

Abstract
Abundant natural flavonoids can induce nuclear factor-erythroid 2 related factor 2 (Nrf2) and/or AMP-activated protein kinase (AMPK) activation, which play crucial roles in the amelioration of various inflammation- and oxidative stress-induced diseases, including acute lung injury (ALI). Xanthohumol (Xn), a principal prenylflavonoid, possesses anti-inflammation and anti-oxidant activities. However, whether Xn could protect from LPS-induced ALI through inducing AMPK/Nrf2 activation and its downstream signals, are still poorly elucidated. Accordingly, we focused on exploring the protective effect of Xn in the context of ALI and the involvement of underlying molecular mechanisms. Our findings indicated that Xn effectively alleviated lung injury by reduction of lung W/D ratio and protein levels, neutrophil infiltration, MDA and MPO formation, and SOD and GSH depletion. Meanwhile, Xn significantly lessened histopathological changes, reactive oxygen species (ROS) generation, several cytokines secretion, and iNOS and HMGB1 expression, and inhibited Txnip/NLRP3 inflammasome and NF-κB signaling pathway activation. Additionally, Xn evidently decreased t-BHP-stimulated cell apoptosis, ROS generation and GSH depletion but increased various anti-oxidative enzymes expression regulated by Keap1-Nrf2/ARE activation, which may be associated with AMPK and GSK3β phosphorylation. However, Xn-mediated inflammatory cytokines and ROS production, histopathological changes, Txnip/NLRP3 inflammasome and NF-κB signaling pathway in WT mice were remarkably abrogated in Nrf2-/- mice. Our experimental results firstly provided a support that Xn effectively protected LPS-induced ALI against oxidative stress and inflammation damage which are largely dependent upon upregulation of the Nrf2 pathway via activation of AMPK/GSK3β, thereby suppressing LPS-activated Txnip/NLRP3 inflammasome and NF-κB signaling pathway.
Keywords: Xanthohumol, Acute lung injury, Oxidative stress, Inflammation, Nrf2/AMPK
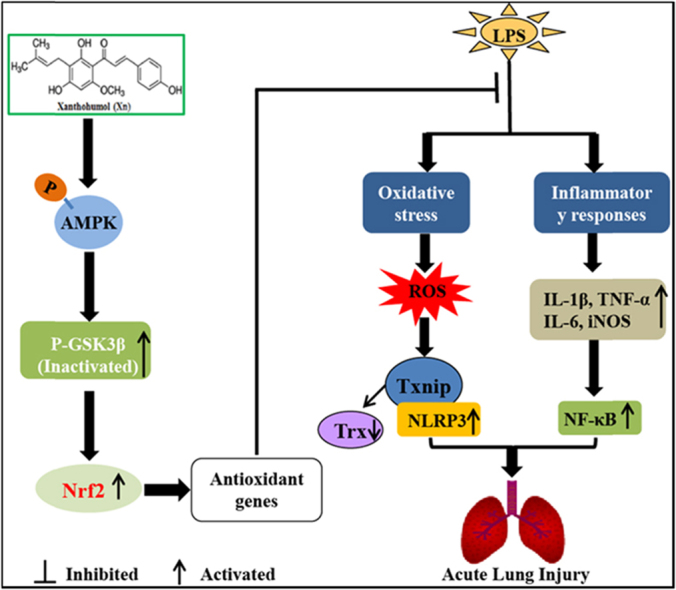
Graphical abstract
Scheme summarizing the protective effects of Xn on LPS-induced acute lung injury via activation of the AMPK/GSK3β-Nrf2 axis.
Highlights
-
•
Xn protects LPS-induced acute lung injury via anti-inflammation and anti-oxidant.
-
•
Xn induces expressions of GCLC, GCLM, HO-1, NQO-1 and Trx-1.
-
•
Xn blocks NF-κB and Txnip/NLRP3 inflammasome activation.
-
•
Xn upregulates AMPK/GSK3β-Nrf2 signal axis.
-
•
Xn-attenuated acute lung injury is effectively abrogated in Nrf2-/- mice.
1. Introduction
Acute lung injury (ALI) and its more serious form, respiratory distress syndrome (ARDS), are regarded as an acute and severe inflammatory process existing in lungs which results from various direct and indirect insults, and still remain high mortality rates [1], [2]. Furthermore, increasing evidence shows that oxidative stress is recognized as key lung injury pathways affecting the severity of ALI [3]. Inflammation and oxidative stress are considered for an extremely related event, which involved in the pathological process of ALI [4], [5], [6]. Accordingly, focusing on the inhibitory of inflammation and/or oxidative stress may be potential strategies for the prevention and treatment of ALI.
Lipopolysaccharide (LPS), derived from the outer membranes of Gram-negative bacteria, exposure to lung elicits many macrophages activation and inflammatory cells leakage, especially neutrophils [7], [8]. Numerous neutrophils influx into the lungs which not only results in the release of uncontrolled inflammatory cytokines, but also leads to the formation of reactive oxygen species (ROS) and oxidative stress [3], [9]. It is of considerable interest that multiple signal pathways are activated or inhibited, which is strongly associated with regulation of inflammatory responses and oxidative stress in process of LPS-induced ALI. Indeed, previous reports have showed that LPS can activate nuclear factor-kappa B (NF-κB), a key nuclear transcription factor that plays a vital role in regulation of inflammation and immune responses [10]. Under physiological condition, heterodimers of NF-κB components, mainly p50/p65, remain in the cytoplasm in an inactive form through their linkage to an inhibitor of κB (IκB) protein [11]. Once activated, however, IκB is phosphorylated by IκB kinase (IKK) and is rapidly degraded, which subsequently dissociates from NF-κB, enabling NF-κB dimers to enter the nucleus and regulate various inflammatory mediators, including tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), IL-6, inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2) [12]. Moreover, high mobility group box 1 (HMGB1), a late inflammatory cytokine, promotes lung vascular hyperpermeability in ventilator-induced lung injury [13]. Importantly, release of these pro-inflammatory cytokines result in the activation of peripheral polymorphonuclear neutrophils (PMNs) and its over-activation further aggravates inflammatory responses and induces oxidative stress [14], the primary sources of which are respiratory burst and degranulation. Oxidative stress leads to overproduction of ROS, myeloperoxidase (MPO) and malondialdehyde (MDA) formation and decreases expression of anti-oxidative enzymes, such as superoxide dismutase (SOD) and reduced glutathione (GSH), protecting lung tissues against oxidative damage in vivo [15]. Intriguingly, increasing evidences shows that thioredoxin-interacting protein (Txnip) is important point linkage oxidative stress to inflammation. In response to ROS, Txnip detaches from thioredoxin (Trx), which is composed of two isoforms, including the cytosolic Trx-1 and mitochondrial Trx-2, and binds to nucleotide-binding domain-like receptor protein 3 (NLRP3), resulting in NLRP3 inflammasome activation [16], [17]. Activation of NLRP3 inflammasome, including NOD like receptor, NLRP3 protein, the adaptor ASC protein and caspase-1, result in the maturation and release of pro-inflammatory cytokines, IL-1β and IL-18 which also plays a significant role in the development of ALI [18], [19].
Nuclear factor-erythroid 2 related factor 2 (Nrf2) plays a crucial role in the amelioration of various inflammatory- and oxidative stress-induced diseases, such as chronic obstructive pulmonary disease (COPD), ALI and asthma [20], [21]. Under unstressed conditions, Nrf2 is bound to Keap1 (Kelch-like ECH-associated protein 1, its repressor protein) in the cytoplasm and degraded through the ubiquitin-proteasome pathways. Upon exposure to stressors and inducers, however, the release of Nrf2 from Keap1 translocates into the nucleus to result in the expression of various cytoprotective genes, such as the glutamate-cysteine ligase modifier (GCLM), the catalytic (GCLC) subunit, heme oxygenase-1 (HO-1), NAD (P) H: quinone oxidoreductase (NQO1) and Trx, dependent upon binding to conserved antioxidant response elements (AREs) [22], [23]. In this process, AMP-activated protein kinase (AMPK) mediates inactivation of glycogen synthase kinase 3β (GSK3β) to increase nuclear accumulation of Nrf2 [24]. AMPK, which is a key energy sensor of cellular metabolism, involves in metabolic stress, including neurodegeneration, inflammation, and oxidative stress [25]. Furthermore, emerging evidence indicates that AMPK, as a negative modulator of inflammatory responses, plays protective roles in a LPS-induced mouse model of ALI [26]. Taken together, to inhibit NF-κB and Txnip/NLRP3 inflammasome activation or/and to activate AMPK and Nrf2 signaling pathway may contribute to anti-oxidant and anti-inflammatory for the attenuation of ALI.
To date, abundant reports have revealed that consumption of natural flavonoids is essential for the prevention of diseases particularly resulted from inflammation and oxidative stress, such as ALI [27]. Xanthohumol (2′, 4′, 4-trihydroxy-6′-methoxy- 3′-prenylchalcone, Xn: Fig. 1A), the principal prenylflavonoid that exists mainly in the hop plants (Humulus lupulus L.), is exploited for preserving and flavoring beer which has been attracted considerable attention because of its various biological activities, including anti-inflammatory and antioxidant properties [28]. Furthermore, recent studies also uncovered that Xn can positively regulate AMPK activity contributing to activate Nrf2 antioxidative signaling pathways, suggesting that it effectively improved oxidative-stress-induced cell injury [29], [30]. Up to now, however, little is known about the protective effect of Xn against LPS-induced damage in vitro or in vivo. Consequently, we investigated the protective effect of Xn on LPS-induced ALI and the mechanisms related to inflammation and oxidative stress associated with the regulation of AMPK/GSK3β-Nrf2 axis.
Fig. 1.

Protective effects of Xn treatment against LPS-induced ALI mice. (A) The hop plants (Humulus lupulus L.). (B) The chemical structure of Xanthohumol (Xn). Xn (10 or 50 mg/kg) or Dex (5 mg/kg) was administered intraperitoneally to mice 1 h prior to LPS (0.5 mg/kg) pretreatment. (C) Lungs (n=5) from each experimental group were processed for histological evaluation at 12 h after the LPS challenge. (D) The lung injury score was determined following a five-point scale from 0 to 4 as follows: 0, l, 2, 3, and 4 represent no damage, mild damage, moderate damage, severe damage, and very severe damage, respectively. Representative histological sections of the lungs were stained with hematoxylin and eosin (H & E staining, magnification ×400). (E) The lung W/D ratio was determined at 12 h after LPS challenge. BALF was collected 12 h after LPS challenge to measure (F) amount of protein or (G) levels of cytokines (TNF-α, IL-6 and IL-1β) secretion. (H–I) Protein of lungs was analyzed by Western blot with specific antibodies (iNOS, COX-2 and HMGB1). Similar results were obtained from three independent experiments. All data are presented as means±SEM (n=5 in each group). ##p<0.01 vs Control group; *p<0.05 and **p<0.01 vs LPS group.
2. Materials and methods
2.1. Reagents and chemical
Xanthohumol (Xn), purity >98%, was provided by the Chengdu Herbpurify CO., LTD (Chengdu, China). LPS (Escherichia coli 055:B5) and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Compound C and Brusatol (specific inhibitors of the AMPK, and Nrf2, respectively), and tert-butyl hydroperoxide (t-BHP) were offered by Sigma-Aldrich (St. Louis, MO). Penicillin and streptomycin, Fetal bovine serum (FBS) and Dulbecco's modified Eagle's medium (DMEM) were acquired from Invitrogen-Gibco (Grand Island, NY). Antibodies against Nrf2, Keap1, GCLC, HO-1, NQO1, GCLM, Trx-1, Txnip, NLRP3, casapase-1, ASC, IL-1β, Lamin B and β-actin were supplied by Cell Signaling (Boston, MA, USA) or Abcam (Cambridge, MA, USA). The Annexin V fluorescein isothiocyanate (FITC)/Propidium Iodide (PI) apoptosis kit were offered from Beyotime Institute of Biotechnology (Jinlin, China). In addition, GSH, MDA, SOD and MPO test kits were obtained from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). All other chemicals were offered by Sigma-Aldrich (St. Louis, MO, USA), if not otherwise indicated.
2.2. Animals
Wild-type (WT) and Nrf2−/−(knockout) C57BL/6 mice were purchased from Liaoning Changsheng Technology Industrial Co., LTD (Certificate SCXK2010-0001; Liaoning, China) and The Jackson Laboratory (Bar Harbor, ME, USA), respectively. All animals were raised under SPF-condition after feeding for several days. All studies were in accordance with the International Guiding Principles for Biomedical Research Involving Animals, which was published by the Council for the International Organizations of Medical Sciences.
2.3. Experimental protocol
WT mice were randomly divided into six groups: Control (saline+0.5% DMSO), Xn only (50 mg/kg, dissolved in 0.5% DMSO), LPS only (0.5 mg/kg, dissolved in saline), Xn (10 or 50 mg/kg)+LPS, and dexamethasone (Dex, as a positive drug, 5 mg/kg dissolved in saline)+LPS. In addition, Nrf2-/- mice were randomly divided into four groups: Control (saline+0.5% DMSO), Xn only (50 mg/kg, dissolved in 0.5% DMSO), LPS only (0.5 mg/kg, dissolved in saline), Xn (50 mg/kg)+LPS. In briefly, Xn (10 or 50 mg/kg) and Dex (5 mg/kg) were administered intraperitoneally. After 1 h, the mice were anesthetized with diethyl ether, and LPS was administered intranasally (i.n.) to induce lung injury. After LPS administration for 12 h, the animals were euthanized by CO2 asphyxiation. Subsequently, lung tissue samples and the bronchoalveolar lavage fluid (BALF) were collected and used for hematoxylin and eosin (H & E) staining, Western blot assay, enzyme-linked immunosorbent assay (ELISA) and flow cytometry assay.
2.4. Cell counting and protein concentration assay in bronchoalveolar lavage fluid (BALF)
After LPS challenge for 12 h, all mice were euthanized for collecting their BALF. The BALF samples were centrifuged to pellet the cells, lysed by ACK Lysis Buffer for 5 min, washed twice with ice-cold PBS and collected, and then again centrifuged for another 5 min, 4 °C. Subsequently, the sedimented cells were resuspended in PBS to obtain the total counts of cells, neutrophils and macrophages were counted using a hemocytometer, and the Wright-Giemsa staining method was used for cytosine staining. In addition, the BALF samples were collected and untreated, and then their protein concentrations were directly measured using a BCA protein assay kit (Beyotime, China).
2.5. Histopathological evaluation
The left lungs of mice were excised at 12 h after the LPS challenge. A histopathological examination was performed on the mice that were not subjected to BALF collection. The lung tissue samples were immersed in normal 10% neutral buffered formalin and fixed for 48 h, dehydrated in a series of graded ethanol, embedded in paraffin wax, and cut into 5-μm-thick sections. The paraffin-embedded sections were stained with hematoxylin and eosin (H & E) for pathological analysis.
2.6. Measurement of MPO, MDA, GSH and SOD levels in lung tissues
All mice were sacrificed using CO2 asphyxiation, and the right lungs were excised after 12 h of LPS-administration. The lung tissues were homogenized and dissolved in extraction buffer for the analysis of MPO, MDA, SOD and GSH content. To examine the accumulation of neutrophils and level of lipid peroxidation in the lung tissue, MPO and MDA content was assessed using commercially available assay kits in accordance with the respective manufacturer's instructions. Furthermore, to further measure the antioxidative enzyme activities in the lung tissue, SOD and GSH levels were detected following the respective manufacturer's instructions.
2.7. Lung wet/dry (W/D) ratios
Lung samples were collected 12 h after LPS stimulation, blotted dry and weighed immediately after removal (wet weight) before being subjected to desiccation in an oven at 80 °C for 48 h to obtain the ‘dry’ weight. The ratio of wet lung weight to dry lung weight was measured by assessment of tissue edema.
2.8. Cell culture and MTT assay
The RAW 264.7 cells (a macrophage line of mouse originated from the China Cell Line Bank, Beijing, China), was cultured in DMEM medium supplemented with 10% fetal bovine serum (FBS), 100 U/mL of penicillin, 100 U/mL of streptomycin and 3 mM glutamine at 37 °C in a humidified atmosphere containing 5% CO2. In all experiments, cells were allowed to acclimate for 24 h before any treatments.
Cell viability was measured by an MTT assay in accordance with the manufacturer's instructions. RAW 264.7 cells were seeded into 96-well plates (2×104 cells/well) for 24 h and treated with different concentrations of Xn (final concentration: 0, 0.625, 1.25, 2.5, 5, 10 or 20 μM) for 1 h, and then exposed to LPS (1 μg/mL) for 24 h. Subsequently, MTT (5 mg/mL) was added to the cells, which were then incubated for an additional 4 h. DMSO was added to dissolve the formazan crystals, and the absorbance was measured at 570 nm.
2.9. Measurement of ROS production
After the BALF samples were lysed by ACK Lysis Buffer, the sedimented cells were resuspended in PBS for detecting ROS generation. Briefly, the cells were stained with 50 μM of DCFH-DA at 37 °C in a dark place for 30 min. DCF fluorescence intensities were detected by flow cytometry and a multi-detection reader (Bio-Tek Instruments Inc.) at an excitation and emission wavelength of 485 nm and 535 nm, respectively. Moreover, for intracellular ROS measurement, RAW 264.7 cells were seeded into 96-well plates (2×104 cells/well) for 24 h incubation, and then recovered in serum-free DMEM and pretreated with Xn for 1 h. Subsequently, cells were subjected to LPS (1 μg/mL) for additional 24 h and washed with PBS twice for DCFH-DA staining. Additionally, RAW 264.7 cells were pretreated with Xn for 18 h and stained with DCFH-DA 30 min prior to stimulation with t-BHP.
2.10. ELISA assay
The BALF was obtained from each sample in vivo, centrifuged, collected supernatants for measurement of the TNF-α, IL-6 and IL-1β secretion using an enzyme-linked immunosorbent assay (ELISA) kit as the manufacturer's instructions (BioLegend, Inc., CA, USA), respectively. In addition, RAW 264.7 macrophages were grown in 24-well plates (2×105 cells/well) for 1 h, and then treated with Xn 1 h prior to exposure to LPS (1 μg/mL). After LPS-stimulation for 24 h, the cell-free supernatants were collected for analysis of the TNF-α, IL-6 and IL-1β secretion. The optical density from each well was detected at 450 nm.
2.11. Apoptosis quantification
RAW 264.7 cells were plated into a 12-well plate (4×105 cells/well) for 24 h and were then pretreated with Xn for 18 h followed by exposure to t-BHP (5 mM) for an additional 30 min. Cells were washed twice with ice-cold PBS, collected and centrifuged at 1500 rpm for 5 min at 4 °C. Next, cells were examined with FITC-labeled annexin V and propidium iodide staining, and the percentages of apoptosis and necrosis were detected using flow cytometry.
2.12. Isolation of nuclear and cytosolic fractions
The cytoplasmic and the nuclear extracts were prepared using an NE-PER Nuclear and Cytoplasmic Extraction Reagents kit (Pierce Biotechnology, Rockford, IL, USA), following the manufacturer's instructions. All steps were performed from on ice or at 4 °C.
2.13. ARE promoter activity
RAW 264.7 cells were seeded in 96-well plates (2×104 cells/well) for incubation. When cell density reached approximately 75% confluence, pGL4.37 and pGL4.74 plasmids were transfected into cells using Viafect transfection reagent in accordance with the manufacturer's protocol (Invitrogen, Carlsbad, CA, USA). After Xn (1.25, 2.5 or 5 μM) treatment for 6 h, we exploited a dual-luciferase reporter assay system (Dual-Glo® Luciferase Assay System) for detecting and analyzing ARE-driven promoter activity.
2.14. Western blot analysis
Lung tissue samples or cells were lysed in a RIPA buffer with protease and phosphatase inhibitors for 30 min. The protein concentrations were measured using a BCA protein assay kit (Beyotime, China), and 40 μg of proteins were electrophoretically transferred onto a PVDF membrane following separation on a 10% SDS-polyacrylamide gel. The membrane was blocked with blocking solution (5% (w/v) nonfat dry milk) for 1 h, followed by an overnight incubation at 4 °C with a specific primary antibody. The following day, the membrane was incubated for an additional 1 h with HRP-conjugated secondary antibody (1:5000 dilution) at room temperature after thoroughly washing three times with PBST. Bands were detected by ECL (Amersham Pharmacia Biotech, Piscataway, NJ) and band intensities were quantified using Image J gel analysis software. All experiments were performed in triplicate.
2.15. Statistical analysis
All data referenced above were expressed as the means±SEM and analyzed using SPSS19.0 (IBM). Comparisons between experimental groups were conducted using one-way ANOVA, whereas multiple comparisons were made using the LSD method. Statistical significance was defined as p<0.05 or p<0.01.
3. Results
3.1. Xn treatment inhibited inflammatory responses and alleviated LPS-induced ALI mice
The histological changes of lung tissues were assessed by light microscopy, as shown in Fig. 1C, LPS induced evidently pathologic changes by increasing accumulation of inflammatory cells and alveolar hemorrhage, when compared with control group and Xn-treated alone group. However, LPS-induced severe histopathological changes were obviously weakened by pretreatment of Xn or Dex, which was measured by the lung injury score (Fig. 1D). Additionally, the severity of edema formation was educated by W/D ratios of lung and protein leakage in BALF. As illustrated in Fig. 1E–F, LPS-stimulated W/D ratio and protein concentration were significantly higher than that of the normal group and Xn-treated alone group. In contrast, pretreatment with Xn and Dex remarkably decreased lung edema and protein leakage. Moreover, to analyze the effects of Xn on LPS-induced inflammatory responses, the secretion of TNF-α, IL-6 and IL-1β in BALF were measured by ELISA as well as the expression of iNOS, COX-2 and HMGB1 protein were determined by Western blot. Our results suggested that Xn pretreatment not only effectively reduced TNF-α, IL-6 and IL-1β production, but also markedly inhibited expression of iNOS and HMGB1 proteins, whereas the expression of COX-2 protein did not exhibit an apparent decrease (Fig. 1G–I).
3.2. Xn treatment reduced total cell number, neutrophils and MPO levels in LPS-induced ALI mice
The BALF from mice exposed to LPS contained more total cells, neutrophils and macrophages were assayed by Wright-Giemsa staining methods. As illustrated in Fig. 2A–C, compared with the control group, LPS exposure to mice dramatically increased the number of total cells and neutrophils, whereas pretreated with Xn and Dex effectively inhibited LPS-induced the increased of these cells. Moreover, increased MPO activity represents polymorphonuclear leukocyte accumulation in the lungs. Analysis of MPO activity was detected according to the manufacturer's instruction. This result indicated that both Xn and Dex significantly decreased the increase in MPO activity that was measured in the LPS-only group (Fig. 2D).
Fig. 2.

Effects of Xn treatment on inflammatory cell count and MPO levels in LPS-induced ALI mice. After LPS challenge for 12 h, all mice were euthanized and their BALF and lungs were collected. (A–C) The total counts of cells, macrophages and neutrophils from the BALF were counted using a hemocytometer, and the Wright-Giemsa staining method was used for cytosine staining (magnification ×400, red arrows: macrophages; green arrows: neutrophils). (D) MPO activity in lung tissues was measured at 12 h after LPS challenge. Similar results were obtained from three independent experiments. All data are presented as means±SEM (n=5 in each group). ##p<0.01 vs Control group; *p<0.05 and **p<0.01 vs LPS group. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.3. Xn treatment alleviated LPS-stimulated oxidative stress in ALI mice
Due to oxidative damage plays a key role in LPS-induced ALI mice, we examined whether Xn pretreatment could inhibit LPS-triggered oxidative stress. Indeed, as shown in Fig. 3, Xn pretreatment not only could dramatically reduce LPS-induced ROS generation and MDA formation, but also obviously lessen SOD and GSH depletion, two antioxidant enzymes which are essential for preventing LPS-induced ALI against oxidative stress.
Fig. 3.

Effects of Xn treatment on LPS-triggered oxidative stress in ALI mice. Xn (10 or 50 mg/kg) was administered intraperitoneally to mice 1 h prior to LPS (0.5 mg/kg) pretreatment, BALF and lungs of mice were collected. (A) ROS generation in BALF was determined using flow cytometry. (B) ROS production in the BALF was analyzed by Fluorence microplate reader. (C–E) Effects of Xn on levels of MDA, GSH and SOD from lung homogenates. Similar results were obtained from three independent experiments. All data are presented as means±SEM (n=5 in each group). ##p<0.01 vs Control group; *p<0.05 and **p<0.01 vs LPS group.
3.4. Xn treatment suppressed LPS-activated Txinp-NLRP3 inflammasome and NF-κB signaling pathways in ALI mice
Given that Xn could effectively decreased LPS-induced ROS generation responsible for activation of Txnip, an inhibitor protein of Trx which participates in NLRP3 inflammasome activation, the study further investigated whether Xn could block Txinp-NLRP3 inflammasome activation. This result indicated that LPS-challenge evidently promoted expression of Txnip, NLRP3, ASC, Caspase-1 and IL-1β protein as well as reduction of Trx-1 protein, whereas these effects were significantly blunted by Xn pretreatment in LPS-stimulated ALI mice and the dosage of 50 mg/kg Xn displayed the more effective than 10 mg/kg (Fig. 4A–F). Moreover, NF-κB signal pathway is a typical inflammatory pathway playing a vital role in LPS-induced ALI mice, our experiment also detected the effects of Xn pretreatment on LPS-activated NF-κB signal pathway. As presented in Fig. 4G–I, Xn pretreatment remarkably suppressed p65 phosphorylation, blocked IκBα phosphorylation and degradation in LPS-exposed ALI.
Fig. 4.

Effects of Xn treatment on LPS-activated Txinp-NLRP3 inflammasome and NF-κB signal pathways in ALI mice. Mice were given different concentrations of Xn (10 or 50 mg/kg) with an intraperitoneal injection 1 h prior to LPS (0.5 mg/kg) pretreatment and lungs of mice were collected. Protein samples were extracted from the lungs and analyzed by Western blot with specific antibodies. (A and D) Protein expressions of Txnip, Trx-1, NLRP3, ASC, caspase-1 and IL-1β were measured by Western blot analysis. (G) Protein expressions of P-p65, p65, P-IκBα, and IκBα were measured by Western blot analysis. (B, C, E, F, H and I) Quantification of relative protein expression was performed by densitometric analysis and β-actin was acted as an internal control. Similar results were obtained from three independent experiments. All data are presented as means±SEM (n=5 in each group). ##p<0.01 vs Control group; *p<0.05 and **p<0.01 vs LPS group.
3.5. Xn treatment induced phosphorylation of AMPK and GSK3β as well as upregulation of Nrf2 in LPS-induced ALI mice
To further investigate the protective mechanism of Xn treatment on LPS-induced ALI, activities of AMPK, GSK3β and Nrf2, which are responsible for improvement of lung injury, were analyzed by Western blot. Our results unveiled that LPS-challenge mildly increased the AMPK and GSK3β phosphorylation and enhanced the Nrf2 expression, whereas these effect were strengthened by Xn pretreatment (Fig. 5), indicating that the protective effects of Xn treatment on LPS-induced ALI may be associated with enhancing phosphorylation of AMPK and GSK3β as well as upregulation of Nrf2.
Fig. 5.

Effects of Xn treatment on AMPK, GSK3β phosphorylation and Nrf2 expression in LPS-induced ALI mice. Mice were given different concentrations of Xn (10 or 50 mg/kg) with an intraperitoneal injection 1 h prior to LPS (0.5 mg/kg) pretreatment and lungs of mice were collected. Protein samples were extracted from the lungs and analyzed by Western blot. Quantification of relative expression of P-AMPK/AMPK, P-GSK3β/GSK3β and Nrf2/β-actin were performed by densitometric analysis. Similar results were obtained from three independent experiments. All data are presented as means±SEM (n=5 in each group). ##p<0.01 vs Control group; *p<0.05 and **p<0.01 vs LPS group.
3.6. Xn exposure blocked LPS-induced inflammatory responses in RAW 264.7 cells
Based on these results in vivo, our further studies investigated whether Xn treatment could inhibit LPS-induced inflammatory responses in RAW 264.7 cells. In this study, the effects of different dosages of Xn and LPS (1 μg/mL) on cell viability were assessed using a MTT assay. The results indicated that LPS (1 μg/mL) plus Xn up to 10 μM were not toxic to RAW 264.7 cells (Fig. 6A). Thus, the dosages of 1.25, 2.5 or 5 μM Xn was chosen for exposure to RAW 264.7 cells for 1 h and then subjected with LPS (1 μg/mL) for another 24 h. As shown in Fig. 6B–C, RAW 264.7 cells were treated with Xn (5 μM) to determine the most effective dosage for inhibiting iNOS and HMGB1, whereas Xn had no significant effect on COX-2 protein expression. Moreover, treatment of RAW 264.7 cells with LPS alone significantly increased production of TNF-α, IL-6, IL-1β and ROS, whereas Xn (5 μM) considerably inhibited LPS-induced the TNF-α, IL-6, IL-1β and ROS generation (Fig. 6D–G). Because ROS generation triggers dissociation of Txnip from Trx and allowing it to bind and activate NLRP3 inflammasome activation which further leads to inflammatory responses. Our results suggested that Xn treatment markedly inhibited LPS-induced expression of Txnip, NLRP3, ASC, Caspase-1 and IL-1β protein and reduction of Trx-1 protein. In addition, NF-κB signaling pathway is one of typical inflammatory signaling pathways, we examined the effects of Xn treatment on LPS-induced NF-κB activation. The result suggested that the Xn pretreatment effectively reduced p65 phosphorylation, blocked IκBα phosphorylation and degradation in LPS-induced RAW 264.7 cells (Fig. 6H–P).
Fig. 6.

Effects of Xn exposure on LPS-induced inflammatory responses in RAW 264.7 cells. RAW 264.7 cells were subjected to various concentrations of Xn for 1 h, and then treated with LPS (1 μg/mL) for another 24 h. (A) Cell viability after LPS exposure was measured by an MTT assay. Cells were subjected to Xn (1.25, 2.5 or 5 μM) for 1 h and then exposed to LPS (1 μg/mL) for 24 h. (B-C) Effects of Xn on the iNOS, COX-2 and HMGB1 protein expression. Levels of TNF-α (D), IL-6 (E) and IL-1β (F) in culture supernatants were measured by ELISA. The ROS generation was measured by according to the Experimental Section. (G) Effect of Xn on LPS-triggered ROS production in RAW 264.7 cells. Additionally, cells were treated with Xn (5 μM) for 1 h and then exposed to LPS (1 μg/mL) for 6 h. (H, I, L, M, N, O and P) Effects of Xn on the Txnip and Trx-1, NLRP3, ASC, Caspase-1, and IL-1β protein expression. Moreover, cells were treated with Xn (5 μM) for 1 h and then exposed to LPS (1 μg/mL) for 45 min. (J-K) Effects of Xn on the P-IκBα, and IκBα protein expression and β-actin was acted as an internal control. Similar results were obtained from three independent experiments. All data are presented as means±SEM (n=5 in each group). ##p<0.01 vs Control group; *p<0.05 and **p<0.01 vs LPS group.
3.7. Xn exposure increased antioxidant enzymes expression to attenuate t-BHP-stimulated oxidative damage in RAW 264.7 cells
t-BHP, a chemical toxin, leads to a variety of cellular injuries by triggering oxidative stress in biological systems. Therefore, to further investigate the potential anti-oxidant effect of Xn, we used t-BHP-induced toxicity in RAW 264.7 cells. Our results suggested that Xn effectively reduced t-BHP-triggered ROS generation, GSH depletion and cell apoptosis in RAW 264.7 (Fig. 7A–E). Given that various antioxidative enzymes, including GCLC, GCLM, HO-1, NQO1 and Trx-1, enhance the resistance of cells to oxidative damage, these enzymes expression were measured by Western blot analysis. In our studies, Xn pretreatment obviously induced GCLC, GCLM, HO-1, NQO1 and Trx-1 protein expression in different dosages and periods (Fig. 7F–I).
Fig. 7.

Effects of Xn exposure ont-BHP-stimulated oxidative damage in RAW 264.7 cells. Cells were subjected to Xn (5 μM) for 18 h, and stained with 50 μM of DCFH-DA for 40 min, and subsequently exposed to t-BHP (5 mM) for 5 min to stimulate the ROS generation. (A–B) Effect of Xn on t-BHP-triggered ROS production in RAW 264.7 cells. In addition, Cells were pretreated with or without Xn (5 μM) for 18 h, then were exposed to t-BHP for additional 1 h. (C) Effect of Xn on t-BHP-stimulated GSH depletion was measured by using a commercial GSH test kit. (D–E) Effects of Xn on t-BHP-stimulated cell apoptosis was determined using flow cytometry. Moreover, (F–G) cells were exposed to Xn (5 μM) for four time points (1, 3, 6 or 18 h) and (H–I) treated with different concentrations of Xn (0.625, 1.25, 2.5 and 5 μM) for 18 h, and then protein expressions of GCLC, GCLM, HO-1, NQO1 and Trx-1 were measured by Western blot analysis. Similar results were obtained from three independent experiments. All data are presented as means±SEM (n=5 in each group). ##p<0.01 vs Control group; $$p<0.01 vs t-BHP group.
3.8. Xn exposure regulated the AMPK/GSK3β and Keap1-Nrf2/ARE signaling pathways in RAW 264.7 cells
Due to the Keap1-Nrf2/ARE signaling pathway plays a key role in regulating numerous antioxidant enzymes expression, we detected whether Xn treatment could upregulate the Keap1-Nrf2/ARE signaling pathway. Our investigations showed that Xn efficiently induced the total protein expression of Nrf2 and Keap1 degradation (Fig. 8A–C). Additionally, our results further suggested that 5 μM of Xn significantly resulted in an increase in the nuclear expression and a concomitant decrease in the cytoplasmic expression of Nrf2 (Fig. 8D–E). Furthermore, because ARE activation is essential for increasing Nrf2 nuclear transcription, Xn-induced changes in luciferase activity acted as a measure of ARE activation. The result discovered that Xn also effectively strengthened ARE-driven luciferase activity in a time-dependent manner (Fig. 8F). Considered that Xn could induce activation of AMPK and inactivation of GSK3β in LPS-induced ALI, our further work explored whether Xn-upregulated Nrf2 is associated with AMPK/GSK3β signaling pathway. In the present study, consistent with results in vivo, Xn treatment significantly induced phosphorylation of AMPK and GSK3β in RAW 264.7 cells (G-I). To further investigate the linkage between Xn-mediated Nrf2 and AMPK/GSK3β, cells were pretreated with Compound C (an inhibitor of AMPK) or Brusatol (an inhibitor of Nrf2). Subsequently, we asked the effect of two inhibitors on Nrf2 expression and AMPK and GSK3β phosphorylation, finding that Xn-mediated Nrf2 activation and GSK3β phosphorylation was effectively blocked by AMPK inhibitor. However, Nrf2 inhibitor had no a significant effect on Xn-mediated AMPK and GSK3β phosphorylation, indicating that AMPK may act as upstream of Nrf2 (Fig. 8J–M). Moreover, we further clarify effects of Xn on Nrf2 expression and AMPK and GSK3β phosphorylation in LPS-induced RAW 264.7. As presented in Fig. 8N–Q, 1 μg/mL of LPS exposure has almost no effect on Nrf2 expression, AMPK and GSK3β phosphorylation, whereas Xn treatment could evidently induced Nrf2 expression, AMPK and GSK3β phosphorylation.
Fig. 8.

Effects of Xn exposure on AMPK/GSK3β and Keap1-Nrf2/ARE signaling pathways in RAW 264.7 cells. (A–C) Cells were treated with Xn (5 μM) for four time points (1, 3, 6 or 18 h), and the total protein of Keap1 and Nrf2 were measured by Western blot analysis. (D-F) Cells were treated with Xn (5 μM) for three time points (1, 3 or 6 h), and the nuclear and cytoplasmic levels of Nrf2 were examined by Western blot analysis. (H) Effect of Xn (5 μM) on ARE luciferase activity was determined by a dual-luciferase reporter assay system. (G-I) Cells were subjected to Xn (5 μM) for four time points (0.5, 1, 3 or 6 h), and effects of Xn on AMPK and GSK3β phosphorylation were measured by Western blot analysis. (J–M) Cells were exposed to CC (Compound C: an inhibitor of AMPK, 3 μM) for 18 h or BT (Brusatol: an inhibitor of Nrf2, 200 nM) for 1 h and then treated with Xn (5 μM) for 3 h. Effects of Xn on AMPK and GSK3β phosphorylation as well as on the expressions of Nrf2 and Trx-1 protein. Moreover, cells were exposed to LPS (1 μg/mL) for 1 h, and were treated with Xn (5 μM) for 3 h. (N–Q) Effects of Xn on AMPK and GSK3β phosphorylation and Nrf2 protein expression. Similar results were obtained from three independent experiments. All data are presented as means±SEM (n=5 in each group). ##p<0.01 vs Control group; *p<0.05 and **p<0.01 vs LPS group; ++p<0.01 vs Xn only group.
3.9. Xn treatment protected from LPS-induced ALI and Nrf2 dependency
To ascertain whether the protective effect of Xn-displayed against LPS-induced ALI is dependent upon Nrf2 activation, WT mice and Nrf2-/- mice were conducted. As shown in Fig. 9, Xn treatment markedly inhibited production of TNF-α, IL-6, IL-1β and ROS in BALF of WT mice, but significantly blocked in BALF of Nrf2-/- mice. Moreover, Xn treatment effectively attenuated severe histopathological changes in WT mice were obviously abrogated in Nrf2-/- mice. Importantly, Xn treatment mediated increases of Nrf2 and Trx-1 as well as decreases of Txnip and NLRP3 protein expressions in WT mice were significantly inhibited in Nrf2-/- mice. Furthermore, Xn treatment remarkably inhibited IκBα phosphorylation and degradation in WT mice were evidently impeded in Nrf2-/- mice. Taken together, our experimental results revealed that Xn plays an important role in the attenuation of LPS-induced oxidative stress and inflammation damage of lungs via inhibiting NF-κB and Txinp/NLRP3 inflammasome activation, which may be dependent upon upregulation of Nrf2.
Fig. 9.

Protective effects of Xn-meditated Nrf2 on LPS-induced ALI mice. WT and Nrf2-/- mice were intraperitoneally injected with Xn (50 mg/kg) prior to LPS (0.5 mg/kg) pretreatment. (A–D) Levels of cytokines (TNF-α, IL-6 and IL-1β) secretion and ROS generation in BALF were measured by ELISA. (E–F) Lungs (n=5) from each experimental group were processed for histological evaluation at 12 h after the LPS challenge (H & E staining, magnification ×100). (G–M) Effects of Xn on expressions of Nrf2, Txnip, Trx-1, NLRP3, P-IκBα and IκBα protein. Similar results were obtained from three independent experiments. All data are presented as means±SEM (n=5 in each group). ##p<0.01 vs Control group; *p<0.05 and **p<0.01 vs LPS group.
4. Discussion
Excessive oxidative stress and/or overwhelmed inflammatory responses are thought to play essential roles in the pathogenesis of ALI [3], [31]. Oxidative stress can exaggerate proinflammatory gene expression, and inflammatory cells can similarly trigger overproduction of reactive oxygen species (ROS), which creates a vicious cycle to provoke the occurrence and development of various diseases, including ALI [32], [33]. Furthermore, Nrf2 has been suggested to play a key coordinator as protecting cells against oxidative and inflammatory insults [34]. Xanthohumol (Xn) has been shown to induce Nrf2 activation in human hepatocytes and PC12 cells [30], [35], and display various biological activities, such as anti-inflammatory and antioxidant properties in streptozotocin-induced diabetic Wistar rats [28]. In the present study, we aimed to investigating whether Xn could mediate Nrf2 signaling pathway, conferring an antioxidant and anti-inflammatory properties to prevent from LPS-induced ALI mice.
Accumulating evidences shows that multiple pathological processes, such as capillary permeability increase, extensive neutrophil infiltration, inflammatory mediators release and edema, are displayed in the pathogenesis of ALI [36]. Our results indicated that Xn alleviated LPS-induced pulmonary edema, coagulation and inflammation cells, reduced neutrophil numbers and MPO activity. Moreover, elevation of lung W/D ratio and protein level could account for an increased pulmonary permeability and lung edema in LPS-instillation lungs [37], which was markedly decreased by Xn pretreatment. Moreover, macrophages activation and neutrophils accumulation in the lung, which not only boost inflammatory cytokines and cells release, but also enhance ROS production, are nearly correlated to the severity of ALI [38], [39]. It is reported that LPS-induced an ALI model is similar with pathological features to ALI in humans by triggering excessive inflammatory mediators secretion, chemokines release and ROS generation [40], [41]. Long-term inflammatory mediators, including TNF-α, IL-6, IL-1β, iNOS, and COX-2, are strongly related to the development of acute and chronic inflammation diseases [42], [43]. HMGB1, a late inflammatory cytokine, irritates the inflammation cascade [44], which aggravates various human diseases. These inflammatory mediators production at the site of injury is one of the hallmarks of ALI. Our findings indicated that Xn treatment effectively reduced TNF-α, IL-6 and IL-1β secretion as well as suppressed HMGB1 and iNOS protein expression in LPS-induced RAW cells and ALI mice. Moreover, the instillation of LPS into the lungs can lead to excessive ROS accumulation and MDA formation, which are reduced by antioxidant enzymes SOD and GSH, protecting various cells against oxidative damage [45], [46]. In our experiments, Xn treatment dramatically induced an increase of SOD and GSH contents and caused a decrease of the levels of MDA and ROS in LPS-induced ALI mice. Our further experimental results discovered that t-BHP significantly induced cell apoptosis, ROS accumulation and GSH depletion, whereas Xn treatment effectively inhibited t-BHP-stimulated these effects in RAW 264.7 cells. Taken together, these investigations implied that Xn is able to mitigate inflammation- and oxidative stress-induced damage in vivo and in vitro.
Based on the above outcome, our further studies explored the mechanism of the protective effect of Xn against LPS-induced inflammation and oxidative stress in ALI mice. Previous abundant reports suggested that NF-κB-mediated various inflammation mediators play a vital role in the pathogenesis of ALI [47]. In this study, Xn treatment efficiently inhibited NF-κB (p65) and IκBα phosphorylation, blocked IκBα degradation in LPS-induced RAW 264.7 cells and ALI mice. Importantly, recent several reports revealed that Txnip is a key point linking oxidative stress to inflammation. In response to ROS, Txnip detaches from Trx and binds to NLRP3, which leads to NLRP3 inflammasome activation [16]. LPS-challenge excited oxidative stress and triggered ROS generation in the lung, following Txnip induction and interaction of Txnip-NLRP3 [48]. Our results similarly discovered that LPS instillation obviously enhanced Txnip protein expression, inhibited Trx-1 protein expression and induced NLRP3 inflammasome activation through increasing the expression of NLRP3, ASC, Caspase-1 and IL-1β protein in the lung and RAW 264.7 cells, whereas these LPS-exhibited effects were blocked by Xn pretreatment. Collectively, these results suggested that Xn-inhibited inflammatory responses may be associated with the suppression of NF-κB and Txnip/NLRP3 inflammasome activation.
In fact, Nrf2, as a multiple signaling pathways coordinator, attenuated hyperoxia-induced ALI by inhibition of inflammation and oxidative stress [49], [50]. In this study, Xn treatment could effectively upregulated Nrf2 protein expression LPS-induced ALI, indicating that the Xn-exhibited protection effect on LPS-induced ALI may be closely correlated to activation of Nrf2. Hence, to further explore the mechanism of Xn-mediated Nrf2 activation, we observed the effect of Xn on the Keap1-Nrf2/ARE antioxidant pathway and regulation of antioxidant enzymes in RAW 264.7 cells. Previous reports showed that activated-Nrf2 is released from Keap1 and translocated into the nucleus, whereupon it sequentially binds to ARE in the promoter region of its target genes, resulting in expressions of various cytoprotective genes and antioxidative enzymes [51]. Our results demonstrated that Xn apparently induced a decrease of Keap1 and an increase of Nrf2 in total cell lysates, which were related to enhance the nuclear translocation of Nrf2 and heighten ARE luciferase activity in a dose-dependent manner in RAW 264.7 cells. Additionally, our findings indicated that various dosages and periods of Xn treatment effectively enhanced expressions of GCLC, GCLM, HO-1, NQO1, and Trx-1 protein in RAW 264.7 cells. More intriguingly, increasing evidences indicates that AMPK, which leads to accumulation of Nrf2 nuclear transcription through phosphorylation in addition to GSK3β inhibition, attenuates LPS-induced ALI [24], [26]. In our study, Xn treatment could significantly resulted in the phosphorylation of AMPK and GSK3β in RAW 264.7 cells and mice. Therefore, to further relationship between AMPK and Nrf2 activated by Xn, cells were pretreatment with Compound C or Brusatol (an inhibitor of AMPK and Nrf2, respectively). Western blot analysis showed that Xn-induced p-AMPK, p-GSK3β and Nrf2 protein expression were mostly blocked by Compound C exposure, whereas Brusatol pretreatment only inhibited Nrf2 protein expression. This results indicated that AMPK may act upstream of Nrf2 in Xn-induced AMPK and Nrf2 activation. Given all of these results, to further illuminate whether Xn-exhibited anti-oxidant and anti-inflammation activities in LPS-induced ALI is directly associated with Nrf2 activation, Nrf2 deficient mice were used as a tool for investigating an underlying connection. Our findings noticed that both Xn-inhibited production of TNF-α, IL-6, IL-1β and ROS and Xn-attenuated severe histopathological changes in WT mice were effectively abrogated in Nrf2-/- mice. Moreover, Xn-mediated increases of Nrf2 and Trx-1 as well as decreases of Txnip and NLRP3 protein expressions in WT mice were significantly inhibited in Nrf2-/- mice. In addition to these, Xn-inhibited IκBα phosphorylation and degradation in WT mice were evidently blocked in Nrf2-/- mice. Collectively, our experimental results provided a support that Xn are crucial for inhibition of LPS-induced oxidative stress and inflammation damage of lungs, which may be associated with upregulation of Nrf2 pathway dependent upon AMPK activation.
In conclusion, as illustrated in Fig. 10, our findings firstly demonstrated that Xanthohumol (Xn) effectively protected acute lung injury against oxidative stress and inflammation damage which was largely dependent on the upregulation of the Nrf2 pathway via activation of AMPK and inhibition of GSK3β, thereby suppressing LPS-activated Txnip/NLRP3 inflammasome and NF-κB signaling pathway. This study provides beneficial evidence for the application of Xn in the prevention of inflammation- and oxidative stress-associated diseases, especially ALI.
Fig. 10.

Scheme summarizing the protective effects of Xn on LPS-induced acute lung injury via activation of the AMPK/GSK3β-Nrf2 axis. Xanthohumol (Xn) treatment effectively protected LPS-induced acute lung injury against oxidative stress and inflammation damage which is largely dependent on upregulation of the Nrf2-mediated anti-oxidant pathway via activation of AMPK/GSK3β, thereby suppressing LPS-activated Txnip/NLRP3 inflammasome and NF-κB signaling pathway.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content of this manuscript.
Acknowledgments
This work was in part supported by the National Natural Science Foundation of China (Grant no. 81603174) and the General Financial Grant from the China Postdoctoral Science Foundation (Grant no. 168847).
References
- 1.Ware L.B., Matthay M.A. The acute respiratory distress syndrome. N. Engl. J. Med. 2000;342:1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 2.Ortiz-Diaz E., Festic E., Gajic O., Levitt J.E. Emerging pharmacological therapies for prevention and early treatment of acute lung injury. Semin. Respir. Crit. Care Med. 2013;34:448–458. doi: 10.1055/s-0033-1351118. [DOI] [PubMed] [Google Scholar]
- 3.Tasaka S., Amaya F., Hashimoto S., Ishizaka A. Roles of oxidants and redox signaling in the pathogenesis of acute respiratory distress syndrome. Antioxid. Redox Signal. 2008;10:739–753. doi: 10.1089/ars.2007.1940. [DOI] [PubMed] [Google Scholar]
- 4.Imai Y., Kuba K., Neely G.G., Yaghubian-Malhami R., Perkmann T., van Loo G., Ermolaeva M., Veldhuizen R., Leung Y.H., Wang H., Liu H., Sun Y., Pasparakis M., Kopf M., Mech C., Bavari S., Peiris J.S., Slutsky A.S., Akira S., Hultqvist M., Holmdahl R., Nicholls J., Jiang C., Binder C.J., Penninger J.M. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell. 2008;133:235–249. doi: 10.1016/j.cell.2008.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang C.H., Yang M.L., Tsai C.H., Li Y.C., Lin Y.J., Kuan Y.H. Ginkgo biloba leaves extract (EGb 761) attenuates lipopolysaccharide-induced acute lung injury via inhibition of oxidative stress and NF-kappaB-dependent matrix metalloproteinase-9 pathway. Phytomedicine. 2013;20:303–309. doi: 10.1016/j.phymed.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 6.Su Z.Q., Mo Z.Z., Liao J.B., Feng X.X., Liang Y.Z., Zhang X., Liu Y.H., Chen X.Y., Chen Z.W., Su Z.R., Lai X.P. Usnic acid protects LPS-induced acute lung injury in mice through attenuating inflammatory responses and oxidative stress. Int. Immunopharmacol. 2014;22:371–378. doi: 10.1016/j.intimp.2014.06.043. [DOI] [PubMed] [Google Scholar]
- 7.Jeyaseelan S., Chu H.W., Young S.K., Worthen G.S. Transcriptional profiling of lipopolysaccharide-induced acute lung injury. Infect. Immun. 2004;72:7247–7256. doi: 10.1128/IAI.72.12.7247-7256.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li C., Yang D., Cao X., Wang F., Jiang H., Guo H., Du L., Guo Q., Yin X. LFG-500, a newly synthesized flavonoid, attenuates lipopolysaccharide-induced acute lung injury and inflammation in mice. Biochem. Pharmacol. 2016;113:57–69. doi: 10.1016/j.bcp.2016.05.007. [DOI] [PubMed] [Google Scholar]
- 9.Goodman R.B., Pugin J., Lee J.S., Matthay M.A. Cytokine-mediated inflammation in acute lung injury. Cytokine Growth Factor Rev. 2003;14:523–535. doi: 10.1016/s1359-6101(03)00059-5. [DOI] [PubMed] [Google Scholar]
- 10.Hayden M.S., Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 11.Collins T., Cybulsky M.I. NF-kappaB: pivotal mediator or innocent bystander in atherogenesis? J. Clin. Investig. 2001;107:255–264. doi: 10.1172/JCI10373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lv H., Yu Z., Zheng Y., Wang L., Qin X., Cheng G., Ci X. Isovitexin exerts anti-inflammatory and anti-oxidant activities on lipopolysaccharide-induced acute lung injury by inhibiting MAPK and NF-kappaB and activating HO-1/Nrf2 pathways. Int. J. Biol. Sci. 2016;12:72–86. doi: 10.7150/ijbs.13188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dong W.W., Liu Y.J., Lv Z., Mao Y.F., Wang Y.W., Zhu X.Y., Jiang L. Lung endothelial barrier protection by resveratrol involves inhibition of HMGB1 release and HMGB1-induced mitochondrial oxidative damage via an Nrf2-dependent mechanism. Free Radic. Biol. Med. 2015;88:404–416. doi: 10.1016/j.freeradbiomed.2015.05.004. [DOI] [PubMed] [Google Scholar]
- 14.Korkmaz A., Kolankaya D. Protective effect of rutin on the ischemia/reperfusion induced damage in rat kidney. J. Surg. Res. 2010;164:309–315. doi: 10.1016/j.jss.2009.03.022. [DOI] [PubMed] [Google Scholar]
- 15.Kuo M.Y., Liao M.F., Chen F.L., Li Y.C., Yang M.L., Lin R.H., Kuan Y.H. Luteolin attenuates the pulmonary inflammatory response involves abilities of antioxidation and inhibition of MAPK and NFkappaB pathways in mice with endotoxin-induced acute lung injury. Food Chem. Toxicol. 2011;49:2660–2666. doi: 10.1016/j.fct.2011.07.012. [DOI] [PubMed] [Google Scholar]
- 16.Zhou R., Tardivel A., Thorens B., Choi I., Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010;11:136–140. doi: 10.1038/ni.1831. [DOI] [PubMed] [Google Scholar]
- 17.Lee S., Kim S.M., Lee R.T. Thioredoxin and thioredoxin target proteins: from molecular mechanisms to functional significance. Antioxid. Redox Signal. 2013;18:1165–1207. doi: 10.1089/ars.2011.4322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jiang L., Zhang L., Kang K., Fei D., Gong R., Cao Y., Pan S., Zhao M., Zhao M. Resveratrol ameliorates LPS-induced acute lung injury via NLRP3 inflammasome modulation. Biomed. Pharmacother. 2016;84:130–138. doi: 10.1016/j.biopha.2016.09.020. [DOI] [PubMed] [Google Scholar]
- 19.dos Santos G., Kutuzov M.A., Ridge K.M. The inflammasome in lung diseases. Am. J. Physiol. Lung Cell Mol. Physiol. 2012;303:L627–L633. doi: 10.1152/ajplung.00225.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rangasamy T., Guo J., Mitzner W.A., Roman J., Singh A., Fryer A.D., Yamamoto M., Kensler T.W., Tuder R.M., Georas S.N., Biswal S. Disruption of Nrf2 enhances susceptibility to severe airway inflammation and asthma in mice. J. Exp. Med. 2005;202:47–59. doi: 10.1084/jem.20050538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Walters D.M., Cho H.Y., Kleeberger S.R. Oxidative stress and antioxidants in the pathogenesis of pulmonary fibrosis: a potential role for Nrf2. Antioxid. Redox Signal. 2008;10:321–332. doi: 10.1089/ars.2007.1901. [DOI] [PubMed] [Google Scholar]
- 22.Kensler T.W., Wakabayashi N., Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharm. Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 23.Niso-Santano M., Gonzalez-Polo R.A., Bravo-San Pedro J.M., Gomez-Sanchez R., Lastres-Becker I., Ortiz-Ortiz M.A., Soler G., Moran J.M., Cuadrado A., Fuentes J.M., Centro de Investigacion Biomedica en Red sobre Enfermedades N. Activation of apoptosis signal-regulating kinase 1 is a key factor in paraquat-induced cell death: modulation by the Nrf2/Trx axis. Free Radic. Biol. Med. 2010;48:1370–1381. doi: 10.1016/j.freeradbiomed.2010.02.024. [DOI] [PubMed] [Google Scholar]
- 24.Joo M.S., Kim W.D., Lee K.Y., Kim J.H., Koo J.H., Kim S.G. AMPK facilitates nuclear accumulation of Nrf2 by phosphorylating at serine 550. Mol. Cell Biol. 2016;36:1931–1942. doi: 10.1128/MCB.00118-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carling D., Thornton C., Woods A., Sanders M.J. AMP-activated protein kinase: new regulation, new roles? Biochem. J. 2012;445:11–27. doi: 10.1042/BJ20120546. [DOI] [PubMed] [Google Scholar]
- 26.Zhao X., Zmijewski J.W., Lorne E., Liu G., Park Y.J., Tsuruta Y., Abraham E. Activation of AMPK attenuates neutrophil proinflammatory activity and decreases the severity of acute lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 2008;295:L497–L504. doi: 10.1152/ajplung.90210.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yeh C.H., Yang J.J., Yang M.L., Li Y.C., Kuan Y.H. Rutin decreases lipopolysaccharide-induced acute lung injury via inhibition of oxidative stress and the MAPK-NF-kappaB pathway. Free Radic. Biol. Med. 2014;69:249–257. doi: 10.1016/j.freeradbiomed.2014.01.028. [DOI] [PubMed] [Google Scholar]
- 28.Costa R., Negrao R., Valente I., Castela A., Duarte D., Guardao L., Magalhaes P.J., Rodrigues J.A., Guimaraes J.T., Gomes P., Soares R. Xanthohumol modulates inflammation, oxidative stress, and angiogenesis in type 1 diabetic rat skin wound healing. J. Nat. Prod. 2013;76:2047–2053. doi: 10.1021/np4002898. [DOI] [PubMed] [Google Scholar]
- 29.Zimmermann K., Baldinger J., Mayerhofer B., Atanasov A.G., Dirsch V.M., Heiss E.H. Activated AMPK boosts the Nrf2/HO-1 signaling axis – a role for the unfolded protein response. Free Radic. Biol. Med. 2015;88:417–426. doi: 10.1016/j.freeradbiomed.2015.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yao J., Zhang B., Ge C., Peng S., Fang J. Xanthohumol, a polyphenol chalcone present in hops, activating Nrf2 enzymes to confer protection against oxidative damage in PC12 cells. J. Agric. Food Chem. 2015;63:1521–1531. doi: 10.1021/jf505075n. [DOI] [PubMed] [Google Scholar]
- 31.Matthay M.A., Ware L.B., Zimmerman G.A. The acute respiratory distress syndrome. J. Clin. Investig. 2012;122:2731–2740. doi: 10.1172/JCI60331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li S., Hong M., Tan H.Y., Wang N., Feng Y. Insights into the role and interdependence of oxidative stress and inflammation in liver diseases. Oxid. Med. Cell Longev. 2016;2016:4234061. doi: 10.1155/2016/4234061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu M., Cao F.L., Zhang Y.F., Shan L., Jiang X.L., An X.J., Xu W., Liu X.Z., Wang X.Y. Tanshinone IIA therapeutically reduces LPS-induced acute lung injury by inhibiting inflammation and apoptosis in mice. Acta Pharmcol. Sin. 2015;36:179–187. doi: 10.1038/aps.2014.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Balligand J.L. Reducing damage through Nrf-2. Cardiovasc. Res. 2013;100:1–3. doi: 10.1093/cvr/cvt204. [DOI] [PubMed] [Google Scholar]
- 35.Krajka-Kuzniak V., Paluszczak J., Baer-Dubowska W. Xanthohumol induces phase II enzymes via Nrf2 in human hepatocytes in vitro. Toxicol. In Vitro. 2013;27:149–156. doi: 10.1016/j.tiv.2012.10.008. [DOI] [PubMed] [Google Scholar]
- 36.Zhang X., Huang H., Yang T., Ye Y., Shan J., Yin Z., Luo L. Chlorogenic acid protects mice against lipopolysaccharide-induced acute lung injury. Injury. 2010;41:746–752. doi: 10.1016/j.injury.2010.02.029. [DOI] [PubMed] [Google Scholar]
- 37.Tsai C.L., Lin Y.C., Wang H.M., Chou T.C. Baicalein, an active component of Scutellaria baicalensis, protects against lipopolysaccharide-induced acute lung injury in rats. J. Ethnopharmacol. 2014;153:197–206. doi: 10.1016/j.jep.2014.02.010. [DOI] [PubMed] [Google Scholar]
- 38.Tsushima K., King L.S., Aggarwal N.R., De Gorordo A., D'Alessio F.R., Kubo K. Acute lung injury review. Intern. Med. 2009;48:621–630. doi: 10.2169/internalmedicine.48.1741. [DOI] [PubMed] [Google Scholar]
- 39.Grommes J., Soehnlein O. Contribution of neutrophils to acute lung injury. Mol. Med. 2011;17:293–307. doi: 10.2119/molmed.2010.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee W.L., Downey G.P. Neutrophil activation and acute lung injury. Curr. Opin. Crit. Care. 2001;7:1–7. doi: 10.1097/00075198-200102000-00001. [DOI] [PubMed] [Google Scholar]
- 41.Rojas M., Woods C.R., Mora A.L., Xu J., Brigham K.L. Endotoxin-induced lung injury in mice: structural, functional, and biochemical responses. Am. J. Physiol. Lung Cell Mol. Physiol. 2005;288:L333–L341. doi: 10.1152/ajplung.00334.2004. [DOI] [PubMed] [Google Scholar]
- 42.Murakami A., Ohigashi H. Targeting NOX, INOS and COX-2 in inflammatory cells: chemoprevention using food phytochemicals. Int. J. Cancer. 2007;121:2357–2363. doi: 10.1002/ijc.23161. [DOI] [PubMed] [Google Scholar]
- 43.Fairweather D., Rose N.R. Inflammatory heart disease: a role for cytokines. Lupus. 2005;14:646–651. doi: 10.1191/0961203305lu2192oa. [DOI] [PubMed] [Google Scholar]
- 44.Lin Y., Chen L., Li W., Fang J. Role of high-mobility group box-1 in myocardial ischemia/reperfusion injury and the effect of ethyl pyruvate. Exp. Ther. Med. 2015;9:1537–1541. doi: 10.3892/etm.2015.2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Victor V.M., Rocha M., Esplugues J.V., De la Fuente M. Role of free radicals in sepsis: antioxidant therapy. Curr. Pharm. Des. 2005;11:3141–3158. doi: 10.2174/1381612054864894. [DOI] [PubMed] [Google Scholar]
- 46.Ryter S.W., Kim H.P., Hoetzel A., Park J.W., Nakahira K., Wang X., Choi A.M. Mechanisms of cell death in oxidative stress. Antioxid. Redox Signal. 2007;9:49–89. doi: 10.1089/ars.2007.9.49. [DOI] [PubMed] [Google Scholar]
- 47.Kang J.L., Lee H.W., Lee H.S., Pack I.S., Chong Y., Castranova V., Koh Y. Genistein prevents nuclear factor-kappa B activation and acute lung injury induced by lipopolysaccharide. Am. J. Respir. Crit. Care Med. 2001;164:2206–2212. doi: 10.1164/ajrccm.164.12.2104017. [DOI] [PubMed] [Google Scholar]
- 48.Jiang L., Fei D., Gong R., Yang W., Yu W., Pan S., Zhao M., Zhao M. CORM-2 inhibits TXNIP/NLRP3 inflammasome pathway in LPS-induced acute lung injury. Inflamm. Res. 2016;65:905–915. doi: 10.1007/s00011-016-0973-7. [DOI] [PubMed] [Google Scholar]
- 49.Mann G.E., Forman H.J. Introduction to special issue on 'Nrf2 regulated redox signaling and metabolism in physiology and medicine. Free Radic. Biol. Med. 2015;88:91–92. doi: 10.1016/j.freeradbiomed.2015.08.002. [DOI] [PubMed] [Google Scholar]
- 50.Cho H.Y., Jedlicka A.E., Gladwell W., Marzec J., McCaw Z.R., Bienstock R.J., Kleeberger S.R. Association of Nrf2 polymorphism haplotypes with acute lung injury phenotypes in inbred strains of mice. Antioxid. Redox Signal. 2015;22:325–338. doi: 10.1089/ars.2014.5942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nguyen T., Sherratt P.J., Huang H.C., Yang C.S., Pickett C.B. Increased protein stability as a mechanism that enhances Nrf2-mediated transcriptional activation of the antioxidant response element. Degradation of Nrf2 by the 26 S proteasome. J. Biol. Chem. 2003;278:4536–4541. doi: 10.1074/jbc.M207293200. [DOI] [PubMed] [Google Scholar]