduce conflicts and to provide more realistic support values in phylogenomic analyses [28,84]. The 16S rRNA gene sequence showed the same separation as found in the MLSA and the nucleotide-based GBDP, but this node achieved only low branch support. The nucleotide-based GBDP analysis showed that in addition to the strains which were separated in the MLSA and amino acid-based GBDP (ARhD1T, ALJDT, HL-EbGr7T and ALJ17), the strains ARh1T and ALEN2T were also separated from the other Thioalkalivibrio. However, neither the relevant subtree of the four strains (ARhD1T, ALJDT, HL-EbGr7T and ALJ17) nor of ARh1T and ALEN2T was sufficiently supported in this analysis.
In the 16S rRNA gene sequence analysis, the MLSA and the amino acid-based GBDP, the genus Thioalkalivibrio is split into two groups by Trs. sibirica and E. haloalkaliphila. However, in the nucleotide-based GBDP, Ths. halophila is found instead of E. haloalkaliphila in between the two Thioalkalivibrio groups. The bacteria Trs. sibirica and E. haloalkaliphila are both anaerobic and haloalkaliphilic purple sulfur bacteria isolated from soda lakes [85,86]. However, due to the fact that Trs. sibirica and E. haloalkaliphila have a different energy metabolism [85,86], they do not adhere to the description of the Thioalkalivibrio genus, which is obligatory chemotrophic [87]. Ths. halophila is a chemolithoautotrophic and haloneutrophilic sulfur oxidizing bacterium which originates from hypersaline inland lakes. Furthermore, the Thiohalospira genus also contains the facultatively alkaliphilic species Ths. alkaliphila [88]. Physiologically, the four separated Thioalkalivibrio strains are closer to the Thiohalospira genus with the exception of their alkaliphilic nature [14,19,20,88].
A taxonomic genus must be monophyletic by definition [25,89]. In the case of a monophyletic group, all members share a common ancestor and therefore, it is possible to detach the group from the tree with a single cut [90]. For this reason, the four strains (HL-EbGr7T, ALJ17, ALJDT, ARhD1T) of Thioalkalivibrio which are separated from the major group of Thioalkalivibrio that contain the type strain Tv. versutus AL2T, cannot remain within the same genus and need to be reclassified into a new genus. However, no fixed and commonly accepted boundary for genus delineation exists, which could be used to clarify the genus boundary in Thioalkalivibrio. This is a known circumstance in microbial taxonomy which is primarily due to the missing ultrametricity [34] in such biological data, especially regarding ranks above species level. In the “All-Species Living Tree Project”, a minimal identity value of the 16S rRNA gene sequence for the separation of two genera was proposed at 94.8% ± 0.25 [91]. Applying this value to the 16S rRNA gene sequence analysis of Thioalkalivibrio (S3 Table), the splitting of the two groups in the phylogenetic tree was confirmed (92.95–94.92%; mean = 93.82%) (S3 Table). Furthermore, the identity values between the four outliers (HL-EbGr7T, ALJ17, ALJDT, ARhD1T) and Ths. alkaliphila are also below this value (91.86–92.22%) (S3 Table). Other findings from the “All-Species Living Tree Project” demonstrate that several genera as Eubacterium, Bacillus, Pseudomonas, Desulfotomaculum [26], Enterococcus, Rhizobium, Clostridium and Lactobacillus [27] are paraphyletic or polyphyletic. These examples indeed visualize that misclassifications are not an uncommon problem, especially when species descriptions were ultimately based on unresolved, hence uninterpretable, 16S rRNA gene sequence trees.
On the basis of their phenotypic characteristics, the outliers also showed differences to the core group of Thioalkalivibrio. The ability of growing at higher salinity ranges of up to 5 M of Na+ is linked to many genomic species in the core group containing the type species, Tv. versutus, whereas the type strains Tv. nitratireducens ALEN2T, Tv. paradoxus ARh1T, Tv. sulfidiphilus HL-EbGr7T, Tv. denitrificans ALJDT and Tv. thiocyanodenitrificans ARhD1T which are genetically further away from their type species, do not have an adaptation to high salt concentrations [14–20].
Biogeography
Given the currently available Thioalkalivibrio sequences, we were able to infer a relation between the geographic origin and the genomic relatedness of the strains with the results of this study (Figs 1–4, S1 Fig). The strains were isolated from soda lakes including Kenya (24 strains), Egypt (23 strains), Buriatia (Russia)(3 strains), Kulunda Steppe (Altai, Russia)(15 strains), Transbaikal region (Russia)(1 strains), North-eastern Mongolia (6 strains), Mono and Searles Lakes in California (USA)(2 strains), as well as from a haloalkaline H2S-removing bioreactor (2 strains).
Based on the set of genome sequences used in this study, some genomic species groups might be suggested to have a candidate endemic biogeographic distribution [44], such as the genomic species group 1 (Tv. versutus), which has so far only been isolated from Central Asian soda lakes, group 16 (Tv. halophilus), which comes from south-western Siberia, as well as the genomic species groups 5 (Egypt), 6 (Egypt) and 9 (Kenya). Other genomic species contain strains that are geographically widely separated from each other. Therefore, it was suggested to classify those in a candidate disjunct distribution [44]. The genomic species groups 11 (Tv. nitratis), 14 (Tv. thiocyanoxidans) and 17 are primarily found in one area, but also included isolates from other distant locations. Different isolation locations are also observed in the genomic species groups 12, 13, 14, 15 and 17, which contain only two or three strains, and therefore, no statement regarding their dispersion can be made. Nevertheless, using our dataset, it can generally be concluded that most genomic species tend to occur in one geographical region such as Central Asia (Mongolia and south Siberian steppes), Kenya or Egypt. The preference for specific locations might correspond to a better adaptation to certain local environmental conditions. Obvious characteristics distinguishing the different locations might be the fluctuations in temperature and the incoming freshwater during the year, as well as the ratio between sodium carbonate and sodium chloride. In particular, the Central Asian soda lakes are characterized by hot summer, freezing winter and a significant brine dilution due to snow melting in spring time. The Wadi Natrun and Searles lakes are characterized by a domination of chlorides over carbonates.
Several studies reported endemicity in different bacterial groups including Hyphomonas [73], Tenacibaculum [92], fluorescent Pseudomonas strains [93], 3-chlorobenzoate-degrading soil bacteria [94], hot spring cyanobacteria [95] and the hyperthermophilic Archaea Sulfolobus [96,97]. [61] studied the genomic diversity and the biogeography by means of rep-PCR and found that most genotypes were bound to a specific region for which an endemic distribution was suggested. However in our results, a disjunct distribution is seen for most Thioalkalivibrio species. It is important to note that only 29 strains were in common in both analyses and thus, a different picture of the geographical dispersion can be produced. Comparing the clustering of the strains common in both studies, the same structure was generally observed. However, some differences are still present as for example the splitting of the genomic species groups 1 (Tv. versutus) and 11 (Tv. nitratis) in the clustering constructed by the rep-PCR profile. Thus, until now, these results provide no clear conclusion on the biogeography of the Thioalkalivibrio genus yet.
Soda lakes are remotely located extreme habitats. To allow migration and dispersion of Thioalkalivibrio in between the different lakes, bird migration or transportation by particles of sand, salt or dust might be used [61]. For these journeys, they need to be equipped against drought and starvation by forming a resting cell form, called cyst-like refractile cells [98], as well as by producing a yellow pigmentation protecting against UV light [71], high salinity and oxidative stress [70]. However, these types of transportation are likely limited to locations in each area and between the African and Asian continent, while the American continent is further isolated from the African and Asian isolation sites. Nevertheless, Tv. jannaschii ALM2T isolated from Mono Lake (USA) presents high genomic relatedness to Tv. versutus AL2T isolated from Transbaikal region (Russia), which might be due to a recent separation or a change in the advance of the molecular clock.
However, to obtain a broader and a more robust view on the species dispersion at a worldwide scale and on a possibly endemic, disjunct or cosmopolitan distribution, the number of studied strains should be considerably increased for example by using metagenomic datasets and their origins should be chosen more homogeneously on a world-wide scale.
Conclusions
The genus Thioalkalivibrio is more diverse at its species and subspecies level than known before. We discovered 15 novel genomic species and 16 genomic subspecies in addition to the ten already described species. Furthermore, the non-described strains were successfully classified into the different genomic species. The analyses also revealed that Thioalkalivibrio is not a monophyletic genus, because other genera of haloalkaliphilic sulfur bacteria clearly separate four Thioalkalivibrio strains from the core group clustering around the type species Tv. versutus AL2T. Therefore, these four outliers need to be split from the current genus and to be reclassified into a new genus. Furthermore, the different genomic species can either be classified as candidate disjunct or candidate endemic. In this study, we provide a backbone for the genomic classification of currently available Thioalkalivibrio strains, as well as for new strains. In the future, the here proposed new species should be effectively described according to current taxonomic conventions via a polyphasic approach.
Supporting information
De novo species clusters obtained without consideration of type strains. Clusters are indicated by dots (green: ANI > 96% (strains belong to the same genomic species); yellow: 94% < ANI < 96% (strains might belong to the same genomic species); red: ANI < 94% (strains do not belong to the same genomic species). The origin of the strains is indicated with different colors (see legend of Fig 1).
(PDF)
(DOCX)
(DOCX)
(XLSX)
(XLSX)
Strains marked with a (T) are type strains. Genomic species classification based on ANIb and ANIm value (green: ANI > 96% (strains belong to the same genomic species); yellow: 94% < ANI < 96% (strains might belong to the same genomic species); black: ANI < 94% (strains do not belong to the same genomic species). Genomic species classification based on TETRA value (green: TETRA > 0.999% (strains belong to the same genomic species); yellow: 0.989% < TETRA < 0.999% (strains might belong to the same genomic species); black: TETRA < 0.989% (strains do not belong to the same genomic species).
(XLSX)
Strains marked with a (T) are type strains. Genomic species classification based on dDDH shown by dots (green: dDDH ≥ 70% (strains belong to the same genomic species); black: dDDH < 70% (strains do not belong to the same genomic species).
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(PDF)
Acknowledgments
We thank Cherel Balkema for her help in the laboratory, Judith Umbach for her assistance with the ANI analysis and Emily D. Melton for proofreading and helpful comments. The work conducted by the U.S. Department of Energy Joint Genome Institute, a DOE Office of Science User Facility, is supported under Contract No. DE-AC02-05CH11231.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
Financial support for Anne-Catherine Ahn, Lex Overmars and Gerard Muyzer was provided by the ERC Advanced Grant PARASOL (N°322551). Michael Richter has received funding from the European Union Seventh Framework Programme (FP7/2007-2013) under grant agreement no. 311975 (MaCuMBA). Dimitry Sorokin was supported by the RFBR grant 16-04-00035; Tanja Woyke was funded by the U.S. Department of Energy Joint Genome Institute, a DOE Office of Science User Facility, and was supported under Contract No. DE-AC02-05CH11231. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Sorokin DY, Kuenen JG. Haloalkaliphilic sulfur-oxidizing bacteria in soda lakes. FEMS Microbiol Rev. 2005;29: 685–702. 10.1016/j.femsre.2004.10.005 [DOI] [PubMed] [Google Scholar]
- 2.Sorokin DY, Banciu H, Robertson LA, Kuenen JG, Muntyan MS, Muyzer G. Halophilic and Haloalkaliphilic Sulfur-Oxidizing Bacteria from Hypersaline Habitats and Soda Lakes In: Rosenberg E, DeLong EF, Lory S, Stackebrandt E, Thompson F, editors. The Prokaryotes—Prokaryotic Physiology and Biochemistry. Berlin-Heidelberg: Springer-Verlag; 2013. pp. 530–555. [Google Scholar]
- 3.Jones BF, Eugster HP, Rettig SL. Hydrochemistry of the Lake Magadi basin, Kenya. Geochim Cosmochim Acta. 1977;41: 53–72. [Google Scholar]
- 4.Jones BE, Grant WD, Duckworth AW, Owenson GG. Microbial diversity of soda lakes. Extremophiles. 1998;2: 191–200. [DOI] [PubMed] [Google Scholar]
- 5.Melack JM, Kilham P. Photosynthetic Rate of Phytoplankton in East African Alkaline Saline Lakes. Limnol Oceanogr. 1974;19: 743–755. [Google Scholar]
- 6.Roesler CS, Culbertson CW, Etheridge SM, Goericke R, Kiene RP, Miller LG, et al. Distribution, production, and ecophysiology of Picocystis strain ML in Mono Lake, California. Limnol Oceanogr. 2002;47: 440–452. [Google Scholar]
- 7.Kompantseva EI, Komova AV, Rusanov II, Pimenov NV, Sorokin DYu. Primary Production of Organic Matter and Phototrophic Communities in the Soda Lakes of the Kulunda Steppe (Altai Krai). Mikrobiology (Moscow, English translation). 2009;78: 709–715. [Google Scholar]
- 8.Jones BE, Grant WD. Microbial diversity and ecology of the Soda Lakes of East Africa In: Bell CR, Brylinsky M, Johnson-Green P, editors. Microbial Biosystems: New Frontiers. Proceedings of the 8th International Symposium Microbial Ecology. Halifax: Atlantic Canada Society for Microbial Ecology; 1999. p.681–687. [Google Scholar]
- 9.Ma Y, Zhang W, Xue Y, Zhou P, Ventosa A, Grant WD. Bacterial diversity of the Inner Mongolian Baer soda lake as revealed by 16S rRNA gene sequence analyses. Extremophiles. 2004;8: 45–51. 10.1007/s00792-003-0358-z [DOI] [PubMed] [Google Scholar]
- 10.Mesbah NM, Abou-El-Ela SH, Wiegel J. Novel and unexpected prokaryote diversity in water and sediments of the alkaline hypersaline lakes of the Wadi An Natrun, Egypt. Microbial Ecol. 2007;54: 598–617. [DOI] [PubMed] [Google Scholar]
- 11.Lanzen A, Simachew A, Gessesse A, Chmolowska D, Jonassen I, Øvreås L. Surprising prokaryotic and eukaryotic diversity, community structure and biogeography of Ethiopian soda lakes. PLoS ONE. 2013;8: e72577 10.1371/journal.pone.0072577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sorokin DY, Berben T, Melton ED, Overmars L, Vavourakis CD, Muyzer G. Microbial diversity and biogeochemical cycling in soda lakes. Extremophiles. 2014;18: 791–809. 10.1007/s00792-014-0670-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sorokin DY, Banciu HL, Muyzer G. Functional microbiology of soda lakes. Curr Opin Microbiol. 2015;25: 88–96. 10.1016/j.mib.2015.05.004 [DOI] [PubMed] [Google Scholar]
- 14.Sorokin DY, Lysenko AM, Mityushina LL, Tourova TP, Jones BE, Rainey FA, et al. Thioalkalimicrobium aerophilum gen. nov., sp. nov. and Thioalkalimicrobium sibericum sp. nov., and Thioalkalivibrio versutus gen. nov., sp. nov., Thioalkalivibrio nitratis sp.nov., novel and Thioalkalivibrio denitrificancs sp. nov., novel obligately alkaliphilic and obligately chemolithoautotrophic sulfur-oxidizing bacteria from soda lakes. Int J Syst Evol Microbiol. 2001;51: 565–580. 10.1099/00207713-51-2-565 [DOI] [PubMed] [Google Scholar]
- 15.Sorokin DY, Tourova TP, Lysenko AM, Mityushina LL, Kuenen JG. Thioalkalivibrio thiocyanoxidans sp. nov. and Thioalkalivibrio paradoxus sp. nov., novel alkaliphilic, obligately autotrophic, sulfur-oxidizing bacteria capable of growth on thiocyanate, from soda lakes. Int J Syst Evol Microbiol. 2002;52: 657–664. 10.1099/00207713-52-2-657 [DOI] [PubMed] [Google Scholar]
- 16.Sorokin DY, Gorlenko VM, Tourova TP, Tsapin AI, Nealson KH, Kuenen GJ. Thioalkalimicrobium cyclicum sp. nov. and Thioalkalivibrio jannaschii sp. nov., novel species of haloalkaliphilic, obligately chemolithoautotrophic sulfur-oxidizing bacteria from hypersaline alkaline Mono Lake (California). Int J Syst Evol Microbiol. 2002;52: 913–920. 10.1099/00207713-52-3-913 [DOI] [PubMed] [Google Scholar]
- 17.Sorokin DY, Tourova TP, Sjollema KA, Kuenen GJ. Thialkalivibrio nitratireducens sp. nov., a nitrate-reducing member of an autotrophic denitrifying consortium from a soda lake. Int J Syst Evol Microbiol. 2003;53: 1779–1783. 10.1099/ijs.0.02615-0 [DOI] [PubMed] [Google Scholar]
- 18.Banciu H, Sorokin DY, Galinski EA, Muyzer G, Kleerebezem R, Kuenen JG. Thialkalivibrio halophilus sp. nov., a novel obligately chemolithoautotrophic, facultatively alkaliphilic, and extremely salt-tolerant, sulfur-oxidizing bacterium from a hypersaline alkaline lake. Extremophiles. 2004;8: 325–334. 10.1007/s00792-004-0391-6 [DOI] [PubMed] [Google Scholar]
- 19.Sorokin DY, Tourova TP, Antipov AN, Muyzer G, Kuenen JG. Anaerobic growth of the haloalkaliphilic denitrifying sulfur-oxidizing bacterium Thialkalivibrio thiocyanodenitrificans sp. nov. with thiocyanate. Microbiology. 2004;150: 2435–2442. 10.1099/mic.0.27015-0 [DOI] [PubMed] [Google Scholar]
- 20.Sorokin DY, Muntyan MS, Panteleeva AN, Muyzer G. Thioalkalivibrio sulfidiphilus sp. nov., a haloalkaliphilic, sulfur-oxidizing gammaproteobacterium from alkaline habitats. Int J Syst Evol Microbiol. 2012;62: 1884–1889. 10.1099/ijs.0.034504-0 [DOI] [PubMed] [Google Scholar]
- 21.Sorokin DY, Kuenen JG, Muyzer G. The microbial sulfur cycle at extremely haloalkaline conditions of soda lakes. Front Microbiol. 2011; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sorokin DY, Tourova TP, Lysenko AM, Kuenen JG. Microbial thiocyanate utilization under highly alkaline conditions. Appl Environ Microbiol. 2001;67: 528–538. 10.1128/AEM.67.2.528-538.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sorokin DY, Kuenen JG, Jetten M. Denitrification at extremely high pH values by the alkaliphilic, obligately chemolithoautotrophic, sulfur-oxidizing bacterium Thioalkalivibrio denitrificans strain ALJD. Arch Microbiol. 2001b;175: 94–101. [DOI] [PubMed] [Google Scholar]
- 24.Wayne LG, Brenner DJ, Colwell RR, Grimont PAD, Kandler O, Krichevsky MI, et al. Report of the Ad Hoc Committee on Reconciliation of Approaches to Bacterial Systematics. Int. J. Syst. Bacteriol. 1987;37: 463–464. [Google Scholar]
- 25.Thompson CC, Chimetto L, Edwards RA, Swings J, Stackebrandt E, Thompson FL. Microbial genomic taxonomy. BMC Genomics, 2013; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yarza P, Richter M, Peplies J, Euzeby J, Amann R, Schleifer KH, et al. The All-Species Living Tree project: A 16S rRNA-based phylogenetic tree of all sequenced type strains. Syst Appl Microbiol. 2008;31: 241–250. 10.1016/j.syapm.2008.07.001 [DOI] [PubMed] [Google Scholar]
- 27.Yarza P, Ludwig W, Euzéby J, Amann R, Schleifer KH, Glöckner FO, et al. Update of the All-Species Living Tree Project based on 16S and 23S rRNA sequence analyses. Syst Appl Microbiol. 2010;33: 291–299. 10.1016/j.syapm.2010.08.001 [DOI] [PubMed] [Google Scholar]
- 28.Hahnke RL, Meier-Kolthoff JP, García-López M, Mukherjee S, Huntemann M, Ivanova NN, et al. Genome-Based Taxonomic Classification of Bacteroidetes. Front Microbiol. 2016; 7: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chun J, Rainey FA. Integrating genomics into the taxonomy and systematics of the Bacteria and Archaea. Int J Syst Evol Microbiol. 2014;64: 316–324. 10.1099/ijs.0.054171-0 [DOI] [PubMed] [Google Scholar]
- 30.Stackebrandt E, Frederiksen W, Garrity GM, Grimont PA, Kämpfer P, Maiden MC, et al. Report of the ad hoc committee for the re-evaluation of the species definition in bacteriology. Int J Syst Evol Microbiol. 2002;52: 1043–1047. 10.1099/00207713-52-3-1043 [DOI] [PubMed] [Google Scholar]
- 31.Goris J, Konstantinidis KT, Klappenbach JA, Coenye T, Vandamme P, Tiedje JM. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol. 2007;57: 81–91. 10.1099/ijs.0.64483-0 [DOI] [PubMed] [Google Scholar]
- 32.Richter M, Rosselló-Móra R. Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci U S A. 2009;106: 19126–19131. 10.1073/pnas.0906412106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meier-Kolthoff JP, Auch AF, Klenk H-P, Göker M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics. 2013;14: 60–73. 10.1186/1471-2105-14-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meier-Kolthoff JP, Klenk HP, Göker M. Taxonomic use of DNA G+C content and DNA-DNA hybridization in the genomic age. Int J Syst Evol Microbiol. 2014;64: 352–356. 10.1099/ijs.0.056994-0 [DOI] [PubMed] [Google Scholar]
- 35.Auch AF, Von Jan M, Klenk HP, Göker M. Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand Genomic Sci. 2010;2: 117–134. 10.4056/sigs.531120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meier-Kolthoff JP, Hahnke RL, Petersen J, Scheuner C, Michael V, Fiebig A, et al. Complete genome sequence of DSM 30083T, the type strain (U5/41T) of Escherichia coli, and a proposal for delineating subspecies in microbial taxonomy. Stand Genomic Sci. 2014;9: 2–20. 10.1186/1944-3277-9-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meier-Kolthoff JP, Auch AF, Klenk HP, Göker M. Highly parallelized inference of large genome-based phylogenies. Concurr Comput Pract Exp. 2014;26: 1715–1729. [Google Scholar]
- 38.Stackebrandt E. The Richness of Prokaryotic Diversity: There Must Be a Species Somewhere. Food Technol Biotechnol. 2003;41: 17–22. [Google Scholar]
- 39.Janda JM, Abbott SL. 16S rRNA Gene Sequencing for Bacterial Identification in the Diagnostic Laboratory: Pluses, Perils, and Pitfalls. J Clin Microbiol. 2007;45: 2761–2764. 10.1128/JCM.01228-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gevers D, Cohan FM, Lawrence JG, Spratt BG, Coenye T, Feil EJ, et al. Re-evaluating prokaryotic species. Nat Rev Microbiol. 2005;3:733–739. 10.1038/nrmicro1236 [DOI] [PubMed] [Google Scholar]
- 41.Glaeser SP, Kämpfer P. Multilocus sequence analysis (MLSA) in prokaryotic taxonomy. Syst Appl Microbiol. 2015;38: 237–245. 10.1016/j.syapm.2015.03.007 [DOI] [PubMed] [Google Scholar]
- 42.Colwell RR. Polyphasic taxonomy of the genus Vibrio: numerical taxonomy of Vibrio cholerae, Vibrio parahaemolyticus, and related Vibrio species. J Bacteriol. 1970;104: 410–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vandamme P, Peeters C. Time to revisit polyphasic taxonomy. Antonie Van Leeuwenhoek, 2014;106: 57–65. 10.1007/s10482-014-0148-x [DOI] [PubMed] [Google Scholar]
- 44.Morrone JJ, Crisci JV. Historical Biogeography: Introduction to Methods. Annu Rev Ecol Systemat. 1995;26: 373–401. [Google Scholar]
- 45.Markowitz VM, Chen IMA, Palaniappan K, Chu K, Szeto E, Grechkin Y, et al. IMG: the integrated microbial genomes database and comparative analysis system. Nucleic Acids Res. 2012;40: D115–122. 10.1093/nar/gkr1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig WG, Peplies J, Glöckner FO. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucl Acids Res. 2007;35: 7188–7196. 10.1093/nar/gkm864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pruesse E, Peplies J, Glöckner FO. SINA: Accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics. 2012;28: 1823–1829. 10.1093/bioinformatics/bts252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhukumar, et al. ARB: a software environment for sequence data. Nucleic Acids Res. 2004;32: 1363–1371. 10.1093/nar/gkh293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol Biol Evol. 2013;30: 2725–2729. 10.1093/molbev/mst197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004; 32, 1792–1797. 10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Richter M, Rosselló-Móra R, Glöckner FO, Peplies J. JSpeciesWS: a web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics. 2016;32: 929–931. 10.1093/bioinformatics/btv681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Garcia-Vallve S, Palau J, Romeu A. Horizontal gene transfer in glycosyl hydrolases inferred from codon usage in Escherichia coli and Bacillus subtilis. Mol Biol Evol. 1999;16: 1125–1134. [DOI] [PubMed] [Google Scholar]
- 53.Sokal R, Michener C. A statistical method for evaluating systematic relationships. Univ Kans Sci Bull. 1958; 28: 1409–1438. [Google Scholar]
- 54.Huson DH, Scornavacca C. Dendroscope 3: An interactive tool for rooted phylogenetic trees and networks. Syst Biol. 2012;61: 1061–1067. 10.1093/sysbio/sys062 [DOI] [PubMed] [Google Scholar]
- 55.Henz SR, Huson DH, Auch AF, Nieselt-Struwe K, Schuster SC. Whole-genome prokaryotic phylogeny. Bioinformatics. 2005;21: 2329–2335. 10.1093/bioinformatics/bth324 [DOI] [PubMed] [Google Scholar]
- 56.Lefort V, Desper R, Gascuel O. FastME 2.0: A comprehensive, accurate, and fast distance-based phylogeny inference program. Mol Biol Evol. 2015;32: 2798–2800. 10.1093/molbev/msv150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu Y, Lai Q, Göker M, Meier-Kolthoff JP, Wang M, Sun Y, et al. Genomic insights into the taxonomic status of the Bacillus cereus group. Sci Rep. 2015;5: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Göker M, García-Blázquez G, Voglmayr H, Tellería MT, Martín MP. Molecular taxonomy of phytopathogenic fungi: a case study in Peronospora. PLoS ONE. 2009;4: e6319 10.1371/journal.pone.0006319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sokal RR, Sneath PHA. Principles of Numerical Taxonomy. San Francisco: Freeman WH and Company; 1963. [Google Scholar]
- 60.Garrido-Sanz D, Meier-Kolthoff JP, Göker M, Martín M, Rivilla R, Redondo-Nieto M. Genomic and Genetic Diversity within the Pseudomonas fluorescens Complex. PLoS One. 2016;11: e0150183 10.1371/journal.pone.0150183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Foti M, Ma S, Sorokin DY, Rademaker JL, Kuenen JG, Muyzer G. Genetic diversity and biogeography of haloalkaliphilic sulphur-oxidizing bacteria belonging to the genus Thioalkalivibrio. FEMS Microbiol Ecol. 2006;56: 95–101. 10.1111/j.1574-6941.2006.00068.x [DOI] [PubMed] [Google Scholar]
- 62.Achtman M, Wagner M. Microbial diversity and the genetic nature of microbial species. Nature. 2008;6: 431–440. [DOI] [PubMed] [Google Scholar]
- 63.Polz MF, Alm EJ, Hanage WP. Horizontal gene transfer and the evolution of bacterial and archaeal population structure. Trends Genet. 2013;29: 170–175. 10.1016/j.tig.2012.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Soucy SM, Huang J, Gogarten JP. Horizontal gene transfer: building the web of life. Nat Rev Genet. 2015;16: 472–482. 10.1038/nrg3962 [DOI] [PubMed] [Google Scholar]
- 65.Feil EJ, Smith JM, Enright MC, Spratt BG. Estimating Recombinational Parameters in Streptococcus pneumoniae from Multilocus Sequence Typing Data. Genetics. 2000;154: 1439–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Spratt BG, Hanage WP, Feil EJ. The relative contributions of recombination and point mutation to the diversification of bacterial clones. Curr Opin Microbiol. 2001;4: 602–606. [DOI] [PubMed] [Google Scholar]
- 67.Muyzer G, Sorokin DY, Mavromatis K, Lapidus A, Foster B, Sun H. Complete genome sequence of Thioalkalivibrio sp. K90mix. Stand Genomic Sci. 2011;5: 341–355. 10.4056/sigs.2315092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mu T, Zhou J, Yang M, Xing J. Complete genome sequence of Thialkalivibrio versutus D301 isolated from Soda Lake in northern China, a typical strain with great ability to oxidize sulfide. J Biotechnol. 2016; 227: 21–22. 10.1016/j.jbiotec.2016.04.019 [DOI] [PubMed] [Google Scholar]
- 69.Banciu H, Sorokin DY, Kleerebezem R, Muyzer G, Galinski EA, Kuenen JG. Growth kinetics of haloalkaliphilic, sulfur-oxidizing bacterium Thioalkalivibrio versutus strain ALJ 15 in continuous culture. Extremophile. 2004b;8: 185–192. [DOI] [PubMed] [Google Scholar]
- 70.Banciu H, Sorokin DY, Rijpstra WI, Sinninghe Damsté JS, Galinski EA, Takaichi S, et al. Fatty acid, compatible solute and pigment composition of obligately chemolithoautotrophic alkaliphilic sulfur-oxidizing bacteria from soda lakes. FEMS Microbiol Lett. 2005;243: 181–187. 10.1016/j.femsle.2004.12.004 [DOI] [PubMed] [Google Scholar]
- 71.Takaichi S, Maoka T, Akimoto N, Sorokin DY, Banciu H, Kuenen JG. Two novel yellow pigments natronochrome and chloronatronochrome from the natrono(alkali)philic sulfur-oxidizing bacterium Thialkalivibrio versutus strain ALJ 15. Tetrahedron Lett. 2004;45: 8303–8305. [Google Scholar]
- 72.Fox GE, Wisotzkey JD, Jurtshuk P Jr . How close is close: 16S rRNA sequence identity may not be sufficient to guarantee species identity. Int J Syst Bacteriol. 1992;42: 166–170. 10.1099/00207713-42-1-166 [DOI] [PubMed] [Google Scholar]
- 73.Li C, Lai Q, Li G, Liu Y, Sun F, Shao Z. Multilocus Sequence Analysis for the Assessment of Phylogenetic Diversity and Biogeography in Hyphomonas Bacteria from Diverse Marine Environments. PLoS ONE. 2014;9: e101394 10.1371/journal.pone.0101394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lai Q, Liu Y, Yuan J, Du J, Wang L, Sun F, et al. Multilocus Sequence Analysis for Assessment of Phylogenetic Diversity and Biogeography in Thalassospira Bacteria from Diverse Marine Environments. PLoS ONE. 2014;9: e106353 10.1371/journal.pone.0106353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chan JZ-M, Halachev MR, Loman NJ, Constantinidou C, Pallen MJ. Defining bacterial species in the genomic era: insights from the genus Acinetobacter. BMC Microbiol. 2012;12: 302–312. 10.1186/1471-2180-12-302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Brown-Elliott BA, Brown JM, Conville PS, Wallace RJ Jr. Clinical and laboratory features of the Nocardia spp. based on current molecular taxonomy. Clin Microbiol Rev. 2006;19: 259–282. 10.1128/CMR.19.2.259-282.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ventura M, Canchaya C, Del Casale A, Dellaglio F, Neviani E, Fitzgerald GF, et al. Analysis of bifidobacterial evolution using a multilocus approach. Int J Syst Evol Microbiol. 2006;56: 2783–2792. 10.1099/ijs.0.64233-0 [DOI] [PubMed] [Google Scholar]
- 78.Ash C, Farrow JAE, Dorsch M, Stackebrandt E, Collins MD. Comparative analysis of Bacillus anthracis, Bacillus cereus, and related species on the basis of reverse transcriptase sequencing of 16S rRNA. Int J Syst Evol Microbiol. 1991;41: 343–346. [DOI] [PubMed] [Google Scholar]
- 79.Asai T, Zaporojets D, Squires C, Squires CL. An Escherichia coli strain with all chromosomal rRNA operons inactivated: complete exchange of rRNA genes between bacteria. Proc Natl Acad Sci USA. 1999;96: 1971–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yap WH, Zhang Z, Wang Y. Distinct types of rRNA operons exist in the genome of the actinomycete Thermomonospora chromogena and evidence for horizontal transfer of an entire rRNA operon. J Bacteriol. 1999;181: 5201–5209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tian RM, Cai L, Zhang WP, Cao HL, Qian PY. Rare events of intra-genus and intra-species horizontal transfer of the 16S rRNA gene. Genome Biol Evol. 2015;7: 2310–2320. 10.1093/gbe/evv143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Martens M, Dawyndt P, Coopman R, Gillis M, De Vos P, Willems A. Advantages of multilocus sequence analysis for taxonomic studies: a case study using 10 housekeeping genes in the genus Ensifer. Int J Syst Evol Microbiol. 2008;58: 200–214. 10.1099/ijs.0.65392-0 [DOI] [PubMed] [Google Scholar]
- 83.Delamuta JRM, Ribeiro RA, Menna P, Bangel EV, Hungria M. Multilocus Sequence Analysis (MLSA) of Bradyrhizobium Strains: Revealing High Diversity Of Tropical Diazotrophic Symbiotic Bacteria. Braz J Microbiol. 2012;43: 698–710. 10.1590/S1517-83822012000200035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Siddall ME. Unringing a bell: metazoan phylogenomics and the partition bootstrap. Cladistics. 2010; 26: 444–452. [DOI] [PubMed] [Google Scholar]
- 85.Imhoff JF, Süling J. The phylogenetic relationship among Ectothiorhodospiraceae: a reevaluation of their taxonomy on the basis of 16S rDNA analyses. Arch Microbiol. 1996;165: 106–113. [DOI] [PubMed] [Google Scholar]
- 86.Bryantseva I, Gorlenko VM, Kompantseva EI, Imhoff JF, Süling J, Mityushina L. Thiorhodospira sibirica gen. nov., sp. nov., a new alkaliphilic purple sulfur bacterium from a Siberian soda lake. Int J Syst Bacteriol. 1999;49: 697–703. 10.1099/00207713-49-2-697 [DOI] [PubMed] [Google Scholar]
- 87.Sorokin DY, Kuenen JG. Chemolithotrophic haloalkaliphiles from soda lakes. FEMS Microbiology Ecology. 2005;52: 287–295. 10.1016/j.femsec.2005.02.012 [DOI] [PubMed] [Google Scholar]
- 88.Sorokin DY, Tourova TP, Muyzer G, Kuenen GJ. Thiohalospira halophila gen. nov., sp. nov. and Thiohalospira alkaliphila sp. nov., novel obligately chemolithoautotrophic, halophilic, sulfur-oxidizing gammaproteobacteria from hypersaline habitats. Int. J. Syst. Evol. Microbiol. 2008; 58: 1685–1692. 10.1099/ijs.0.65654-0 [DOI] [PubMed] [Google Scholar]
- 89.Wood S. Monophyly and comparisons between trees. Cladistics. 1994;10: 339–346. [Google Scholar]
- 90.Ashlock PD. Monophyly and Associated Terms. Syst Zool. 1971;20: 63–69. [Google Scholar]
- 91.Yarza P, Yilmaz P, Pruesse E, Glöckner FO, Ludwig W, Schleifer KH, et al. Uniting the classification of cultured and uncultured bacteria and archaea using 16S RNA gene sequences. Nat Rev Microbiol. 2014;12: 635–645. 10.1038/nrmicro3330 [DOI] [PubMed] [Google Scholar]
- 92.Habib C, Houel A, Lunazzi A, Bernardet JF, Olsen AB, Nilsen H, et al. Multilocus sequence analysis of the marine bacterial genus Tenacibaculum suggests parallel evolution of fish pathogenicity and endemic colonization of aquaculture systems. Appl Environ Microbiol. 2014;80: 5503–5514. 10.1128/AEM.01177-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cho JC, Tiedje JM. Biogeography and Degree of Endemicity of Fluorescent Pseudomonas Strains in Soil. Appl Environ Microbiol. 2000;66: 5448–5456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Fulthorpe RR, Rhodes AN, Tiedje JM. High levels of endemicity of 3-chlorobenzoate-degrading soil bacteria. Appl Environ Microbiol. 1998;64: 1620–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Papke RT, Ramsing NB, Bateson MM, Ward DM. Geographical isolation in hot spring Cyanobacteria. Environ Microbiol. 2003;5: 650–659. [DOI] [PubMed] [Google Scholar]
- 96.Whitaker RJ, Grogan DW, Taylor JW. Geographic Barriers Isolate Endemic Populations of Hyperthermophilic Archaea. Science. 2003; 301: 976–978. 10.1126/science.1086909 [DOI] [PubMed] [Google Scholar]
- 97.Reno ML, Held NL, Fields CJ, Burke PV, Whitaker RJ. Biogeography of the Sulfolobus islandicus pan-genome. Proc Natl Acad Sci U S A. 2009;106: 8605–8610. 10.1073/pnas.0808945106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Loĭko NG, Soina VS, DIu Sorokin, Mitiushina LL, El'-Registan GI. Resting forms of gram negative chemolithoautotrophic bacteria Thioalkalivibrio versutus and Thioalkalimicrobium aerophilum. Mikrobiology (Moscow, English translation). 2003;72: 328–237. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
De novo species clusters obtained without consideration of type strains. Clusters are indicated by dots (green: ANI > 96% (strains belong to the same genomic species); yellow: 94% < ANI < 96% (strains might belong to the same genomic species); red: ANI < 94% (strains do not belong to the same genomic species). The origin of the strains is indicated with different colors (see legend of Fig 1).
(PDF)
(DOCX)
(DOCX)
(XLSX)
(XLSX)
Strains marked with a (T) are type strains. Genomic species classification based on ANIb and ANIm value (green: ANI > 96% (strains belong to the same genomic species); yellow: 94% < ANI < 96% (strains might belong to the same genomic species); black: ANI < 94% (strains do not belong to the same genomic species). Genomic species classification based on TETRA value (green: TETRA > 0.999% (strains belong to the same genomic species); yellow: 0.989% < TETRA < 0.999% (strains might belong to the same genomic species); black: TETRA < 0.989% (strains do not belong to the same genomic species).
(XLSX)
Strains marked with a (T) are type strains. Genomic species classification based on dDDH shown by dots (green: dDDH ≥ 70% (strains belong to the same genomic species); black: dDDH < 70% (strains do not belong to the same genomic species).
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(PDF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.