

Abstract
Time-resolved temperature-jump (T-jump) coupled with fluorescence and infrared (IR) spectroscopy is a powerful technique for monitoring protein dynamics. Although IR spectroscopy of the polypeptide amide I mode is more technically challenging, it offers complementary information because it directly probes changes in the protein backbone, whereas, fluorescence spectroscopy is sensitive to the environment of specific side chains. With the advent of widely tunable quantum cascade lasers (QCL) it is possible to efficiently probe multiple IR frequencies with high sensitivity and reproducibility. Here we describe a dual time-resolved T-jump fluorescence and IR spectrometer and its application to study protein folding dynamics. A Q-switched Ho:YAG laser provides the T-jump source for both time-resolved IR and fluorescence spectroscopy, which are probed by a QCL and Ti:Sapphire laser, respectively. The Ho:YAG laser simultaneously pumps the time-resolved IR and fluorescence spectrometers. The instrument has high sensitivity, with an IR absorbance detection limit of less than 0.2 mOD and a fluorescence sensitivity of 2% of the overall fluorescence intensity. Using a computer controlled QCL to rapidly tune the IR frequency it is possible to create a T-jump induced difference spectrum from 50 ns to 0.5 ms. This study demonstrates the power of the dual time-resolved T-jump fluorescence and IR spectroscopy to resolve complex folding mechanisms by complementary IR absorbance and fluorescence measurements of protein dynamics.
Keywords: Fluorescence Spectroscopy, Infrared Spectroscopy, Protein Folding, Quantum Cascade Laser, Temperature-Jump, WW domain
Graphical abstract
1. Introduction
The three-dimensional structure of proteins is not static; flexibility is necessary in order for proteins to function properly. Many methods have been developed to study the functional dynamics of proteins, but are limited in their ability to monitor fast dynamics with structural specificity. Experimental techniques such as X-ray crystallography, nuclear magnetic resonance and cryogenic electron microscopy can detect equilibrium fluctuations in protein structures with high structural resolution, but these populations are stochastic and such methods do not usually provide dynamics of the structural fluctuations.1 Even single-molecule and stopped-flow methods, which have millisecond resolution at the cost of reduced structural resolution, are too slow to capture important dynamics.2,3 Submillisecond resolution is necessary to monitor important protein processes including early events in protein folding, fast folding/unfolding of secondary structures, or fast unfolding for degradation and regulation.4,5 Photo-triggers can access faster times, but cannot be generally applied to all proteins.6 Laser-induced T-jump has arisen as a powerful method for investigating fast protein dynamics.7 The free energy of a protein in solution depends on many factors including temperature, pressure, pH, and other solvent conditions. Manipulating one of these factors, temperature in the case of T-jump, changes the relative free energies of multiple alternative structural states, resulting in a new equilibrium distribution among these states. A spectroscopic probe monitors the system as it relaxes to the new equilibrium under the new conditions. Fluorescence is one of the most convenient T-jump probes, because it requires a relatively small amount of material (μM), has a high signal-to-noise ratio and the intrinsic fluorophore tryptophan (Trp) is sensitive to environmental changes. The structural specificity of fluorescence spectroscopy depends on the location of the side chain reporter, which may report on local or global structure, or both. IR spectroscopy of the amide I mode is a complementary method that offers improved structural resolution. It directly probes changes in the protein backbone, and different vibrational modes can be assigned to specific secondary structural elements.
With the advent of the widely tunable quantum cascade laser (QCL) operating in the mid-IR it is possible to efficiently probe multiple frequencies in order to monitor dynamics of multiple protein secondary structure elements. Before the availability of QCL sources, continuous wave lead salt diode lasers that were partially tunable across the amide I′ spectral region were used to obtain time-resolved T-jump IR transients.8 QCLs offer several advantages over diode lasers that make it possible to probe the temporal response as a systematic function of the probe wavelength. Because commercially available lead salt diode lasers do not possess an external cavity and thus no tuning element, they can only be tuned by varying the temperature and current. As a result these lasers exhibit nonlinear tuning, mode-hopping and the tuning parameters change over time. The QCL by contrast has an external cavity and dispersive tuning element that can be computer controlled in a linear and reproducible manner, yielding finer, more rapid and reproducible control of the output wavelength. QCLs also have a broader tuning range, usually between 100–200 cm−1. QCLs have improved amplitude stability, they are less prone to mode-hopping and they are less sensitive to temperature variations.12 QCLs have higher output power than diode lasers, which eases alignment and improves measurement sensitivity.13 The QCL beam diverges much less than diode lasers, and the high power allows the use of liquid crystal paper as a heat sensor to locate, focus and align the beam. The higher power also enables transmission through more optically dense samples, which in turn improves signal levels because higher sample concentrations and longer path lengths are accessible. Finally, the ability to measure multi-wavelength IR absorbance kinetics using the QCL allows us to sort complex, multi-step reactions.
T-jump has been coupled to broadband mid-IR methods, for example step-scan FTIR and 2DIR.9,10 The advantage of broadband IR methods over single wavelength methods is that the entire spectral region of interest is collected simultaneously at a specific time following pump laser excitation, but the associated challenge is that acquisition of broadband data is relatively slow. In order to speed measurements, step-scan probed T-jump measurements are collected at a relatively low spectral resolution, typically 4–8 cm−1 compared to 0.0007 cm−1 accessible by QCL lasers.9 High spectral resolution is desirable to detect changes in different populations; for example there is less than 5 cm−1 difference between the frequency for single and double stranded β-sheets.11 2DIR spectra are often only collected at select time points during broadband 2DIR measurements, and other nonlinear methods are used to collect the kinetic measurements. 2DIR measurements have several additional inherent technical difficulties including synchronization of the nanosecond T-jump laser and the femtosecond laser system, decreased signal-to-noise ratio from low repetition rate data acquisition and maintaining optical alignment as changes in index of refraction are induced by the T-jump.10
Here we have developed a dual T-jump apparatus that is capable of simultaneously pumping transient fluorescence and IR spectroscopy experiments from a single source (Figure 1). Eigen and Hammes pioneered the T-jump technique based on a rapid capacitance discharge to heat a solution held between capacitor plates.14 However, the temporal resolution of this approach was limited to the microsecond timescale by the slow capacitive discharge and optical access to the solution was not possible. These limitations were alleviated by the use of a Q-switched laser characterized by its short pulse to optically heat the solution, with a T-jump response on the order of nanoseconds.15 Typically the T-jump is achieved using a 10 Hz pulsed Q-switched Nd:YAG laser, which has a fundamental output wavelength at 1.064 μm. In order to achieve effective heating this fundamental wavelength must be shifted to match a vibrational absorbance of the solvent. D2O is chosen as the solvent for IR spectroscopy of the protein amide I′ band, because its background absorbance is much lower in this spectral region compared to H2O. The broad D2O overtone absorption at 1.9 μm is selected for optical heating with a short laser pulse to produce the T-jump. Frequency shifting is often accomplished using a Raman shifter with one Stokes shift in 200 psi H2 gas.16 Recently, we and others have demonstrated that a Q-switched Ho:YAG laser, which has a fundamental output wavelength at 2.09 μm, can be used as an IR T-jump source.17,18 The advantages of the Ho:YAG laser over the more widely used Nd:YAG laser/Raman shifter are its stability, beam quality and safety. There are non-linear processes in Raman shifting that result in shot-to-shot fluctuations of the pump pulse of ~15%.8,15 Furthermore, the Raman shifter output has hot spots and is nonuniform due to the nonlinearity of the process and instability of the gas medium. By eliminating the Raman shifter in the Ho:YAG apparatus this variability decreases. The output mode of the Ho:YAG is a Gaussian TEM 00 mode, which produces a stable and uniform heated volume. The T-jump is stabilized by minimizing shot-to-shot fluctuations in the T-jump magnitude and giving a more defined final temperature. Eliminating the Nd:YAG fundamental also improves safety because 1.064 μm is easily focused on the retina whereas the 2.09 μm Ho:YAG is strongly absorbed by water and less likely to reach the retina.19 Our dual T-jump approach uses a commercially available 50 Hz Q-switched Ho:YAG laser, which is pulse picked to provide a pump source for both time-resolved IR and fluorescence measurements. The advantage of this arrangement is that two independent experiments can be run simultaneously, eliminating the cost of time-sharing between the time-resolved infrared and fluorescence spectroscopy experiments or purchasing a second pump laser. In principle, it is possible with this instrument to obtain time-resolved fluorescence and infrared spectra simultaneously on the same sample. In practice, however, the concentration ranges that are optimal for time-resolved infrared (mM) and fluorescence (μM) spectroscopy differ by 3 orders of magnitude, hence using the same concentration for both would sacrifice sensitivity of one of the probes.
Figure 1.

Schematic of the dual time-resolved T-jump fluorescence and IR spectrometer. An AOM pulse picker reduces the repetition rate of the 50 Hz pulsed Q-switched Ho:YAG laser and produces the 2.09 μm pump radiation that is the source for both the IR and fluorescence T-jump. By using multiple quantum cascade lasers, the total frequency range spanned by the IR system is 1000–2250 cm−1. Fluorescence is excited using a mode-locked, frequency tripled Ti:Sapphire laser and emission is collected with a photomultiplier tube and an appropriate band pass filter.
Here we have demonstrated the versatility of the dual T-jump spectrometer in the context of the well studied FBP28-N,Q WW Domain.20 The present study extended previous work on FBP28-N,Q by measuring the folding relaxation kinetics with both fluorescence and infrared probes. We also obtained transient difference infrared spectra (defined as ΔA = At − A0, where At is the absorption at the final temperature at time t and A0 is the absorption at the initial temperature at time zero) spanning the complete amide I′ region for the first time. Because the mid-IR frequency of the QCL can be rapidly tuned by a computer controlled tuning element, it was possible to create a T-jump induced difference spectrum at any time from 50 ns to 0.5 ms. The tunable mid-IR QCL was used to collect T-jump transients at 1 cm−1 resolution across the amide I′ region. A transient difference spectrum was constructed from these data by plotting the ΔA versus frequency at a specific time of interest. The dynamics of specific secondary structure elements, β-sheets, turns, disordered structure, were independently probed by collecting transients at the frequency assigned to each component in the amide I′ region. As expected from previous work, we found that the turns form prior to the β-sheets of the WW domain.20–23 Temperature dependent data were used to determine differences in the barriers to folding of the β-sheets and turns. Formation of the β-sheets was found to be rate limiting and to have a relatively higher barrier to folding. Time-resolved fluorescence transients from the dual T-jump instrument provided a complementary view of the relaxation dynamics. The dynamics of the WW Domain Trps, located in the β-sheets, confirm assignments made by time-resolved IR spectroscopy.
These results demonstrate the power of the dual T-jump to combine IR and fluorescence probes to analyze complex dynamics. The Ho:YAG laser simultaneously pumps the IR and fluorescence T-jump with an IR sensitivity of 0.2 mOD and a fluorescence sensitivity of 2% change in fluorescence intensity. Transients are generated at IR frequencies corresponding to different secondary structures, which is useful for determining the folding mechanism. Fluorescence measurements complement IR measurements and aid in interpretation of complex IR results. Finally, this study demonstrates that the dual time-resolved fluorescence and IR T-jump instrument offers improved sensitivity and versatility over past T-jump methods.
2. Materials and Methods
2.1 Time Resolved T-jump Relaxation Measurements
The principle of the time-resolved T-jump infrared and fluorescence method has been described previously, the major difference here being the source of the heating pulse and the ability to pump both fluorescence and IR experiments simultaneously.16,25 Pulsed laser excitation is used to rapidly perturb the folding equilibrium on a timescale faster than the molecular dynamics of interest. An actively Q-switched Tm:fiber-pumped Ho:YAG laser (IPG Photonics Corp., Oxford, MA), operated at 50 Hz produces a 7 mJ, 10 ns pulse at 2.09 μm that is the T-jump pump source for both time-resolved IR and fluorescence spectroscopy. A home built pulse picker (Gooch & Housego acousto-optic modulator and RF driver, Orlando, FL) directs the pulse and reduces the repetition rate to the optimum level, typically 12.5 or 25 Hz to allow complete cooling between heating pulses.
The T-jump cell consists of two CaF2 windows stacked and separated by a 100 μm Teflon spacer split into two compartments, one for the sample and the other for the reference. Using this design, reference data can be collected under nearly identical conditions as the sample data. The same cells are used for equilibrium FTIR and T-jump experiments. The reference is comprised of a solution of 20 mM potassium phosphate buffer at pD* 7.0 (pD refers to uncorrected pH meter reading) in D2O, the same conditions as the sample solution but without protein. Absorbance changes of the reference solution are due only to changes in D2O buffer absorbance, which is used as an internal thermometer.16 The magnitude of the T-jump is calculated using the change in reference absorbance with temperature. The protein relaxation signal can be extracted by subtracting the change in absorbance of the reference from the sample in response to the T-jump. The initial temperature is maintained by contact with the sample stage, which is temperature controlled by a recirculating water bath.
The change in signal induced by the T-jump is probed in real time by an IR laser with a frequency in the amide I′ spectral region, or in the case of fluorescence, a frequency tripled Ti:Sapphire laser at 285 nm to excite Trp. The broadly tunable fluorescence excitation source provides versatility for complementary fluorescence probes including Trp and NADH.24 The mid-IR probe beam is generated by a continuous wave QCL (Daylight Solutions Inc., San Diego, CA) with a tunable output range of 1570–1730 cm−1. A QCL with a different tuning range can be easily swapped into the instrument, providing near continuous coverage of the amide and CO/CN/CD regions from 1000–2250 cm−1. The transient transmission of the IR probe beam through the sample is measured using a fast, 100 MHz, photovoltaic MCT IR detector/preamplifier (Kolmar Technologies, Newburyport, MA). Transient signals are digitized and signal averaged (1000 shots) using a Wavesurfer 62Xs-B oscilloscope (Teledyne LeCroy, Chestnut Ridge, NY). Data collection is managed by an in-house routine using the LabVIEW computer program (National Instruments, Austin, TX).
Trp fluorescence is excited at 285 nm and collected from 320–370 nm. A Verdi V22 DPSS High-Power continuous wave laser (Coherent, Santa Clara, CA) is used to pump a Mira 900 Ti:Saph laser (Coherent, Santa Clara, CA) which produces a quasi-continuous beam at 845 nm. The beam is then passed through a second and third harmonic generator (Coherent, Santa Clara, CA) to produce the fluorescence probe beam at 285 nm. The back-emitted fluorescence light induced by the 285 nm laser is detected using a Hamamatsu R7518 photomultiplier tube (PMT, Hamamatsu Photonics K.K., Hamamatsu City, Japan), digitized and signal averaged (5000 shots) using a Wavesurfer 62Xs-B oscilloscope (Teledyne LeCroy, Chestnut Ridge, NY). The shortest possible time response of the T-jump experiment is given by the width of the heating pulse (10 ns), which is the same for both the infrared and fluorescence experiments (Figure 2). The observed response time of the fluorescence instrument varies, however, depending on the size of the terminating resistance on the PMT detector. The signal voltage level (and S/N of the measurement) is increased by using a larger terminating resistance (Ohm’s law); however, this increase in resistance also increases the RC time constant and the response of the system is slowed (τ = 0.5 RC). Our experiments were conducted using terminating resistors of 10,630 Ω, 500 Ω or 200 Ω, which gave response times of 4 μs, 0.5 μs and 200 ns, respectively. A 400 μM Trp solution in D2O is used as a reference to measure the magnitude of the T-jump and to determine the temperature dependence of the Trp signal. Instrument control and data collection are controlled using a LabVIEW computer program (National Instruments, Austin, TX).
Figure 2.

Difference between two signal averaged D2O reference transients. (A) Difference IR absorbance transient of 20 mM potassium phosphate buffer (pD 7) detected at 1619 cm−1 with a 1000 shot average. (B) Difference fluorescence transient of 400 μM Trp in 20 mM potassium phosphate buffer (pD 7) excited at 285 nm and monitored at 350 nm using a 200 Ω resistor and a 5000 shot average.
2.2 Protein Synthesis and Purification
FBP28-N,Q was synthesized via standard 9-fluorenylmethyloxycarbonyl (Fmoc) based solid-phase chemistry on a Liberty1 microwave peptide synthesizer (CEM, Matthews, NC).20 Fmoc-PAL-PS resin (Applied Bio-systems, Foster City, CA) was used to form a peptide amide. The peptide was purified by reverse-phase chromatography (C18 column) using a water/acetonitrile gradient with 0.1% trifluoroacetic acid (TFA) as the counter ion. TFA interferes in the Amide-I IR measurements at 1672 cm−1, so we remove it by anion exchange. The peptide was lyophilized and dissolved in a 2 mM HCl solution to allow exchange of the TFA counter-ion for chloride.26 The identity of the peptide was confirmed by matrix-assisted laser desorption ionization time-of-flight mass spectrometry. The peptide was dissolved in D2O to allow deuterium hydrogen exchange of the amide protons. The peptide was lyophilized a second time and re-suspended in D2O buffer with 20 mM potassium phosphate buffer at pD* 7.0. Sample concentrations of 0.5–2.0 mM were prepared for both IR and fluorescence experiments.
2.3 FTIR Spectroscopy
The equilibrium melting behavior was monitored on a Varian Excalibur 3100 FTIR spectrometer (Varian Inc., Palo Alto, USA) using a temperature controlled IR cell. No aggregation was observed in the amide I′ at the reported concentrations. The IR spectrum is highly sensitive to protein aggregation because the amide I′ band of an aggregated protein is dominated by an intense, sharp band at ~1620 cm−1 and a coupled peak that is also sharp but not as intense at ~1680 cm−1, due to dipolar coupling of the C=O groups in protein aggregates.27 Furthermore, the T-jump experiments are also very sensitive to aggregation, because protein aggregates serve as nucleation sites for cavitation, leading to large cavitation artifacts in the IR transients, which were not observed in our experiments. The absorbance spectra are constructed by subtracting the spectrum of reference buffer solution without protein from sample solution with protein. The temperature-dependent difference spectrum was then generated by subtracting the spectrum with a temperature corresponding to the initial T-jump temperature from the spectrum with a temperature corresponding to the final T-jump temperature.
2.4 Analysis of Kinetics Data
The peptide relaxation kinetics must be deconvolved from the observed kinetics. Accurate deconvolution is possible as the instrument response is determined from the reference measurement under the exact conditions of the sample measurements. In order to minimize detector artifacts, the reference is scaled prior to subtraction from the sample. The decay function is an exponential decay with the formula:
| (1) |
where A0 is an offset, n is the number of exponentials to fit, An is a preexponential factor, τn is the relaxation lifetime of the sample and x0 is the time offset. In order to best fit the data, the minimum number of exponentials with unique relaxation lifetimes was selected. IR data are fit over the interval from 90 ns to 0.4 ms, with the endpoint an order of magnitude greater than the lifetime of the slowest exponential. Fluorescence data are fit from 5 μs to 0.4 ms, an order of magnitude in time before and after the observed exponential lifetime. The data analysis was performed in IGOR PRO (WaveMetrics, Lake Oswego, OR).
3. Results and Discussion
3.1 Determination of Instrument Sensitivity
The signal-to-noise (S/N) ratio of the IR and fluorescence T-jump was determined by comparing the difference between two reference transients. The transient absorbance dynamics of 20 mM potassium phosphate buffer pD 7 at 1619 cm−1, in the amide I region, was induced by T-jump and signal averaged for 1000 shots. The difference was generated by subtracting the two reference transients (Figure 2A). The fluctuation of the difference between the two transients is ±0.2 mOD, giving a noise level on the order of 10−4 ΔOD. The minimum detection signal level under these conditions would be 0.2 ΔmOD giving a S/N ratio of ~1. This is consistent with the S/N ratio of previous time-resolved T-jump IR spectrometers both in our lab and reported by others.18,28 Equilibrium data can be used to determine the anticipated change in IR absorbance between two temperatures. Smaller signals can be detected by additional signal averaging, with a practical limit near 0.5×10−5 ΔOD.
The same method was used to determine the S/N ratio of the fluorescence T-jump. Fluorescence kinetics of 400 μM Trp in 20 mM potassium phosphate buffer pD 7 were excited at 285 nm and monitored at 350 nm using a 200 Ω resistor. The 200 Ω resistor provides the maximum time resolution with the least signal amplification. For a given number of shots the Johnson-Nyquist noise is the same regardless of the resistor, so the 200 Ω resistor provides the worst detection sensitivity of the available resistors. The signal was averaged for 5000 shots and the percent fluorescence was calculated as the change in fluorescence signal induced by the T-jump divided by the total observed fluorescence signal pre-jump. The difference was generated by subtracting two reference transients (Figure 2B). The fluctuation of the difference between the two transients is ±2 %. The number of shots can be optimized based on the anticipated change in fluorescence intensity determined from equilibrium fluorescence experiments. The detection limit can also be improved by selection of a larger resistor. The slight drift in the baseline arises from differences in saturation of the photomultiplier tube at long time. In summary, both the time-resolved T-jump fluorescence and IR spectrometers offer high sensitivity and reproducibility.
A computer controlled tuning element provides a fast and highly reproducible method for tuning the probe frequency across the available range of a specific QCL. The percent transmittance of the QCL through a pinhole was measured to determine the reproducibility of the probe beam alignment across the frequency tuning range of the QCL (Figure 3). The QCL signal was chopped and signal averaged to determine the total signal and recorded in 1 cm−1 steps across the available range. Without modifying the beam alignment, a 100 μm pinhole was placed in the path of the beam and the signal transmitted through the pinhole was measured at each frequency using the same method. The average percent transmittance through the pinhole was 84 ± 4 %, with 92% of the frequencies falling in this range. The small variance of the transmittance through the 100 μm pinhole indicates that the alignment and focus of the probe laser does not change as the probe frequency is tuned. Most of the frequencies that do not fall within this range are near a water vapor line (Figure 3). At these frequencies, slight differences in probe frequency (0.01 cm−1) can make a large difference in the signal level between measurements due to differences in background absorbance of the very narrow atmospheric water vapor lines.
Figure 3.

Percent transmittance of the QCL probe beam through a 100 μM pinhole as a function of probe frequency. Error bars represent the standard deviation of three trials. Red lines represent one standard deviation from the average percent transmission through the pinhole. The FTIR absorbance spectrum of water vapor is overlaid for comparison (blue line, spectrum scaled for best comparison with QCL transmittance spectrum).
3.2 T-jump Relaxation Kinetics
With the dual T-jump it is possible to simultaneously monitor the probe dependent relaxation kinetics by IR and fluorescence spectroscopy. To test the ability of the dual T-jump to measure the relaxation kinetics of protein folding/unfolding transitions, a well studied protein, FBP28-N,Q WW Domain, was selected as a model system.20 The complementary IR and fluorescence probes of the dual T-jump system are essential to sort out the complex folding mechanism of this WW domain.
Using the QCL it is possible to construct the time-dependent IR absorbance in the amide I′ region with a frequency resolution of 1 cm−1 or better. Figure 4A displays a 3D image of the frequency dependent relaxation kinetics of FBP28-N,Q following jumps from 15 to 30 °C. The x and y axes are frequency and time, respectively, and the third dimension is the change in absorbance (ΔA), represented by a color scale. From this 3-D landscape it is possible to make some preliminary assessments about the system. There are two main times of interest, a fast event around 10−7 s and a slow event between 10−5–10−3 s. Because there are multiple times where changes in absorbance occur, this suggests that FBP28-N,Q folding cannot be explained by a simple two-state model. There are three regions of interest in the amide I′ absorption band, negative difference regions between 1625–1650 cm−1 and at 1680 cm−1 and a positive difference region around 1660 cm−1. It is possible to expand on these observations by taking a slice across a specific time or frequency of interest.
Figure 4.

(A) Wavelength dependent time-resolved T-jump IR spectroscopy of FBP28-N,Q monitored in the amide I′ spectral region following a T-jump from 15 to 30 °C. Individual T-jump transients were collected at 1 cm−1 resolution from 1550 to 1690 cm−1. The change in absorbance (mOD) is indicated by the color. (B) Representative IR relaxation kinetics monitored at 1619, 1634 and 1660 cm−1. (C) T-jump induced difference absorbance spectrum of FBP28-N,Q recorded at 50 ns, 70 μs and 400 μs after the pump laser. (D) Equilibrium difference spectra obtained by subtracting the equilibrium FTIR spectrum at 15 °C from the spectrum at 30 °C.
The T-jump induced transient IR absorbance spectra, obtained by plotting the IR absorbance at 50 ns, 70 μs and 400 μs after the laser pulse, are shown in Figure 4C. The transient IR spectra are compared to the equivalent equilibrium FTIR difference spectrum (Figure 4D) generated by subtracting the FTIR spectrum at 15 °C from the spectrum at 30 °C. Negative peaks in the FTIR difference spectrum correspond to specific structures or interactions present in the folded state, and positive peaks correspond to new interactions with solvent in the unfolded state. Analysis of the equilibrium difference spectra reveals four main components of the amide I′ band centered at ~1619, 1634, 1660 and 1680 cm−1. These peaks have previously been observed in other WW domains.20,29,30 The peaks in the transient difference spectra are sharper, slightly shifted in frequency and have intensities that change with time, but generally agree with the peaks identified in the equilibrium difference spectra. Improved resolution of IR bands by time-resolved T-jump IR spectroscopy over equilibrium FTIR has been reported for a similar T-jump apparatus.18 The likely explanation for this and for the slightly shifted maxima is that there is a slower phase (>0.5 ms) associated with the broad peak at 1660 cm−1 that is not captured by the transients at early time points. In particular the peak at 1680 cm−1, arising from out-of-phase coupling of carbonyls in the β-sheets, is relatively weak in the FTIR data but strong in the kinetic IR data. In the equilibrium data, this peak appears as a small negative peak on top of the broad positive peak characteristic of disordered polypeptides (Figure 4D).8,31,32 When the disordered peak is present it masks the smaller peak. This observation supports our assessment that the slow phase associated with the disordered peak has not been captured by the transients at these early times. The reason for this is that the laser induced T-jump persists for a few ms, then the solution cools back to the initial temperature. Therefore any events that occur more slowly (>1 ms) are not captured by this method. In summary, using the QCL probe, the dual T-jump is capable of monitoring changes in the IR difference spectra with time from 10−7 to 10−2 s.
Wavelength dependent measurements reveal differences in the dynamics of specific secondary structure elements. The transient kinetics acquired at 1619 cm−1, 1634 cm−1 and 1660 cm−1, assigned to the WW-Domain turn, β-sheets and disordered structure, respectively, are shown in Figure 4B.20,30,33,34 The slow (ms) return of the signal to the initial baseline, which is observed for all transients in Figure 4B, corresponds to re-cooling of the sample to the initial temperature. The heated region cools down by thermal conduction, on a timescale proportional to the square of its diameter. For a typical heated region the size of our pinhole, 100 μm, cooling is gradual and would take ~10 ms. Therefore, it is not possible to study fast re-folding dynamics during this cooling process. The apparent difference in the cooling between the 1634 cm−1 probe compared to the other two frequencies is caused by instrumental noise, a mismatch in low-frequency oscillations between the sample and reference caused by laser fluctuations and mechanical vibrations. Another example of this type of artifact is a small hiccup in the transient collected at 1660 cm−1 between 10−3 and 10−2 s. As observed in previous experiments the folding kinetics are multi-exponential, and can be fit by a triple exponential (Equation 1) with a fast 100’s of ns phase, a μs phase and a slower ~100 μs phase.20 The peak at 1619 cm−1 appears as a shoulder on the 1634 cm−1 peak in the difference spectra (Figure 4D). Probes at either 1619 or 1634 cm−1 are sensitive to dynamics associated with both locations because these peaks overlap one another.
It is also likely that the spectral responses are coupled such that formation of one structure affects the others. Differences in the intensity of each of the phases for the probed frequency reveal differences in the local folding of the turn and sheets. In Figure 4B, the amplitude of the ΔA of the fast 100 ns phase is the same for the 1619 and 1634 cm−1 probe, but the overall ΔA of the 1634 cm−1 is twice that of the 1619 cm−1 probe. There is a higher percentage of the overall intensity change in the fast 100 ns phase of the 1619 cm−1 probe, so the fast phase can be assigned to the turn. There is a higher percentage of the overall intensity change in the slow 100 μs phase of the 1634 cm−1 probe, so the 100 μs phase can be assigned to formation of hydrogen bonds in the β-sheet. The intermediate 10 μs phase is thought to arise from development of the cross-strand interactions of hairpin 1 to form the first half of the WW Domain. While this assignment cannot be made from the presented data alone, we have conducted two previous IR spectroscopy studies that confirm this assignment. In the first study the relative stability of the two hairpins was perturbed, resulting in characteristic IR peaks that could be assigned to each of the two hairpins.29 In the other, an aspartic acid side chain found in the turn of the first hairpin was monitored and found to change conformation on the same timescale as the intermediate phase.35 These studies have provided evidence that the intermediate phase arises from formation of the β-sheet of the first hairpin. These assignments support a model where the turns form prior to the β-sheets, which has been predicted from previous studies.20–23 The kinetics at 1660 cm−1 corresponds to formation of the disordered polypeptide upon unfolding, and cannot be assigned to a specific local structure. However, the kinetics measured at this frequency is a useful control, as it should reflect changes on similar time-scales to those recorded for the other frequencies. In summary, varying the dual T-jump QCL probe frequency can be used to reveal subtle differences in the protein folding mechanism.
The dual T-jump can also determine the dependence of the relaxation rates on the final temperature following the T-jump (Figure 5A). In these experiments the magnitude of the T-jumps was kept constant while varying the final temperature, with all final temperatures below the melting transition. The observed kinetic rates are dominated by the folding rate. Figure 5A displays the relaxation kinetics probed by IR spectroscopy of the peptide following 15 °C jumps with final temperatures between 30 and 45 °C. The T-jump is monitored at 1626 cm−1, the average of 1619 and 1634 cm−1, as a representative frequency of the structured amide I′ region. The same three phases evident in the 1619 and 1634 cm−1 transients (Figure 4A) are present at all temperatures in the 1626 cm−1 transients. As the final temperatures becomes closer to the Tm, 52 °C, the magnitude of the ΔA increases.20 The kinetics of the fast phase is relatively temperature independent with a similar intensity and relaxation lifetime regardless of temperature probed. This suggests that formation of the turn, assigned to the fast phase, requires little or no activation energy. The kinetics of the slow phase is highly temperature dependent, increasing in intensity and speeding up from 100’s to 10’s of μs. Formation of the β-sheets, the slow step, is the rate-limiting step and has a relatively higher activation barrier. Temperature dependent measurements can be used to further expand our understanding of protein folding by providing information about the activation energies of each of the folding steps identified by frequency dependent measurements.
Figure 5.

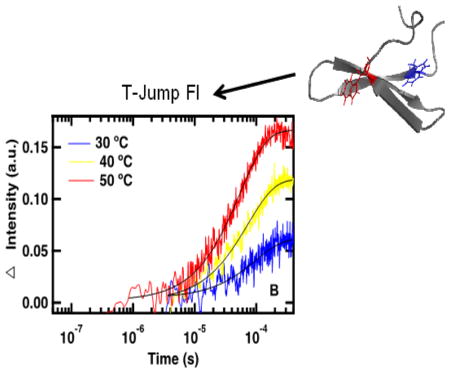
Representative IR absorbance (A) and fluorescence (B) relaxation kinetics of FBP28-N,Q following T-jumps to the listed final temperatures. IR absorbance relaxation kinetics were monitored in the amide I′ spectral region at 1626 cm−1 following a 15 °C T-jump. Fluorescence relaxation kinetics were excited at 285 nm and monitored at 350 nm following a 10 °C T-jump. Differences in fluorescence response times are due to resistor selection. Data at 30 and 40 °C were collected using a 10,630 Ω resistor and data at 50 °C were collected using a 500 Ω resistor. A triple exponential fit is overlaid on the IR absorbance transients and a single exponential fit is overlaid on the fluorescence transients (Equation 1).
Complementary dual T-jump experiments can be probed by time-resolved fluorescence simultaneously to IR absorbance measurements (Figure 5B). There are two Trps in FBP28-N,Q, one located on the outside of each β-sheet, so Trp fluorescence intensity is sensitive to changes in the environment of the β-sheets of the WW-domain. Unlike the IR absorbance measurements, which are sensitive to the peptide backbone, fluorescence measurements monitor packing of the Trp side chains into the hydrophobic core. Therefore, one advantage of the fluorescence measurements is that the kinetics is dominated by a single molecular process, the formation of the hydrophobic core. Because of this each transient can be fit to a single exponential (Equation 1).20 The fits of the fluorescence transients correspond to the slowest IR probed relaxation lifetime, supporting assignment of this kinetic phase to formation of the β-sheets. IR absorbance measurements offer a level of detail not available from the fluorescence experiments. Fluorescence measurements can be used to interpret IR absorbance measurements. Dual T-jump allows simultaneous collection of complementary time-resolved IR and fluorescence spectroscopy, which can be used to determine detailed mechanisms of protein folding.
4. Conclusions
We have developed a dual T-jump apparatus with a Ho:YAG laser as the pump source for simultaneous time-resolved laser-induced T-jump fluorescence and IR spectroscopy. The time-resolved T-jump IR spectroscopy has a sensitivity of 10−4 mOD and the fluorescence spectroscopy has a sensitivity of 2% of the total fluorescence signal. A QCL offers a highly flexible and reproducible IR probe source. As an example of the power of this technique we have characterized the folding mechanism of the well studied protein, FBP28-N,Q WW domain. Using the dual T-jump it is possible to create an IR difference spectrum at any time of interest within 0.5 ms. Transients at frequencies corresponding to different secondary structures can be used to determine the folding mechanism. Fluorescence measurements complement IR absorbance measurements and can aid interpretation of complex IR absorbance results. Temperature dependent data can be used to determine the energy barriers associated with each phase. The high sensitivity of the dual T-jump to both fluorescence and IR absorbance measurements will be useful for determining protein-folding mechanisms.
Highlights.
Dual time-resolved T-jump spectrometer simultaneously measures infrared and fluorescence transients with high sensitivity.
Quantum cascade laser (QCL) infrared probe enables rapid acquisition of complete time-resolved IR spectrum with high temporal and spectral resolution.
Complementary IR and fluorescence spectroscopic probes simplify assignment of complex kinetics observed in protein folding reactions.
Acknowledgments
This work was supported by a grant from the National Institutes of Health (NIH R01 GM53640) to R.B.D.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Yang LW, Eyal E, Chennubhotla C, Jee J, Gronenborn AM, Bahar I. Structure. 2007;15:741. doi: 10.1016/j.str.2007.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jamin M, Baldwin RL. Nat Struct Biol. 1996;3:613. doi: 10.1038/nsb0796-613. [DOI] [PubMed] [Google Scholar]
- 3.Roy R, Hohng S, Ha T. Nat Methods. 2008;5:507. doi: 10.1038/nmeth.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dyer RB. Curr Opin Struct Biol. 2007;17:38. doi: 10.1016/j.sbi.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 5.Eaton WA, Munoz V, Hagen SJ, Jas GS, Lapidus LJ, Henry ER, Hofrichter J. Annu Rev Biophys Biomol Struct. 2000;29:327. doi: 10.1146/annurev.biophys.29.1.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kolano C, Helbing J, Bucher G, Sander W, Hamm P. J Phys Chem B. 2007;111:11297. doi: 10.1021/jp074184g. [DOI] [PubMed] [Google Scholar]
- 7.Serrano AL, Waegele MM, Gai F. Protein Sci. 2012;21:157. doi: 10.1002/pro.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Williams S, Causgrove TP, Gilmanshin R, Fang KS, Callender RH, Woodruff WH, Dyer RB. Biochemistry. 1996;35:691. doi: 10.1021/bi952217p. [DOI] [PubMed] [Google Scholar]
- 9.Wang J, El-Sayed MA. Biophys J. 1999;76:2777. doi: 10.1016/S0006-3495(99)77431-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chung HS, Khalil M, Smith AW, Tokmakoff A. Rev Sci Instrum. 2007;78:063101. doi: 10.1063/1.2743168. [DOI] [PubMed] [Google Scholar]
- 11.Kubelka J, Keiderling TA. J Am Chem Soc. 2001;123:12048. doi: 10.1021/ja0116627. [DOI] [PubMed] [Google Scholar]
- 12.Mezzapesa FP, Columbo LL, Brambilla M, Dabbicco M, Borri S, Vitiello MS, Beere HE, Ritchie DA, Scamarcio G. Opt Express. 2013;21:13748. doi: 10.1364/OE.21.013748. [DOI] [PubMed] [Google Scholar]
- 13.Kalchmair S, Blanchard R, Mansuripur TS, de Naurois GM, Pfluegl C, Witinski MF, Diehl L, Capasso F, Loncar M. Opt Express. 2015;23:15734. doi: 10.1364/OE.23.015734. [DOI] [PubMed] [Google Scholar]
- 14.Eigen M, Hammes GG. Journal of the American Chemical Society. 1960;82:5951. [Google Scholar]
- 15.Kubelka J. Photochem Photobiol Sci. 2009;8:499. doi: 10.1039/b819929a. [DOI] [PubMed] [Google Scholar]
- 16.Williams S, Causgrove TP, Gilmanshin R, Fang KS, Callender RH, Woodruff WH, Dyer RB. Biochemistry. 1996;35:691. doi: 10.1021/bi952217p. [DOI] [PubMed] [Google Scholar]
- 17.Reddish MJ, Vaughn MB, Fu R, Dyer RB. Biochemistry. 2016;55:1485. doi: 10.1021/acs.biochem.5b01364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li D, Li Y, Li H, Wu X, Yu Q, Weng Y. Rev Sci Instrum. 2015;86:053105. doi: 10.1063/1.4921473. [DOI] [PubMed] [Google Scholar]
- 19.Henderson AR, Shulmeister K. Laser safety. Institute of Physics; Bristol: Philadelphia: 2004. [Google Scholar]
- 20.Davis CM, Dyer RB. Biochemistry. 2014;53:5476. doi: 10.1021/bi500556h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deechongkit S, Nguyen H, Powers ET, Dawson PE, Gruebele M, Kelly JW. Nature. 2004;430:101. doi: 10.1038/nature02611. [DOI] [PubMed] [Google Scholar]
- 22.Petrovich M, Jonsson AL, Ferguson N, Daggett V, Fersht AR. J Mol Biol. 2006;360:865. doi: 10.1016/j.jmb.2006.05.050. [DOI] [PubMed] [Google Scholar]
- 23.Jager M, Nguyen H, Crane JC, Kelly JW, Gruebele M. J Mol Biol. 2001;311:373. doi: 10.1006/jmbi.2001.4873. [DOI] [PubMed] [Google Scholar]
- 24.Nie B, Lodewyks K, Deng H, Desamero RZ, Callender R. Biochemistry. 2016;55:3803. doi: 10.1021/acs.biochem.6b00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gulotta M, Deng H, Deng H, Dyer RB, Callender RH. Biochemistry. 2002;41:3353. doi: 10.1021/bi016009a. [DOI] [PubMed] [Google Scholar]
- 26.Andrushchenko VV, Vogel HJ, Prenner EJ. J Pept Sci. 2007;13:37. doi: 10.1002/psc.793. [DOI] [PubMed] [Google Scholar]
- 27.Jackson M, Mantsch HH. Can J Chem. 1991;69:1639. [Google Scholar]
- 28.Callender R, Dyer RB. Curr Opin Struct Biol. 2002;12:628. doi: 10.1016/s0959-440x(02)00370-6. [DOI] [PubMed] [Google Scholar]
- 29.Davis CM, Dyer RB. J Am Chem Soc. 2013;135:19260. doi: 10.1021/ja409608r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang T, Xu Y, Du D, Gai F. Biopolymers. 2004;75:163. doi: 10.1002/bip.20101. [DOI] [PubMed] [Google Scholar]
- 31.Dyer RB, Gai F, Woodruff WH. Accounts Chem Res. 1998;31:709. [Google Scholar]
- 32.Werner JH, Dyer RB, Fesinmeyer RM, Andersen NH. Journal of Physical Chemistry B. 2002;106:487. [Google Scholar]
- 33.Hilario J, Kubelka J, Keiderling TA. J Am Chem Soc. 2003;125:7562. doi: 10.1021/ja030039e. [DOI] [PubMed] [Google Scholar]
- 34.Maness SJ, Franzen S, Gibbs AC, Causgrove TP, Dyer RB. Biophys J. 2003;84:3874. doi: 10.1016/S0006-3495(03)75115-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Davis CM, Dyer RB. J Am Chem Soc. 2016;138:1456. doi: 10.1021/jacs.5b13201. [DOI] [PMC free article] [PubMed] [Google Scholar]