

Abstract
Objective
Anti-atherosclerotic effects of TNFα blockade in patients with systemic inflammatory states are not conclusively demonstrated, which suggests that effects depend on the cause of inflammation. Macrophage LRP1 and apoE contribute to inflammation through different pathways. We studied the anti-atherosclerosis effects of TNFα blockade in hyperlipidemic mice lacking either LRP1 (MΦLRP1−/−) or apoE from macrophages.
Approach and Results
Lethally irradiated LDLR−/− mice were reconstituted with bone marrow from either wild type (WT), MΦLRP1−/−, apoE−/−, or apoE−/−/MΦLRP1−/−(DKO) mice, and then treated with the TNFα inhibitor adalimumab while fed a western-type diet. Adalimumab reduced plasma TNFα concentration, suppressed blood ly6Chi monocytes levels and their migration into the lesion, and reduced lesion cellularity and inflammation in both WT→LDLR−/− and apoE−/−→LDLR−/− mice. Overall adalimumab reduced lesion burden by 52%–57% in these mice. Adalimumab reduced TNFα and blood ly6Chi monocytes levels in MΦLRP1−/−→LDLR−/− and DKO→LDLR−/− mice, but it did not suppress ly6Chi monocyte migration into the lesion or atherosclerosis progression.
Conclusions
Our results show, for the first time, that TNFα blockade exerts anti-atherosclerotic effects that are dependent on the presence of macrophage LRP1.
Keywords: Macrophage, Inflammation, TNFα, Atherosclerosis
Graphical abstract: Schematic representation of the effects of TNFα blockade and loss of macrophage LRP1 on atherogenesis
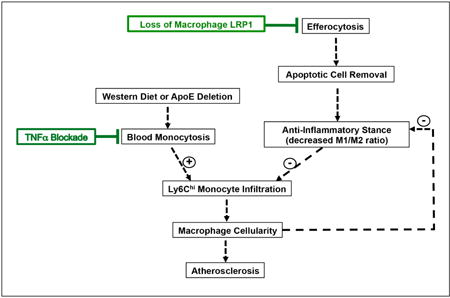
Western diet, alone or together with apoE deletion, increases circulating inflammatory monocytes. Efferocytosis causes apoptotic cell removal, limits M1 and promotes M2 macrophage polarization, reduces post-apoptotic necrosis, and improves inflammatory resolution. Both paths influence the ability of Ly6Chi monocytes to infiltrate the intima, rebalance plaque cellularity, and modulate atherogenesis. The TNFα antibody suppresses blood monocytosis but does not affect efferocytosis. Loss of macrophage LRP1 drastically impairs efferocytosis, thus promoting the maintenance of an inflammatory response that is not sensitive to the anti-atherosclerotic effects of TNFα blockade.
Introduction
Inflammation plays a fundamental role in all stages of atherosclerosis,1 from the early lesion to the complex plaque.2 Hyperlipidemia induces inflammation and is atherogenic. In addition, systemic inflammatory states such as rheumatoid arthritis (RA), confer predisposition to atherosclerosis.2 A meta-analysis of observational studies shows that RA patients have increased cardiovascular mortality compared with the general population.3 Apolipoprotein E (ApoE) induces an anti-inflammatory phenotype in mice by suppressing macrophage migration and chemokine secretion4. Deficiency of ApoE in macrophages causes systemic inflammation with monocytosis and promotes atherosclerosis.5–7 Local inflammation in the artery wall recruits monocyte/macrophages and drives atherosclerosis.1, 8 Cholesterol loading of macrophages in the atherosclerotic plaque activates macrophage cytokine secretion9 and causes endoplasmic reticulum stress and apoptosis of lesion foam cells10. These apoptotic macrophages are then cleared by active phagocytes in a process called efferocytosis.1, 8. Efficient efferocytosis prevents the release of inflammatory factors from dead cells and limits inflammation.11 In advanced atherosclerotic lesions with increased macrophage apoptosis, efferocytosis becomes inefficient. This inefficient efferocytosis increases cytokine secretion, amplifies inflammatory responses, eventually leading to plaque progression, disruption and atherothrombosis. 8
Macrophage LDL receptor-related protein 1 (LRP1) is a membrane receptor linked to inflammatory signaling and efferocytosis.12 LRP1 mediates the induction of focal adhesion disassembly13 and is involved in triggering engulfment during phagocytosis.14 LRP1 acts in conjunction with calreticulin serving as a recognition receptor to mediate apoptotic cell uptake.15–17 Lack of LRP1 impairs macrophage phagocytic function and impairs efferocytosis both in-vivo and in-vitro.18–20 Macrophage-specific deficiency of LRP1 (MΦLRP1−/−) increases atherosclerosis in hyperlipidemic mice, an effect associated with defective efferocytosis and increased inflammation.20–22
Tumor necrosis factor α (TNFα) is a master regulator of inflammatory responses which accelerates atherosclerosis.23 TNFα enhances murine hematopoietic stem and progenitor cell (HSPC) proliferation24 and macrophage differentiation,25 which may cause monocytosis and increase plaque burden.26 Anti-atherosclerotic effects of TNFα blockade in RA patients have been reported.27, 28 However, these effects are not conclusive.2 To investigate whether or not the protective role of TNFα blockade in atherosclerosis is dependent on the cause of inflammation, we used a commercially available therapeutic antibody (adalimumab) and evaluated its effects on atherosclerosis in LDLR−/− mice reconstituted with bone marrow from either wild-type (WT) mice or from mice carrying either macrophage deletion of LRP1 (MΦLRP1−/−), global deletion of apoE (apoE−/−), or both (DKO). We show that adalimumab suppresses TNFα levels and Ly6Chi monocytosis in all mice, but only reduces atherosclerotic burden in mice expressing macrophage LRP1. Mice without macrophage LRP1 showed defective efferocytosis and increased atherosclerosis burden, which were not improved by TNFα blockade.
Materials and Methods
Materials and Methods are available in the online-only Data Supplement.
Results
TNFα antibody reduces atherosclerosis in WT→LDLR−/−, but not in MΦLRP1−/−→LDLR−/− mice
To investigate the anti-atherosclerotic effects of TNFα blockade in different inflammation models, 12-week old female LDLR−/− mice received lethal irradiation and were reconstituted with bone marrow from WT or MΦLRP1−/− mice. Four weeks after bone marrow transplant (BMT), recipient mice were treated with the TNFα antibody adalimumab or human IgG (control) by intraperitoneal injection twice weekly for 10 weeks while on a western-type diet. Mice receiving MΦLRP1−/− bone marrow (MΦLRP1−/−→LDLR−/−) showed a 150% increase in atherosclerosis (P<0.001, Figure 1, A–B) compared with WT bone marrow recipients (WT→LDLR−/−). Plasma cholesterol levels were unchanged but TG levels were slightly increased in MΦLRP1−/−→LDLR−/− mice (Figure 1, D–E). TNFα blockade suppressed aortic sinus area atherosclerosis by 52% in WT→LDLR−/− (193.9±31.6 vs. 93.3±19.6 ×103μm2, P<0.05) but did not significantly change lesion size (296.1±18.9 vs. 319±30.2 ×103μm2) in MΦLRP1−/−→LDLR−/− mice (Figure 1, A–B). Adalimumab suppressed plasma TNFα levels in both groups of mice (Figure 1C), without affecting plasma cholesterol or TG levels (Figure 1, D–E).
Figure 1. Adalimumab limits atherosclerosis in WT→LDLR−/−, but not in MΦLRP1−/−→LDLR−/− mice.

After BMT, mice were treated with adalimumab or human IgG as described in Methods, and euthanized after 10 weeks of treatment and western-type diet. Cross-sections of aortic sinus area from WT→LDLR−/− and MΦLRP1−/−→LDLR−/− mice were stained with oil-red-O (A) and quantified (B). Plasma levels of TNFα (C), cholesterol (D), and triglycerides (E) in adalimumab treated and control mice. At least two sections were analyzed for each mouse, and all the mice (n=5–7) from each group were included. * P<0.05; & P<0.001 (2-way ANOVA with Bonferroni’s post-test).
TNFα antibody suppresses apoptotic cell accumulation and reduces necrosis in atherosclerotic lesions of WT→LDLR−/−, but not in lesions of MΦLRP1−/−→LDLR−/− mice
Accumulation of apoptotic macrophages in the lesion has been shown to cause inflammatory responses and contribute to atherosclerosis progression.8, 29 We previously reported that MΦLRP1−/− increased lesion cellularity and apoptotic cells.20 To evaluate if this effect is dependent on TNFα, we quantified the number of apoptotic cells in the plaque of adalimumab treated and control BMT mice. The apoptotic cell burden increased by 130% in lesions of MΦLRP1−/−→LDLR−/− mice compared with those of WT→LDLR−/− mice (P<0.05, Figures 2, A and C). Interestingly, adalimumab reduced apoptotic cells by 36% in WT→LDLR−/− mice but did not have any effect in MΦLRP1−/−→LDLR−/− mice (P<0.05, Figure 2, A and C). Accumulation of apoptotic cells in the lesion contributes to necrosis.1 We evaluated lesion necrosis by staining sections with H&E as previously described,20 and found a 180% increase in necrosis in MΦLRP1−/−→LDLR−/− mice compared with WT→LDLR−/− mice (P<0.001, Figure 2, B and D). Adalimumab reduced necrosis in WT→LDLR−/− mice by 59% (P<0.001) but did not have any effect in MΦLRP1−/−→LDLR−/− mice, consistent with the results we obtained for apoptotic cells in lesions (Figure 2, B and D).
Figure 2. Ex vivo macrophage apoptosis and necrotic core area analysis.


A. Apoptotic cells either extracellular (“free”, yellow arrows) or associated with macrophages (white arrows) were visualized using CD68 and TUNEL staining and quantified (C, E). Sections of aortic sinus area were stained with hematoxylin and eosin (B), and the necrotic core area was quantified (D). Examples of necrotic area are indicated by black arrows. At least two sections were analyzed for each mouse, and all mice in each group (n=5–7) were included. *, P < 0.05, and ***, P<0.001 significance of differences with adalimumab treatment; &, P<0.001 for differences between macrophage genotypes (2-way ANOVA with Bonferroni’s post-test).
Macrophage LRP1 in plaques causes efficient efferocytosis and limits inflammatory response, and TNFα blockade has no effect on efferocytosis in lesions
Clearance of apoptotic cells limits post-necrotic inflammation and promotes inflammation resolution. The relative proportion of apoptotic cells internalized by macrophages is a measure of efferocytosis efficiency,20, 22, 30–32 and increase in the percentage of “free” apoptotic cells is an index of inefficient efforocytosis. The percentage of “free” apoptotic cells in lesions of MΦLRP1−/−→LDLR−/− mice was 280% larger than that in lesions of WT→LDLR−/− mice (P<0.001, Figure 2, A and E), suggesting impaired efferocytosis in the absence of macrophage LRP1. Adalimumab did not affect efferocytosis in either mouse type (Figure 2, A and E).
Efficient efferocytosis promotes production of anti-inflammatory cytokines and induces anti-inflammatory macrophage (M2) polarization.11, 33 To evaluate inflammatory response, we stained lesion macrophages for CD68 and used arginase-1 (Arg1) as anti-inflammatory (M2) marker and arginase-2 (Arg2) as pro-inflammatory (M1) marker. As shown in Figure 3, total macrophage (CD68+) numbers increased by 130% (P<0.001) and M1 macrophages (Arg2+, CD68+) increased by 130% (P<0.001) in MΦLRP1−/−→LDLR−/− mice compared with WT→LDLR−/− mice. Adalimumab suppressed total macrophages by 18% (P<0.05) and M1 macrophages by 24% (P<0.05) in WT→LDLR−/− mice, but did not change total macrophage and M1 macrophages in MΦLRP1−/−→LDLR−/− mice (Figure 3, A–F). Macrophage LRP1 deletion reduced M2 macrophages (Arg1+, CD68+) by 30% (P<0.001) compared with WT→LDLR−/− mice. However, adalimumab treatment did not affect M2 contribution to the lesion in either WT→LDLR−/− mice or MΦLRP1−/−→LDLR−/− mice (Figure 3, A–G).
Figure 3. Ex vivo analysis of M1 and M2 macrophages in lesions in WT→LDLR−/− and MΦLRP1−/−→LDLR−/− mice.



A–D. Lesion macrophages were distinguished with CD68 staining. Arginase-1 (Arg1) was used as macrophage M2 marker and arginase-2 (Arg2) as M1 marker. Nuclei were counterstained with Hoechst. E. Quantification of total macrophage (CD68+) numbers in necrosis-free areas. F. Quantification of M1 macrophages (Arg2+ and CD68+) in necrosis-free areas. G. Quantification of M2 macrophages (Arg1+ and CD68+) in necrosis-free areas. H. Ratio of M1 to M2 macrophages in lesions. At least two sections were analyzed for each mouse, and all mice in each group (n=5~7) were included. *, Statistically significant differences for treatment with adalimumab (P < 0.05); #, P <0.01 and &, P<0.001 for differences between macrophage genotypes (2-way ANOVA with Bonferroni’s post-test).
TNFα blockade suppressed inflammation response in lesions and reduced lesion size in WT→LDLR−/− mice. Consistent with a previous report,21 we show that deletion of macrophage LRP1 increased lesion cellularity, caused defective efferocytosis and increased atherosclerosis burden. These effects could not be improved by TNFα blockade in the absence of LRP1.
Adalimumab suppresses atherosclerosis in apoE−/−→LDLR−/− mice, but this effect is reduced if macrophage LRP1 is also absent
ApoE deletion promotes systemic inflammatory responses.5, 34 Adalimumab suppresses inflammation due to hyperlipidemia in WT BMT mice. To test the anti-atherosclerotic effects of TNFα blockade in the absence of macrophage apoE−/−, we used LDLR−/− mice as recipients of bone marrow from mice lacking either apoE (apoE−/−→LDLR−/−) or apoE and macrophage LRP1 (DKO→LDLR−/−). As expected from our previous work,35 apoE−/−→LDLR−/− mice showed a 160% increase in atherosclerotic burden compared to WT→LDLR−/− mice (301.1±19.22 vs. 193.9±31.6 ×103 μm2, P<0.001). The atherosclerotic burden was 140% greater in DKO→LDLR−/− mice than in apoE−/−→LDLR−/− mice (418.0±31.2 vs. 301.1±19.22×103 μm2, P<0.001, Figure 4). Adalimumab reduced atherosclerosis by 57% (P<0.001) in apoE−/− BMT mice, but only by 19% (P<0.05) in DKO→LDLR−/− mice (Figure 4, A–B). Similar to what has been reported for WT→LDLR−/− and MΦLRP1−/− →LDLR−/− mice, DKO→LDLR−/− mice had a slight increase in plasma TG compared with apoE−/−→LDLR−/− mice (Figure 4, D and E). Adalimumab did not affect plasma cholesterol or TG levels in either mouse group. Circulating TNFα levels were increased by 200% (P<0.001) in apoE−/−→LDLR−/− compared with WT→LDLR−/− and did not further increase in DKO→LDLR−/− mice. Adalimumab suppressed TNFα levels by 34% in apoE−/−→LDLR−/− mice and by 27% in DKO→LDLR−/− mice (Figure 4C).
Figure 4. Effects of adalimumab on atherosclerosis, plasma TNFα and lipids in apoE−/−→LDLR−/− and DKO→LDLR−/− mice.


Cross sections of aortic sinus area from apoE−/− and DKO bone marrow reconstituted mice was stained with oil-red-O (A) and quantified (B). Plasma levels of TNFα (C), cholesterol (D), and triglyceride (E) in adalimumab treated and control mice. At least two sections were analyzed for each mouse, and all mice in each group (n=5–7) were included. *, P < 0.05, **, P<0.01, and ***, P<0.001 significance of differences with adalimumab treatment; &, P<0.001 for differences between macrophage genotypes (2-way ANOVA with Bonferroni’s post-test).
Deficiency of macrophage LRP1 decreases efferocytosis and reduces the ability of TNFα antibody to suppress apoptosis and necrosiss
Total apoptotic cells increased by 160% in DKO→LDLR−/− mice compared with apoE−/−→LDLR−/− mice (90.2±5.6 vs. 55.9±2.7 cells/mm2, P<0.001, Figure 5, A and C). Adalimumab reduced apoptotic cells by 34% in lesions of apoE−/−→LDLR−/− mice (P<0.01) and by 29% in lesions of DKO→LDLR−/− mice (P<0.05, Figure 5). Plaque necrosis increased by 140% in DKO→LDLR/− mice compared with apoE−/−→LDLR−/− mice (P<0.001, Figure 5, B and D). Adalimumab reduced plaque necrosis by 39% (P<0.01) in apoE−/−→LDLR−/− mice and by 25% in DKO→LDLR−/− mice (P<0.01, Figure 5, B and D). Percentage of “free” apoptotic cells increased 160% in DKO→LDLR−/− mice compared with apoE−/−→LDLR−/− mice (P<0.001, Figure 5, A and E). Adalimumab did not improve efferocytosis in either apoE−/−→LDLR−/− or DKO→LDLR−/− mice (Figure 5, A and E).
Figure 5. Effects of adalimumab on efferocytosis and necrosis in atherosclerotic lesions in apoE−/−→LDLR−/− and DKO→LDLR−/− mice.


A. Free (yellow arrows) and macrophage associated (white arrows) apoptotic cells were visualized using CD68 and TUNEL staining and quantified (C, E). Sections of aortic sinus area were stained with hematoxylin and eosin to evaluate necrotic core area (B) and then quantified (D). Examples of necrotic area were indicated by black arrows. At least two sections were analyzed for each mouse, and all mice from each group (n=5–7) were included; *, P < 0.05, and **, P<0.01 significance of differences with adalimumab treatment. &, P<0.001 for differences by macrophage genotypes (2-way ANOVA with Bonferroni’s post-test).
Adalimumab reduces macrophage cellularity and decreases inflammatory responses in the lesion, which depends on macrophage LRP1
Coincident with the increased lesion size, total macrophage (CD68+) numbers increased by 160% in apoE−/−→LDLR−/− compared with WT→LDLR−/− mice (624.1±36.4 vs. 370.9±17.3 cells/mm2, P<0.001), and a similar change affected M1 (arg2+, CD68+) macrophages (154.1±6.9 vs. 39.7±2.1 cells/mm2, P<0.001). Total macrophages increased by 130% (P<0.01) and M1 macrophages increased by 170% (P<0.01) in DKO→LDLR−/− relative to apoE−/−→LDLR−/− mice. Adalimumbab suppressed total macrophages and M1 macrophages in apoE−/−→LDLR−/− mice and was less effective in DKO→LDLR−/− mice (Figure 6, A–F).
Figure 6. Ex vivo analysis of CD68+ macrophage and inflammatory status in lesions from apoE−/−→LDLR−/− and DKO→LDLR−/− mice.



A–D. Lesions were stained with CD68 antibody to visualize macrophages. M2 macrophages were defined as Arg1+ and CD68+ double positive. M1 macrophages were defined as Arg2+ and CD68+ double positive. Nuclei were counterstained with Hoechst. E. Quantification of CD68+ macrophages in necrosis-free areas. F. Quantification of M1 macrophages in necrosis-free areas. G. Quantification of M2 macrophages in necrosis-free areas. H. Ratio of M1 to M2 macrophages in lesions. At least two sections were analyzed for each mouse, and all mice from each group (n=5–7) were included. *, P < 0.05, and **, P<0.01 significance of differences with adalimumab treatment; #, P <0.05 and &, P<0.001 for differences between macrophage genotypes (2-way ANOVA with Bonferroni’s post-test).
M2 macrophage (arg1+, CD68+) numbers increased by 155% in apoE−/−→LDLR−/− mice compared with WT→LDLR−/− mice (110.3±9.4 vs. 70.9±4.2 cells/mm2, P<0.01, Figure 6G), and increased by 146% in DKO−/−→LDLR−/− mice compared with MΦLRP1−/−→LDLR−/− mice (67.3±8.3 vs. 45.9±2.8 cells/mm2, P<0.01, Figure 6G). However, the percentage of M2 macrophages relative to total macrophages was lower in apoE−/−→LDLR−/− mouse lesion than in WT→LDLR−/− mouse lesion (18.45±2.2% vs. 22.1±4.7% P<0.05). The ratio of M2 macrophages to total macrophages was not different between DKO→LDLR−/− and MΦLRP1−/−→LDLR−/− mice (8.6±0.8% vs. 9.8±0.6%, n.s.). Both numbers and the proportion of anti-inflammatory M2 macrophages were lower in DKO→LDLR−/− mice than in apoE−/−→LDLR−/− mice (number: 110.3 ±9.4 vs. 67.3±8.3 cells/mm2, P<0.01; percentage: 17.3±0.7% vs. 8.6±0.8%, P<0.01). The administration of adalimumab did not affect M2 macrophage abundance or percentage in the lesion (Figure 6G). These data indicate that deletion of apoE in bone marrow derived cells or deletion of macrophage LRP1 reduces M2 polarization. Furthermore, TNFα blockade with adalimumab does not affect M2 macrophage polarization.
Macrophage LRP1 is required for the suppression of Ly6C cells in the lesion by Adalimumab
Blood ly6Chi monocytes give rise to inflammatory macrophages in the atherosclerotic plaque7. Ly6C expression is down-regulated during monocytes differentiation into macrophages in the lesion, but is still detectable within 48 hr of monocyte migration36. We evaluated Ly6Chi monocytes in blood and ly6C positive cells in lesions in all BMT mice. Blood Ly6Chi monocytes were similar for WT→LDLR−/− and MΦLRP1−/−→LDLR−/− mice (Figure 7B, Supplemental Figure II). Adalimumab reduced blood ly6Chi monocytes by 42% in WT→LDLR−/− mice (P<0.01) and by 34% in MΦLRP1−/−→LDLR−/− mice (P<0.05) (Figure 7B). Blood ly6Chi monocytes increased by 250% in apoE−/−→LDLR−/− mice compared with WT→LDLR−/− mice (2.1±0.2 vs. 0.8±0.1 ×104/ml, P<0.001). Blood ly6Chi monocytes were not further increased in DKO→LDLR−/− mice (Figure 7B). Adalimumab significantly suppressed blood ly6Chi monocytes in apoE−/−→ and DKO→LDLR−/− mice (Figure 7B). Lesion ly6C positive cells increased by 130% in MΦLRP1−/−→LDLR−/− compared with WT→LDLR−/− mice (P<0.001, Figure 7, A and C). Adalimumab reduced lesion ly6C positive cells by 54% in WT→LDLR−/− mice (139.8±11.9 vs. 92.4±6.9 cells/mm2, P<0.01) but did not alter lesion ly6C positive cells in MΦLRP1−/−→LDLR−/− mice (184.7±12.7 vs. 157.6±7.7, n.s., Figure 7, A and C). Lesion ly6C positive cells increased by 150% in apoE−/−→LDLR−/− compared with WT→LDLR−/− mice (210.3±9.3 vs 139.8±11.9 cells/mm2, P<0.01, Figure 7, A and C). Lesion ly6C positive cells were further increased by 130% in DKO→LDLR−/− mice compared with apoE−/−→LDLR−/− mice (P<0.001, Figure 7, A and C). There was no significant difference in ly6C cells between MΦLRP1−/−→LDLR−/− and apoE−/−→LDLR−/− mice (Figure 7C). Adalimumab reduced lesion ly6C cells by 35% in apoE−/−→LDLR−/− mice (P<0.01, Figure 7, A and C) and was less effective for the reductions of ly6c cells in DKO→LDLR−/− mice (by 27%, P<0.05, Figure 7, A and C).
Figure 7. Analyses of Ly6Chi monocytes in blood and Ly6C macrophages in atheroma.



Ly6C positive cells in lesions were visualized by immunofluorescence (A) and quantified (C). Blood Ly6Chi monocytes were identified by flow-cytometry analysis (B; gating and representative graphs in Supplemental Figure II). Immunofluorescence staining of endothelial MCP-1 expression (E) and quantification (D). White arrows indicate the endothelial side of the lesion. At least two sections were analyzed for each mouse, and all mice from each group (n=5–7) were included. *, P < 0.05, and **, P<0.01 significance of differences with adalimumab treatment; &, P<0.001 significance of differences by macrophage genotypes (2-way ANOVA with Bonferroni’s post-test).
Migration of inflammatory monocytes from blood to the lesion is driven both by monocytosis and by chemoattractant cytokines secreted by endothelial cells. To better understand the mechanisms by which adalimumab fails to suppress inflammation in the absence of LRP1, we evaluated endothelial expression of monocyte chemoattractant protein 1 (MCP-1) and vascular cell adhesion molecule 1 (VCAM-1) in the atherosclerotic lesion. Immunostaining density of MCP-1 in endothelial cells in lesions of MΦLRP1−/−→LDLR−/− mice increased by 171% compared with WT→LDLR−/− mice (19.2±1.3 vs 11.2±1.2, P<0.05, Figure 7, D and E). Adalimumab suppressed MCP-1 staining by 61% in lesions of WT→LDLR−/− mice but not in MΦLRP1−/−→LDLR−/− mice (Figure 7, D and E). Immunostaining density of MCP-1 in endothelial cells in lesions of DKO→LDLR−/− mice increased by 145% compared with apoE−/−→LDLR−/− mice (37.3±1.4 vs 25.7±14.6, P<0.01, Figure 7, D and E). Adalimumab suppressed MCP-1 staining by 44% in lesions of apoE−/−→LDLR−/− mice and by 21% in lesions of DKO→LDLR−/− mice (Figure 7, D and E). There was no significant difference in MCP-1 expression between MΦLRP1−/−→LDLR−/− mice and apoE−/−→LDLR−/− mice, suggesting both macrophage LRP1 and apoE are important for limiting the recruitment of monocytes. In addition, expression of VCAM-1 in endothelial cells of the lesion had the similar pattern as the expression of MCP-1 for all groups of mice (unpublished data). Together with results for atherosclerotic inflammation (Figures 3 and 6) and systemic inflammation (Figure 7B), these data show that: 1) deletion of macrophage LRP1 increases both atherosclerotic plaque inflammation and MCP-1 mediated recruitment of ly6Chi monocytes in atherosclerotic lesions; 2) systemic inflammation due to macrophage apoE deficiency increases ly6Chi monocyte migration into the lesion, plaque inflammation, and MCP-1 expression; and 3) adalimumab reduces blood ly6Chi monocytes, plaque MCP-1 expression, and plaque inflammation in WT BMT mice or mice lacking macrophage ApoE, but not in mice lacking macrophage LRP1 (Figure 8). Overall, these data indicate that loss of apoE and LRP1 in macrophages has synergistic effects to increase atherosclerosis burden, and that adalimumab reduces atherosclerosis through a pathway that requires macrophage LRP1.
Figure 8. Schematic representation of the effects of TNFα blockade and loss of macrophage LRP1 on atherogenesis.

Western diet, alone or together with apoE deletion, increases circulating inflammatory monocytes. Efferocytosis causes apoptotic cell removal, limits M1 and promotes M2 macrophage polarization, reduces post-apoptotic necrosis, and improves the resolution of inflammation. Both paths influence the ability of Ly6Chi monocytes to infiltrate the intima and modulate atherogenesis. TNFα blocking antibodies suppress blood monocytosis, migration of pro-inflammatory monocytes, lesion cellularity, and atherosclerosis, but do not affect efferocytosis. Loss of macrophage LRP1 drastically impairs efferocytosis, thus promoting inflammatory responses, stimulating endothelium chemoattractant cytokine expression, increasing pro-inflammatory monocyte migration and atherosclerosis. These effects are not responsive to the anti-atherosclerotic action of TNFα blockade.
Discussion
We examined atherosclerotic burden in hyperlipidemic mice known to have defective efferocytosis (MΦLRP1−/−→LDLR−/−), increased inflammation (apoE−/−→LDLR−/−), or both (DKO→LDLR−/−). We also examined whether suppressing inflammation using adalimumab, a blocking TNFα antibody, could reduce atherosclerotic burden in these models. Adalimumab suppressed cytokine production and blood ly6Chi monocytes in all recipients of BMT. In WT→LDLR−/− mice, the adalimumab-mediated reduction in blood pro-inflammatory ly6Chi monocytes was associated with reduced MCP-1 expression, reduced ly6C positive cells in the lesion, and decreased atherosclerosis. MΦLRP1−/−→LDLR−/− mice showed increased necrosis and apoptosis as well as defective efferocytosis and increases in inflammation in the lesion, which could not be suppressed by adalimumab. Despite reduced blood ly6Chi monocytes, adalimumab failed to reduce MCP-1 expression and ly6C positive cells in the lesion and failed to prevent atherosclerosis progression in MΦLRP1−/−→LDLR−/− mice. Deletion of apoE in bone marrow derived cells in apoE−/−→LDLR−/− mice promoted systemic inflammation and blood ly6Chi monocytosis. Adalimumab-mediated reduction in blood ly6Chi monocytes was associated with reduced MCP-1 expression and ly6C positive cells in the lesion and reduced atherosclerosis. Similarly to MΦLRP1−/−→LDLR−/− mice, despite reduced blood ly6Chi, adalimumab was less effective to suppress atheroslecrosis in the absence of macrophage LRP1 in DKO→LDLR−/− mice.
We show that TNFα blockade is effective in suppressing atherosclerosis caused by macrophage ApoE deletion, but not effective in suppressing atherosclerosis caused by macrophage LRP1 deletion. Loss of macrophage ApoE may cause atherosclerosis through increased systemic inflammation. Loss of macrophage LRP1, however, causes atherosclerosis via reduced macrophage efferocytosis. Although studies in mice have shown that genetic deletion of TNFα reduces atherosclerosis,37, 38 the anti-atherosclerotic effects of monoclonal antibody-mediated blocking of systemic TNFα have not been conclusively demonstrated. In humans, protective effects of adalimumab for cardiovascular disease have been reported in patients with RA or psoriasis.27, 28 Adalimumab reduced vascular inflammation and improved endothelial function in these patients, reducing carotid atherosclerosis and arterial stiffness.27, 28 However, other studies have not been able to detect a significant effect of TNFα inhibitors on the risk of cardiovascular events.2 Our data indicate that adalimumbab therapy may be effective in reducing risk of atherosclerosis in cases of increased systemic inflammation involving a pathway requiring macrophage LRP1 expression.
Ly6Chi monocytosis is an important contributor to atherosclerosis by fueling lesion cellularity.39, 40 Migration of inflammatory monocytes to the lesion is determined by the secretion of chemoattractant cytokines, such as MCP-1, and by the expression of adhesion molecules, such as VCAM-1, by endothelial cells on the artery wall.41 We demonstrate that TNFα blockade suppressed blood ly6Chi monocytes and prevents atherosclerosis in an LRP1-dependent pathway. In the absence of LRP1, increased plaque inflammation enhances the MCP-1 expression and increases recruitment of ly6Chi monocytes to the atherosclerotic lesion. TNFα blockade, despite the suppression of systemic Ly6C monocytes, did not reduce monocyte recruitment in the absence of macrophage LRP1. These results support a scenario where the increased inflammation afforded by MΦLRP1−/− promotes overexpression of MCP-1 and VCAM-1 by adjacent endothelial cells, which further promotes monocyte/macrophage infiltration and local inflammation and could not be suppressed by adalimumab.
After migration into the atheroma, ly6Chi monocytes preferentially differentiate into the pro-inflammatory M1 phenotype, down-regulating ly6C expression and up-regulating CD68 expression.36 Our results showing a decrease in ly6C positive cells in the lesion suggest a suppression of the migration of ly6Chi cells to the arterial wall. Furthermore, in vitro 3H-thymidine labeling experiments showed no differences in uptake between macrophages of WT, MΦLRP1, apoE−/−, and DKO genotypes (not shown) indicating that the decrease found in total and M1 macrophages in lesions of adalimumab treated mice were not due to altered proliferation. Systemic inflammation was elicited by western diet in WT→LDLR−/− mice and was further stimulated in apoE−/−→LDLR−/− mice (Figures 1C and 4C). Consistent with the study reported by Murphy et al. 42, absence of macrophage apoE increased blood ly6Chi monocytes in apoE−/−→LDLR−/− and DKO−/−→LDLR−/− mice (Figure 7) due to the elevated proliferation of hematopoietic stem and progenitor cell (HPSC) under western-type diet. Increases in blood ly6Chi drove the monocyte migration to the atherosclerotic lesion. HSPC cell proliferation was not changed by macrophage LRP1 deletion in vivo, likely because LRP1 was not expressed in bone marrow cells (Supplemental Figure III).
Macrophage apoptosis is a fundamental process in the development of the atherosclerotic plaque.29 Oxidative stress, abnormal secretion of proinflammatory cytokines and endoplasmic reticulum stress are among the most common inducers of apoptotic cell death.43–45 We showed that adalimumab was effective in reducing apoptotic cell content only in WT→LDLR−/− mice but not in MΦLRP1−/−→LDLR−/− mice. Similarly, adalimumab reduced apoptosis in the absence of apoE in bone marrow derived cells but was less effective when macrophage LRP1 was also absent. In early lesions, efficiency of apoptotic cell clearance is associated with a decrease in lesion cellularity and reduction of atherosclerosis progression. However, in advanced lesions, the progressive inefficiency of apoptotic cell clearance by phagocytes leads to increases in necrosis, with consequent increase in plaque inflammation and enlargement of the necrotic core.46 Similarly to apoptosis, we demonstrated that adalimumab significantly reduced necrosis, but only in the presence of MΦLRP1. When apoE was absent, adalimumab improved necrosis, but was less effective when both macrophage apoE and LRP1 were deficient. This indicates that the effects of TNFα blockade on apoptosis and necrosis depend on LRP1.
Macrophage LRP1 acts as a recognition receptor for apoptotic cell removal,13, 15–17 and defective efferocytosis in hyperlipidemic mice with macrophage LRP1 deletion has been reported.18–20 In this study, the percentage of “free” apoptotic cells in lesions was higher in BMT mice carrying macrophages without LRP1, showing defective efferocytosis in those mice (Figures 2 and 5). The TUNEL positive staining that was not co-localized with CD68 were considered as “free” apoptotic cells and has been used as a measurement of efferocytosis efficiency frequently.20, 22, 30–32 The limitation of this method is that when a TUNEL positive staining close to a nucleus that not merged with CD68+ staining, there is possibility that this apoptosis cell might be cleared by other kind of phagocytes. However, as shown in Figures 3 and 6, more than 90% of cells in lesion were CD68 positive. In addition, our previous work showed that lymphocytes and dendritic cells don’t express LRP1 in mouse.47 Thus, we estimate that the contribution of other cell types such as dendritic cells would be minimal. Of interest, Eriksson et al reported an increase in LRP1 (CD91) expression in CD3-expressing T-lymphocytes in patients who did not respond to anti-TNFα antibody treatment.48 Although the mechanism was not clear, the authors proposed that the increased CD91 expression was caused by the over-stimulation of T-cells.48 Defective efferocytosis is closely associated with inflammation.8, 11 However, despite the effect of adalimumab on inflammation, it did not affect efferocytosis in any of the treated groups in our study.
Our results show that anti-TNFα therapy inhibits atherosclerosis induced by western-type diet or macrophage apoE deletion in hyperlipidemic mice. The effect is due to improved local inflammatory response, with fewer Ly6Chi monocytes in the circulation and reduced monocyte recruitment into the atheroma. Despite the reduction of systemic levels of TNFα and Ly6Chi monocytes, adalimumab was ineffective in inhibiting the local inflammatory response in the absence of LRP1, indicating that macrophage LRP1 is necessary for the anti-inflammatory effect of TNFα blockade.
Supplementary Material
Highlights.
Atherosclerosis is an inflammatory process but anti-inflammatory drugs have not proven to reduce CVD rates.
Vascular inflammation in atherosclerosis has the unique component of lipid-laden cells undergoing apoptosis and necrosis.
It is not clear if cell death and removal of cellular debris (efferocytosis) contribute to vascular inflammation.
Monoclonal antibody that reduces inflammation via TNFα blockade also reduces atherosclerosis in a murine model but only if the receptor LRP1 is present on the macrophage.
Absence of LRP1 results in defective efferocytosis and persistent inflammation, thus suggesting that local cell survival and recruitment of circulatory inflammatory cells are driving factors of inflammation in the atheroma.
Acknowledgments
Source of funding: This study was funded by NIH R01 grant HL057986 to Dr. Fazio. Dr. Ormseth was supported by NIH K23 grant AR068443. Dr. Stafford was supported by the Department of Veteran’s Affairs.
Abbreviations
- Arg-1 or Arg-2
arginase-1 or arginase-2
- BMT
bone marrow transplant
- DKO
apoE and macrophage LRP1 double knockout
- LDLR
low density lipoprotein receptor
- LRP1
low density lipoprotein receptor related protein 1
- MΦLRP1−/−
macrophage LRP1 deletion
- MCP-1
monocyte chemoattractant protein 1
- VCAM-1
vascular cell adhesion molecule 1
Footnotes
Disclosure: None
References
- 1.Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105:1135–1143. doi: 10.1161/hc0902.104353. [DOI] [PubMed] [Google Scholar]
- 2.McKellar GE, McCarey DW, Sattar N, McInnes IB. Role for tnf in atherosclerosis? Lessons from autoimmune disease. Nature reviews. Cardiology. 2009;6:410–417. doi: 10.1038/nrcardio.2009.57. [DOI] [PubMed] [Google Scholar]
- 3.Avina-Zubieta JA, Choi HK, Sadatsafavi M, Etminan M, Esdaile JM, Lacaille D. Risk of cardiovascular mortality in patients with rheumatoid arthritis: A meta-analysis of observational studies. Arthritis and rheumatism. 2008;59:1690–1697. doi: 10.1002/art.24092. [DOI] [PubMed] [Google Scholar]
- 4.Baitsch D, Bock HH, Engel T, Telgmann R, Muller-Tidow C, Varga G, Bot M, Herz J, Robenek H, von Eckardstein A, Nofer JR. Apolipoprotein e induces antiinflammatory phenotype in macrophages. Arteriosclerosis, thrombosis, and vascular biology. 2011;31:1160–1168. doi: 10.1161/ATVBAHA.111.222745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gaudreault N, Kumar N, Posada JM, Stephens KB, Reyes de Mochel NS, Eberle D, Olivas VR, Kim RY, Harms MJ, Johnson S, Messina LM, Rapp JH, Raffai RL. Apoe suppresses atherosclerosis by reducing lipid accumulation in circulating monocytes and the expression of inflammatory molecules on monocytes and vascular endothelium. Arteriosclerosis, thrombosis, and vascular biology. 2012;32:264–272. doi: 10.1161/ATVBAHA.111.238964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grainger DJ, Reckless J, McKilligin E. Apolipoprotein e modulates clearance of apoptotic bodies in vitro and in vivo, resulting in a systemic proinflammatory state in apolipoprotein e-deficient mice. J Immunol. 2004;173:6366–6375. doi: 10.4049/jimmunol.173.10.6366. [DOI] [PubMed] [Google Scholar]
- 7.Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, Pittet MJ. Ly-6chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. The Journal of clinical investigation. 2007;117:195–205. doi: 10.1172/JCI29950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tabas I. Macrophage death and defective inflammation resolution in atherosclerosis. Nature reviews. Immunology. 2010;10:36–46. doi: 10.1038/nri2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li Y, Schwabe RF, DeVries-Seimon T, Yao PM, Gerbod-Giannone MC, Tall AR, Davis RJ, Flavell R, Brenner DA, Tabas I. Free cholesterol-loaded macrophages are an abundant source of tumor necrosis factor-alpha and interleukin-6: Model of nf-kappab- and map kinase-dependent inflammation in advanced atherosclerosis. The Journal of biological chemistry. 2005;280:21763–21772. doi: 10.1074/jbc.M501759200. [DOI] [PubMed] [Google Scholar]
- 10.Devries-Seimon T, Li Y, Yao PM, Stone E, Wang Y, Davis RJ, Flavell R, Tabas I. Cholesterol-induced macrophage apoptosis requires er stress pathways and engagement of the type a scavenger receptor. The Journal of cell biology. 2005;171:61–73. doi: 10.1083/jcb.200502078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thorp E, Tabas I. Mechanisms and consequences of efferocytosis in advanced atherosclerosis. Journal of leukocyte biology. 2009;86:1089–1095. doi: 10.1189/jlb.0209115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boucher P, Herz J. Signaling through lrp1: Protection from atherosclerosis and beyond. Biochemical pharmacology. 2011;81:1–5. doi: 10.1016/j.bcp.2010.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Orr AW, Elzie CA, Kucik DF, Murphy-Ullrich JE. Thrombospondin signaling through the calreticulin/ldl receptor-related protein co-complex stimulates random and directed cell migration. Journal of cell science. 2003;116:2917–2927. doi: 10.1242/jcs.00600. [DOI] [PubMed] [Google Scholar]
- 14.Lillis AP, Van Duyn LB, Murphy-Ullrich JE, Strickland DK. Ldl receptor-related protein 1: Unique tissue-specific functions revealed by selective gene knockout studies. Physiological reviews. 2008;88:887–918. doi: 10.1152/physrev.00033.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, Bratton DL, Oldenborg PA, Michalak M, Henson PM. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of lrp on the phagocyte. Cell. 2005;123:321–334. doi: 10.1016/j.cell.2005.08.032. [DOI] [PubMed] [Google Scholar]
- 16.Ogden CA, deCathelineau A, Hoffmann PR, Bratton D, Ghebrehiwet B, Fadok VA, Henson PM. C1q and mannose binding lectin engagement of cell surface calreticulin and cd91 initiates macropinocytosis and uptake of apoptotic cells. The Journal of experimental medicine. 2001;194:781–795. doi: 10.1084/jem.194.6.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vandivier RW, Ogden CA, Fadok VA, Hoffmann PR, Brown KK, Botto M, Walport MJ, Fisher JH, Henson PM, Greene KE. Role of surfactant proteins a, d, and c1q in the clearance of apoptotic cells in vivo and in vitro: Calreticulin and cd91 as a common collectin receptor complex. J Immunol. 2002;169:3978–3986. doi: 10.4049/jimmunol.169.7.3978. [DOI] [PubMed] [Google Scholar]
- 18.Derocq D, Prebois C, Beaujouin M, Laurent-Matha V, Pattingre S, Smith GK, Liaudet-Coopman E. Cathepsin d is partly endocytosed by the lrp1 receptor and inhibits lrp1-regulated intramembrane proteolysis. Oncogene. 2012;31:3202–3212. doi: 10.1038/onc.2011.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yahiro K, Satoh M, Nakano M, Hisatsune J, Isomoto H, Sap J, Suzuki H, Nomura F, Noda M, Moss J, Hirayama T. Low-density lipoprotein receptor-related protein-1 (lrp1) mediates autophagy and apoptosis caused by helicobacter pylori vaca. The Journal of biological chemistry. 2012;287:31104–31115. doi: 10.1074/jbc.M112.387498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yancey PG, Blakemore J, Ding L, Fan D, Overton CD, Zhang Y, Linton MF, Fazio S. Macrophage lrp-1 controls plaque cellularity by regulating efferocytosis and akt activation. Arteriosclerosis, thrombosis, and vascular biology. 2010;30:787–795. doi: 10.1161/ATVBAHA.109.202051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Overton CD, Yancey PG, Major AS, Linton MF, Fazio S. Deletion of macrophage ldl receptor-related protein increases atherogenesis in the mouse. Circulation research. 2007;100:670–677. doi: 10.1161/01.RES.0000260204.40510.aa. [DOI] [PubMed] [Google Scholar]
- 22.Yancey PG, Ding Y, Fan D, Blakemore JL, Zhang Y, Ding L, Zhang J, Linton MF, Fazio S. Low-density lipoprotein receptor-related protein 1 prevents early atherosclerosis by limiting lesional apoptosis and inflammatory ly-6chigh monocytosis: Evidence that the effects are not apolipoprotein e dependent. Circulation. 2011;124:454–464. doi: 10.1161/CIRCULATIONAHA.111.032268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beutler B, Cerami A. Tumor necrosis, cachexia, shock, and inflammation: A common mediator. Annual review of biochemistry. 1988;57:505–518. doi: 10.1146/annurev.bi.57.070188.002445. [DOI] [PubMed] [Google Scholar]
- 24.Fahlman C, Jacobsen FW, Veiby OP, McNiece IK, Blomhoff HK, Jacobsen SE. Tumor necrosis factor-alpha (tnf-alpha) potently enhances in vitro macrophage production from primitive murine hematopoietic progenitor cells in combination with stem cell factor and interleukin-7: Novel stimulatory role of p55 tnf receptors. Blood. 1994;84:1528–1533. [PubMed] [Google Scholar]
- 25.Witsell AL, Schook LB. Tumor necrosis factor alpha is an autocrine growth regulator during macrophage differentiation. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:4754–4758. doi: 10.1073/pnas.89.10.4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stewart RA, White HD, Kirby AC, Heritier SR, Simes RJ, Nestel PJ, West MJ, Colquhoun DM, Tonkin AM. White blood cell count predicts reduction in coronary heart disease mortality with pravastatin. Circulation. 2005;111:1756–1762. doi: 10.1161/01.CIR.0000160924.73417.26. [DOI] [PubMed] [Google Scholar]
- 27.Kerekes G, Soltesz P, Szucs G, Szamosi S, Der H, Szabo Z, Csathy L, Vancsa A, Szodoray P, Szegedi G, Szekanecz Z. Effects of adalimumab treatment on vascular disease associated with early rheumatoid arthritis. Isr Med Assoc J. 2011;13:147–152. [PubMed] [Google Scholar]
- 28.Bissonnette R, Tardif JC, Harel F, Pressacco J, Bolduc C, Guertin MC. Effects of the tumor necrosis factor-alpha antagonist adalimumab on arterial inflammation assessed by positron emission tomography in patients with psoriasis: Results of a randomized controlled trial. Circ Cardiovasc Imaging. 2013;6:83–90. doi: 10.1161/CIRCIMAGING.112.975730. [DOI] [PubMed] [Google Scholar]
- 29.Kockx MM, Herman AG. Apoptosis in atherosclerosis: Beneficial or detrimental? Cardiovascular research. 2000;45:736–746. doi: 10.1016/s0008-6363(99)00235-7. [DOI] [PubMed] [Google Scholar]
- 30.Aprahamian T, Rifkin I, Bonegio R, Hugel B, Freyssinet JM, Sato K, Castellot JJ, Jr, Walsh K. Impaired clearance of apoptotic cells promotes synergy between atherogenesis and autoimmune disease. The Journal of experimental medicine. 2004;199:1121–1131. doi: 10.1084/jem.20031557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schrijvers DM, De Meyer GR, Kockx MM, Herman AG, Martinet W. Phagocytosis of apoptotic cells by macrophages is impaired in atherosclerosis. Arteriosclerosis, thrombosis, and vascular biology. 2005;25:1256–1261. doi: 10.1161/01.ATV.0000166517.18801.a7. [DOI] [PubMed] [Google Scholar]
- 32.Taylor PR, Carugati A, Fadok VA, Cook HT, Andrews M, Carroll MC, Savill JS, Henson PM, Botto M, Walport MJ. A hierarchical role for classical pathway complement proteins in the clearance of apoptotic cells in vivo. The Journal of experimental medicine. 2000;192:359–366. doi: 10.1084/jem.192.3.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu Y, Li D, Chen J, Xie J, Bandyopadhyay S, Zhang D, Nemarkommula AR, Liu H, Mehta JL, Hermonat PL. Inhibition of atherogenesis in ldlr knockout mice by systemic delivery of adeno-associated virus type 2-hil-10. Atherosclerosis. 2006;188:19–27. doi: 10.1016/j.atherosclerosis.2005.10.029. [DOI] [PubMed] [Google Scholar]
- 34.Ali K, Middleton M, Pure E, Rader DJ. Apolipoprotein e suppresses the type i inflammatory response in vivo. Circulation research. 2005;97:922–927. doi: 10.1161/01.RES.0000187467.67684.43. [DOI] [PubMed] [Google Scholar]
- 35.Fazio S, Babaev VR, Murray AB, Hasty AH, Carter KJ, Gleaves LA, Atkinson JB, Linton MF. Increased atherosclerosis in mice reconstituted with apolipoprotein e null macrophages. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:4647–4652. doi: 10.1073/pnas.94.9.4647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dal-Secco D, Wang J, Zeng Z, Kolaczkowska E, Wong CH, Petri B, Ransohoff RM, Charo IF, Jenne CN, Kubes P. A dynamic spectrum of monocytes arising from the in situ reprogramming of ccr2+ monocytes at a site of sterile injury. The Journal of experimental medicine. 2015;212:447–456. doi: 10.1084/jem.20141539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ohta H, Wada H, Niwa T, Kirii H, Iwamoto N, Fujii H, Saito K, Sekikawa K, Seishima M. Disruption of tumor necrosis factor-alpha gene diminishes the development of atherosclerosis in apoe-deficient mice. Atherosclerosis. 2005;180:11–17. doi: 10.1016/j.atherosclerosis.2004.11.016. [DOI] [PubMed] [Google Scholar]
- 38.Branen L, Hovgaard L, Nitulescu M, Bengtsson E, Nilsson J, Jovinge S. Inhibition of tumor necrosis factor-alpha reduces atherosclerosis in apolipoprotein e knockout mice. Arteriosclerosis, thrombosis, and vascular biology. 2004;24:2137–2142. doi: 10.1161/01.ATV.0000143933.20616.1b. [DOI] [PubMed] [Google Scholar]
- 39.Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: A dynamic balance. Nature reviews. Immunology. 2013;13:709–721. doi: 10.1038/nri3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nagareddy PR, Murphy AJ, Stirzaker RA, Hu Y, Yu S, Miller RG, Ramkhelawon B, Distel E, Westerterp M, Huang LS, Schmidt AM, Orchard TJ, Fisher EA, Tall AR, Goldberg IJ. Hyperglycemia promotes myelopoiesis and impairs the resolution of atherosclerosis. Cell Metab. 2013;17:695–708. doi: 10.1016/j.cmet.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakashima Y, Raines EW, Plump AS, Breslow JL, Ross R. Upregulation of vcam-1 and icam-1 at atherosclerosis-prone sites on the endothelium in the apoe-deficient mouse. Arteriosclerosis, thrombosis, and vascular biology. 1998;18:842–851. doi: 10.1161/01.atv.18.5.842. [DOI] [PubMed] [Google Scholar]
- 42.Murphy AJ, Akhtari M, Tolani S, Pagler T, Bijl N, Kuo CL, Wang M, Sanson M, Abramowicz S, Welch C, Bochem AE, Kuivenhoven JA, Yvan-Charvet L, Tall AR. Apoe regulates hematopoietic stem cell proliferation, monocytosis, and monocyte accumulation in atherosclerotic lesions in mice. The Journal of clinical investigation. 2011;121:4138–4149. doi: 10.1172/JCI57559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dickhout JG, Hossain GS, Pozza LM, Zhou J, Lhotak S, Austin RC. Peroxynitrite causes endoplasmic reticulum stress and apoptosis in human vascular endothelium: Implications in atherogenesis. Arteriosclerosis, thrombosis, and vascular biology. 2005;25:2623–2629. doi: 10.1161/01.ATV.0000189159.96900.d9. [DOI] [PubMed] [Google Scholar]
- 44.Canault M, Peiretti F, Kopp F, Bonardo B, Bonzi MF, Coudeyre JC, Alessi MC, Juhan-Vague I, Nalbone G. The tnf alpha converting enzyme (tace/adam17) is expressed in the atherosclerotic lesions of apolipoprotein e-deficient mice: Possible contribution to elevated plasma levels of soluble tnf alpha receptors. Atherosclerosis. 2006;187:82–91. doi: 10.1016/j.atherosclerosis.2005.08.031. [DOI] [PubMed] [Google Scholar]
- 45.Seimon TA, Nadolski MJ, Liao X, Magallon J, Nguyen M, Feric NT, Koschinsky ML, Harkewicz R, Witztum JL, Tsimikas S, Golenbock D, Moore KJ, Tabas I. Atherogenic lipids and lipoproteins trigger cd36-tlr2-dependent apoptosis in macrophages undergoing endoplasmic reticulum stress. Cell metabolism. 2010;12:467–482. doi: 10.1016/j.cmet.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schrijvers DM, De Meyer GR, Herman AG, Martinet W. Phagocytosis in atherosclerosis: Molecular mechanisms and implications for plaque progression and stability. Cardiovascular research. 2007;73:470–480. doi: 10.1016/j.cardiores.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 47.Covarrubias R, Wilhelm AJ, Major AS. Specific deletion of ldl receptor-related protein on macrophages has skewed in vivo effects on cytokine production by invariant natural killer t cells. PloS one. 2014;9:e102236. doi: 10.1371/journal.pone.0102236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eriksson C, Rantapaa-Dahlqvist S, Sundqvist KG. T-cell expression of cd91 - a marker of unresponsiveness to anti-tnf therapy in rheumatoid arthritis. APMIS. 2010;118:837–845. doi: 10.1111/j.1600-0463.2010.02677.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.