

Abstract
Human African Trypanosomiasis is a neglected tropical disease that is lethal if left untreated. Existing therapeutics have limited efficacy and severe associated toxicities. 2-(2-(((3-((1H–Benzo[d]imidazol-2-yl)amino)propyl)amino)methyl)-4,6-dichloro-1H–indol-1-yl)ethan-1-ol (NEU-1053) has recently been identified from a high throughput screen of >42,000 compounds as a highly potent and fast acting trypanocidal agent capable of curing a blood stream infection of T. brucei in mice. We have designed a library of analogs to probe the SAR and improve the predicted CNS exposure of NEU-1053. We report the activity of these inhibitors of Trypanosoma brucei, the efficacy of NEU-1053 in a murine CNS model of infection, and identification of the target of NEU-1053 via X-ray crystallography.
Keywords: Trypanosoma brucei, methionyl-tRNA synthetase, medicinal chemistry
Graphical Abstract

INTRODUCTION
Insect-borne trypanosomal diseases are a menace to human health. Human African trypanosomiasis (HAT, or sleeping sickness) is a neglected tropical disease caused by two subspecies of Trypanosoma brucei for which current therapeutics are toxic and inconvenient. Though there are two compounds in clinical trials, SCYX-71581 and fexinidazole,2 given the clinical failure rate for infectious diseases,3 it is prudent to continue the search for new drugs.
To that end, we recently described a high-throughput screening campaign performed as part of an industry-academic partnership between GlaxoSmithKline, Spanish National Research Council (CSIC), and Northeastern University, in which we uncovered 798 inhibitors of T. brucei cellular proliferation. We also reported in vitro drug metabolism, physicochemical properties, and pharmacokinetics data, plus kinase selectivity data for key analogs.4 Included in that report was NEU-1053 (SB-443342, 1), a singleton compound identified in the screen that showed rapid and irreversible proliferation inhibition of T. brucei, showed good plasma exposure, and cured a bloodstream infection in a T. brucei rhodesiense mouse model of HAT. As a singleton hit with no other analogs included in the screening campaign, there was no SAR information apparent from the HTS. Besides looking to better understand the SAR of this series, we wished to explore the various structural regions of the compound in order to identify effective analogs with more attractive physicochemical properties. We report those efforts here, definitively demonstrate the mechanism of action for 1, and describe the results of an in vivo efficacy experiment for this compound in a murine model of Stage II HAT.
RESULTS
The overall strategy for the SAR exploration of 1 is shown in Figure 1. First, noting that the chlorine atoms on the indole provide substantial contribution to molecular weight and lipophilicity, we were interested to understand their importance. Second, the hydroxyethyl substituent on the indole nitrogen seemed to be a potential metabolic liability, and exploration of this region was needed. Third, the linker between the two aromatic systems in the molecule needed to be probed in terms of length, vector, and rigidity. Lastly, looking to reduce the size of the molecule, we wished to better understand the requirements of the 2-aminobenzimidazole eastern end by replacing the benzimidazole functionality. We first describe the synthesis of these analogs, and will then discuss the impact on potency and properties for this series.
Figure 1.

1 SAR regions of interest
The synthesis of 1 and its 3,5-dichloroindole analogs commenced with the condensation of hydrazine 2 and ethyl pyruvate to yield a mixture of E and Z isomers of 3 (Scheme 1). Cyclization of 3 generated 3,5-dichloroindole 4, which could be recrystallized from H2O/ethanol. Alkylation with the appropriate alkyl halides or tosylates5, 6 and subsequent reduction with DIBAL gave corresponding N-substituted 2-(hydroxymethyl)indoles 6a–f in good to high yields. Oxidation of the benzylic alcohol with freshly prepared MnO2 following the Attenburrow procedure7 produced aldehydes 7a–f in 50–71% yield. The oxidation of diol 6a also generated lactone 8 in 20% yield, likely stemming from over-oxidation of the intermediate lactol. This material could be reverted to 6a via reduction or used for further analog synthesis (vide infra). The non-halogenated 13 was synthesized in a similar manner from the commercially available indole 9 (Scheme 2).
Scheme 1. Synthesis of the 3,5-dichloroindole western end of 1 and its analogs.

Reagents and conditions: (a) ethyl pyruvate, EtOH, reflux, 1 hr; (b) PPA, 130 °C, 10 min; (c) methyl bromoacetate, K2CO3, DMF, rt, o.n.; (d) NaH, DMF, 0 °C, 30 min; then CH3I, 0 °C→rt, 3 hrs; (e) NaH, DMF, 0 °C, 30 min; then CH3OCH3Cl , 0 °C→rt, 3 hrs; (f) NaH, DMF, 0 °C, 30 min; then CH3OCH2CH2Br , 0 °C→rt, 18 hrs; (g) K2CO3, DMF, BocNH(CH2)2OTs, rt, o.n.; (h) DIBAL, THF, rt, 1 hr; (i) MnO2, THF, rt, 2 hrs.
Scheme 2. Synthesis of the western end of dechloro-1 analogs.

Reagents and conditions: (a) H2SO4, EtOH, reflux, o.n.; (b) methyl bromoacetate, K2CO3, DMF, rt, 3 hrs; (c) LiAlH4, THF, 0 °C, 30 min, rt, 1.5 hrs; (d) MnO2, CH2Cl2, rt, o.n.
The alcohol moiety of compound 6b was protected as the TBS ether, and the sulfonylation of this compound was performed using the same protocol for 5c–f; this reaction also effected desilylation, providing 16 in 31% yield (Scheme 3). The free alcohol was oxidized to the corresponding aldehyde with MnO2 in modest yield.
Scheme 3. TBS protected N-substituted indole synthesis.

Reagents and conditions: (a) TBSCl, imidazole, CH2Cl2, rt, 1 hr; (b) NaH, DMF, rt, 30 min; CH3SO2Cl, rt, 1 hr; (c) MnO2, CH2Cl2, rt, 2 hrs.
Aminoalkyl-2-aminobenzimidazole intermediates were prepared as shown in Schemes 4–7. Briefly, 20 was formed via heating of 18 in the presence of urea followed by deoxy-chlorination with POCl3. Mono-Boc protected diamines 22a–d, 26a–b, 29, and 32, prepared according to known procedures8–13 or commercially available, were coupled to 20 using microwave irradiation at 150 °C. Removal of the Boc group was effected under either acidic conditions to yield the bis-TFA salt, or via microwave promoted thermolysis to give the free base. Attempts to couple 20 with 21a directly were successful in generating product, though the purification of 24a was complicated due to its high water solubility. This was overcome by the introduction a Boc group, allowing purification via extraction followed by chromatography. The isolation of 24 was accomplished by simple concentration of the reaction medium.
Scheme 4. Chloro-1H-benzo[d]imidazole synthesis.

Reagents and conditions: (a) urea, (CH2OH)2, 140 °C, 1 hr, 170 °C, 7 hrs; (b) POCl3, reflux, 2 hrs.
Scheme 7. 1 cyclic linker analog synthesis.

Reagents and conditions: (b) 20, toluene, µW, 150 °C, 6 hrs; (b) TFA, CH2Cl2, rt, o.n.
2-(aminophenyl)aminobenzimidazoles 36a–c were prepared and purified without Boc protection under microwave heating (Scheme 8). Reaction times were reduced to 15 minutes by increasing the temperature from 150 °C to 200 °C.
Scheme 8. Phenylenediamine linker synthesis.

Reagents and conditions: (a) 20, xylenes, µW, 200 °C, 15 min.
Reductive amination of 2-indolecarboxaldehydes 7a–f, 13, and 17 with 24a was carried out with NaBH3CN under either mildly basic conditions with the bis-TFA salt of 24a, or mildly acidic conditions with the free base (Scheme 9, product structures in Table 1 and 3). The same reaction conditions were used to couple 7a with amines 24, 28, 31, 34, and 36 (Table 4).
Scheme 9. Synthesis of 1 analogs via reductive amination.

Reagents and conditions: (a) amine, KOAc, NaBH3CN, CH3OH; (b) amine, AcOH, NaBH3CN, CH3OH
Table 1.
Activity of modified indole analogs of 1 against T. brucei
 | |||||
|---|---|---|---|---|---|
| Compound | R | X |
T. brucei EC50 (nM)a |
MRC5-SV2 EC50 (µM)b |
Selectivity (TC50/EC50) |
| 1 | Cl | CH2 | 0.96 | 12 | |
| 39 | Cl | CO | 500 | 18 | 36 |
| 46 | H | CH2 | 75 | 27c | 360 |
SD <0.20.
SD <1.5 unless otherwise noted.
SD=9.3
Table 3.
Activity of indole N-substituted analogs of 1 against T. brucei
 | ||||
|---|---|---|---|---|
| Compound | R |
T. brucei EC50 (nM)a |
MRC5-SV2 TC50 (µM)b |
Selectivity (TC50/EC50) |
| 47a | H | 9.0 | 8.8 | 978 |
| 47b | CH3 | 5.0 | 9.8 | 1960 |
| 47c | 8.0 | 7.4 | 925 | |
| 47d | 620 | 10 | 16 | |
| 47e | 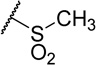 |
340 | 1.4 | 4.1 |
| 47f | 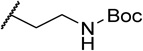 |
790 | 8.1 | 10 |
| 47g | 200 | 8.4 | 42 | |
SD <0.20.
SD <3.0.
Table 4.
Activity of linker analogs of 1 against T brucei
 | |||||
|---|---|---|---|---|---|
| Compound | R1 | R2 |
T. brucei EC50 (nM)a |
MRC5-SV2 TC50 (µM)b |
Selectivity (TC50/EC50) |
| 48a | 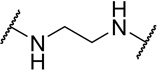 |
H | 5.0 | 26c | 5200 |
| 48b | 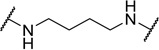 |
H | 5.0 | 11 | 2200 |
| 48c | 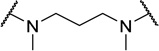 |
H | 980 | 22 | 22 |
| 48d | 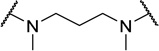 |
CH3 | 2200 | 10 | 4.5 |
| 48e | 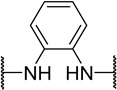 |
H | 420 | 25 | 60 |
| 48f | 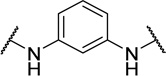 |
H | 960 | 21 | 22 |
| 48g | 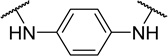 |
H | 5500 | 23 | 4.2 |
| 48h | 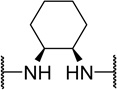 |
H | 680 | 14 | 21 |
| 48i | 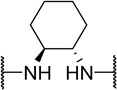 |
H | 340 | 17 | 50 |
| 48j | 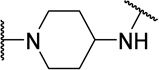 |
H | 400 | 12 | 30 |
| 48k | 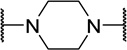 |
H | >5000 | 50 | -- |
SD <0.55.
SD <3.0 unless otherwise noted.
SD=8.8
The synthesis of analogs 38, substituting the dichloroindole scaffold with chlorophenyl rings, are shown in Scheme 10, via reductive amination from commercially available chlorobenzaldehydes 37.
Scheme 10. Synthesis of chlorophenyl 1 analogs.

Reagents and conditions: (a) 24a, AcOH, NaBH3CN, CH3OH
The side product 8 (Scheme 1) was used to produce 39 via ester aminolysis with the free base of 24a and microwave irradiation (Scheme 11).
Scheme 11. Synthesis of 38 via ester-amide exchange.

Reagents and conditions: (a) 24a, DMF, µW, 150 °C, 1 hr
To synthesize analogs varying the benzimidazole of 1, aldehyde 7a was first reductively aminated with 22a (Scheme 12). Exchange of the Boc protecting group for the acid-stable N-(1-(4,4-dimethyl-2,6-dioxocyclohexylidene)ethyl) (Dde) protecting group provided 41 in high yield. The secondary amine was Boc protected and the enamine removed upon heating in the presence of hydrazine monohydrate. The free primary amine of 43 was then reacted with phenylisocyanate to provide 44a, 2-chlorobenzoxazole (44b), or 4-chloropyrimidine (44c). Deprotection of the Boc-protected secondary amines under acidic conditions gave the final analogs in modest yields.
Scheme 12. Heterocyclic replacements on the eastern half of 1.

Reagents and conditions: (a) 22a, AcOH, NaBH3CN, CH3OH, rt, 12 hr; (b) 3M aq. HCl, THF, rt, 1 hr, 80 °C, 1 hr; then DDE-OH, (i-Pr)2NEt, CH2Cl2, CH3OH, rt, 12 hr; (c) Boc2O, K2CO3, CH3CN, H2O, rt, 12 hr; (d) H2NNH2H2O, CH3CN, 45 °C, 3 hr; (e) PhCNO, CH3CN, rt, 12 hr; (f) 2-chlorobenzoxazole, (i-Pr)2NEt, CH3CN, 80 °C, 12 hr; (g) 4-chloropyrimidine, (i-Pr)2NEt, CH3CN, µW, 150 °C, 2 hr; (h) 4M HCl in 1,4-dioxane, rt, 6 hr.
In vitro assessment against T. brucei
Indole halogens
Removal of both chlorides on the indole ring resulted in an 78x loss in potency (46, EC50: 75 nM, Table 1). A significant reduction in MW (432→364) and clogP (3.60→2.39) was effected however, resulting in the highest CNS multiparameter optimization (MPO) score14 among these analogs. CNS exposure is essential to effectively treat stage II HAT when the parasites have invaded the CNS and crossed the blood brain barrier (BBB). Noting that a CNS MPO score ≥4 is suggestive of CNS penetration, 46 score was calculated to be 4.3. This compound also retained a high lipophilic ligand efficiency (LLE, pEC50-cLogP) of 4.71 (the desirable range for LLE is ≥4).15 Replacement of the aminomethyl group of 1 with an amide (39) resulted in a 520-fold loss in activity. This correlates well with the conclusions from the linker SAR in that the basicity of the dialkylamine is essential for high potency.
Table 2 shows the results of replacing the dichloroindole scaffold with simplified chlorophenyl rings. Compound 38c, retaining the 3,5-dichloro motif showed only a slight reduction in potency (EC50=16 nM). Removal of one of the chlorine atoms (38a) resulted in an 8-fold loss in activity (EC50=0.15 µM) compared to 38c and migration of the chloride to the para position (38b) resulted in a further 6-fold loss in activity (EC50=0.96 µM). Our observation that the indole moiety in this class of compounds can be replaced with a simple dichlorophenyl group may provide a useful path forward in the event that toxicity due to indole metabolism is observed.
Table 2.
Activity of chlorophenyl analogs of 1 against T. brucei
 | ||||
|---|---|---|---|---|
| Compound | R |
T. brucei EC50 (nM)a |
MRC5-SV2 TC50 (µM)b |
Selectivity (TC50/EC50) |
| 38a | 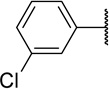 |
150 | 22 | 147 |
| 38b | 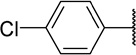 |
960 | 22 | 23 |
| 38c | 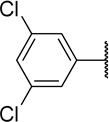 |
16 | 22 | 1375 |
SD <0.20.
SD <3.0.
Indole N-substituents
Modifications to the 2-hydroxyethyl group of 1 displayed mixed results (Table 3). Truncations (47a, 47b) or a shift of the oxygen atom to an internal position (47c) resulted in less than a 10-fold loss in activity. Larger groups (47d, 47e, 47f) comparatively caused a far larger loss in activity (>300x). Replacement of the alcohol with an amine (47g) led to around a 200-fold loss in activity. MPO scoring for these compounds was similar (≤Δ 0.3) to 1 except for the large increase resulting from addition of the methanesulfonamide group (47e, MPO=4.1) and large decrease apparent in the Boc-protected 47f (MPO=1.5). Generally, the smaller indole N-substituents remained highly active while those that increased steric bulk resulted in a drop in potency by 2–3 orders of magnitude.
Linker
Contracting or extending the diamino linker by a methylene unit (48a and 48b) did not lead to a loss in potency (EC50=5.0 nM each, Table 4) compared to 1. Compound 48a did however possess a slightly improved MPO score (3.8). Capping both amines of the linker with methyl groups was not tolerated (48c, EC50=0.98 µM). The trimethylated compound (48d) showed approximately an additional 3-fold loss in activity (EC50=2.2 µM).
The tolerance for a chain of 2–4 carbons between amines suggests adequate space within the biological target(s) of action for conformational variations of the linker. The para-, meta-, and ortho- phenylenediamine linkers, designed to restrict the conformation of the linker, all showed a dramatic loss in activity. The ortho-linked 48e was the most active (EC50=0.42 µM), followed closely by the meta-linked 48f (EC50=0.96 µM). The para-linked compound, 48g, was an additional order of magnitude less active (EC50=5.5 µM). These data suggested either that (1) a bent orientation of the linker is closest to the active conformation of 1, or (2) the basicity of these nitrogens is important.
Compounds 48h–k further probed the tolerance for reduced linker flexibility and diamine orientation. Three of the four saturated cyclic linked analogs, 48h–j, remained sub-micromolar in activity. Compounds 48h and 48i were within 2-fold of one another,showing no preference for cis or trans substitution of the cyclohexyl ring, and were essentially equipotent to the aromatic analog 48e. Compound 48k, bearing a piperazinyl linker, was >5 µM, compared to the 4-aminopiperadinyl linked 48j (EC50=0.40 µM). This difference in activity is likely not a result of linker reduction as 48a retained essentially all activity compared to 1. Rather, taken together with the poor activity of 48c and 48d, the reduced activity of 48k illustrates the importance of (1) the hydrogen bonding donor motif and (2) a high degree of flexibility in the linker for high potency.
Benzimidazole replacements
Any replacements for the benzimidazole ring that lacked a hydrogen bond donor were not tolerated (Table 5, 45b and 45c). Specifically, changing to an isosteric benzoxazole (45b) led to around an 800-fold loss in activity compared to 1. Compound 45c, also devoid of a hydrogen bond donating 4-pyrimidine ring, possessed activity approximating 45b (0.79 µM vs. 0.27 µM). Reintroduction of a hydrogen bond donor with a urea N-H (45a), which could recapitulate that of the benzimidazole, regained the high potency observed for 1.
Table 5.
Activity of chlorophenyl analogs of 1 against T. brucei
 | ||||
|---|---|---|---|---|
| Compound | R |
T. brucei EC50 (nM)a |
MRC5-SV2 TC50 (µM)b |
Selectivity (TC50/EC50) |
| 45a | 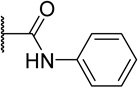 |
2.0 | 14 | 7000 |
| 45b | 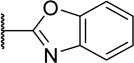 |
790 | 26 | 33 |
| 45c | 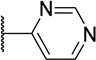 |
270 | >50 | -- |
SD <0.20.
SD <3.0.
All analogs of 1 were screened against MRC5-SV2 human lung cells to assess toxicity. Most compounds possessed a selectivity index in the 10–50x range. Compounds in the single digit nanomolar range against T. brucei displayed a higher selectivity index in the range of 900–7,000x.
Physicochemical and ADME properties
The calculated physicochemical properties of compounds 1, 38a–c, 39, 45a–c, 46, 47a–g, and 48a–k are presented in Table 6, color coded in terms of desirability. Compound 47e is noteworthy among analogs replacing the 2-hydroxyethyl group for its high MPO score above 4.0, due to its reduced clogP and pKa (calculated using JChem for Excel, Chemaxon, Inc.) Analogs 48g, 48f, 48e, 48h, and 48i bearing phenylenediamine and 1,2-diaminocyclohexyl linkers possess poor MPO scores as a result of high molecular weights, clogP, and clogD values.
Table 6.
Physicochemical properties of 1 and its analogs
| Cmpd | MW (g/mol)a |
clogPa,c | clogDa,d | H-bond donorsa |
pKab,c | TPSA (Å2)bc |
MPO Scoree |
LE | LLE |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 432.4 | 3.60 | 1.59 | 4 | 9.29 | 82.48 | 3.5 | 0.31 | 5.57 |
| 38a | 314.8 | 3.44 | 1.57 | 3 | 9.15 | 57.32 | 4.4 | 0.31 | 3.38 |
| 38b | 314.8 | 3.44 | 1.53 | 3 | 9.19 | 57.32 | 4.4 | 0.27 | 2.49 |
| 38c | 349.3 | 4.04 | 2.33 | 3 | 8.98 | 57.32 | 4.0 | 0.33 | 3.65 |
| 39 | 446.3 | 3.11 | 2.98 | 4 | 6.98 | 94.97 | 3.7 | 0.21 | 3.19 |
| 45a | 435.4 | 3.28 | 1.36 | 4 | 9.34 | 82.90 | 3.7 | 0.30 | 5.42 |
| 45b | 433.3 | 3.66 | 1.78 | 3 | 9.29 | 79.83 | 3.7 | 0.21 | 2.45 |
| 45c | 394.3 | 2.30 | 0.42 | 3 | 9.29 | 79.58 | 4.3 | 0.25 | 4.27 |
| 46 | 363.5 | 2.39 | 0.36 | 4 | 9.31 | 82.48 | 4.3 | 0.26 | 4.71 |
| 47a | 388.3 | 4.06 | 2.06 | 4 | 9.28 | 73.11 | 3.6 | 0.31 | 4.04 |
| 47b | 402.3 | 4.29 | 2.28 | 3 | 9.29 | 62.25 | 3.4 | 0.31 | 4.11 |
| 47c | 432.4 | 4.35 | 2.36 | 3 | 9.27 | 71.48 | 3.2 | 0.28 | 3.75 |
| 47d | 446.4 | 4.24 | 2.23 | 3 | 9.29 | 71.48 | 3.2 | 0.21 | 1.96 |
| 47e | 466.4 | 2.88 | 2.08 | 3 | 8.01 | 96.39 | 4.1 | 0.22 | 3.59 |
| 47f | 531.5 | 4.98 | 2.97 | 4 | 9.29 | 100.58 | 1.5 | 0.17 | 1.12 |
| 47g | 431.4 | 3.49 | −0.73 | 4 | 9.83 | 89.89 | 3.3 | 0.23 | 3.21 |
| 48a | 418.3 | 3.54 | 1.88 | 4 | 8.97 | 82.48 | 3.8 | 0.30 | 4.76 |
| 48b | 446.4 | 4.11 | 1.82 | 4 | 9.60 | 82.48 | 3.0 | 0.28 | 4.39 |
| 48c | 460.4 | 4.62 | 3.43 | 2 | 8.54 | 61.52 | 3.0 | 0.20 | 1.48 |
| 48d | 474.4 | 4.84 | 3.66 | 1 | 8.54 | 50.66 | 3.0 | 0.18 | 0.81 |
| 48e | 466.4 | 5.34 | 5.07 | 4 | 7.38 | 79.15 | 2.2 | 0.20 | 1.06 |
| 48f | 466.4 | 5.34 | 5.06 | 4 | 7.39 | 79.15 | 2.2 | 0.19 | 0.76 |
| 48g | 466.4 | 5.34 | 5.04 | 4 | 7.42 | 79.15 | 2.2 | 0.16 | −0.14 |
| 48h | 472.4 | 4.95 | 2.94 | 4 | 9.35 | 82.48 | 2.1 | 0.20 | 1.51 |
| 48i | 472.4 | 4.95 | 2.94 | 4 | 9.35 | 82.48 | 2.1 | 0.19 | 1.22 |
| 48j | 458.4 | 3.93 | 3.61 | 3 | 7.38 | 70.31 | 3.2 | 0.21 | 2.47 |
| 48k | 444.4 | 4.38 | 4.30 | 2 | 6.73 | 60.32 | 3.2 | 0.18 | 0.92 |
Values that are within the MPO more desired ranges are green and outside the less desired range are red.
Calculated using ChemAxon JChem for Excel.
Calculated at pH 7.4 using ChemAxon JChem for Excel.
Calculated using MPO calculator previously reported.14
An excellent MPO score (4.3) is calculated for 46, which resulted from reduced molecular weight compared to 1 (363.5, Δ68.9), clogP (2.39, Δ1.21), and clogD (0.36, Δ1.23); this results simply from the removal of the chlorine atoms from the indole. In addition to the improved MPO scores of many compounds in this series, several compounds retained LE values ≥0.30 and LLE values ≥4.0, indicating a good balance of size and lipophilicity.
In vitro absorption, distribution, metabolism, and excretion (ADME) properties were collected for several compounds and are tabulated in Table 7. Few compounds improved the aqueous solubility to an appreciable extent, with 46 as the only significant exception at 298 µM. Compound 46 was also the only analog to show reduced human plasma protein binding (PPB) to <99%, or to reduce the logD7.4 below 3.0 among those tested. Of the 16 compounds assessed for human liver microsome clearance (HLM Clint), only 4 analogs possessed lower clearance than 1. Compounds 47a and 47b displayed the lowest human microsomal clearance, implicating the 2-hydroxyethyl group may indeed be a metabolic handle as we expected. Others with high microsomal clearance are 47d, 47e, 47f, 48c, 48e, and 48h; the Boc protected analog 47f and the dimethylamino analog 48c both showing rapid metabolism (142 and 135 µL/min/mg respectively). Similarly, few compounds possessed good rat hepatocyte clearance, with only 47g (Clint = 7.96 µL/min/106) and 48k (Clint = 12.8 µL/min/106) showing any significant improvement over 1.
Table 7.
ADME measurements of 1 and analogs
| Compound | Aqueous Solubility (uM) |
Human PPB (%) |
HLM Clint (µL/min/mg) |
HLM t1/2 (min) |
Rat Hepatocyte Clint (µL/min/106) |
Rat Hepatocyte t1/2 (min) |
logD 7.4 |
|---|---|---|---|---|---|---|---|
| 1 | 6.33 | > 99.9 | 24 | * | 24.9 | * | 4.0 |
| 46 | 298 | 98.61 | 22.6 | * | 81.4 | * | 2.0 |
| 47a | 4.02 | 99.8 | 15.5 | * | 17.2 | * | 4.3 |
| 47b | 12.2 | 99.8 | 12.9 | * | 29.3 | * | 4.5 |
| 47c | < 1 | 99.9 | 34.2 | * | 33.6 | * | 4.0 |
| 47d | 3.77 | > 99.9 | 43.8 | * | 49.7 | * | 4.1 |
| 47e | 19 | * | 80.9 | 8.6 | 98.8 | 7 | * |
| 47f | 32 | * | 142 | 4.9 | 33.3 | 20.8 | * |
| 47g | * | * | * | * | 7.96 | 87.1 | * |
| 48a | 21 | 99.8 | 36 | * | 30.7 | * | 4.3 |
| 48b | 4.26 | 99.9 | 22.5 | * | 17 | * | 3.7 |
| 48c | 11.7 | > 99.9 | 135 | * | 61.7 | * | 4.0 |
| 48d | * | * | * | * | > 300 | < 2.3 | * |
| 48e | * | > 99.9 | 51.6 | * | 100 | * | 4.9 |
| 48f | < 1 | > 99.9 | 33.9 | * | 22.5 | * | 4.3 |
| 48g | 1.39 | > 99.9 | 27.3 | * | 18.7 | * | 4.7 |
| 48h | 11 | > 100 | 64.2 | * | 115 | * | 4.0 |
| 48j | 0.3 | 100 | * | * | 38.8 | * | 4.4 |
| 48k | < 1 | 99.0 | 21.1 | * | 12.8 | * | 4.9 |
Not obtained
Pharmacokinetics study
Though we previously reported plasma pharmacokinetics, we did not determine CNS drug levels. Thus, female BALB/c mice were injected with a single, 10 mg/kg intraperitoneal dose of 1, and plasma and brain levels were measured, showing a brain-to-plasma ratio of 0.39, with drug levels of 31.6 ng/g of brain tissue measured at the 24 hour time point. (The PK parameters are summarized in the Supporting Information, Tables S1–S3 and Figure S1). With this evidence in hand, we proceeded to assess this compound in a mouse model of Stage II HAT.
Murine CNS infection model
We advanced 1 to a mouse model of a CNS infection with T. b. brucei (GVR 35 strain). On day 0, 2 groups of 5 mice each were infected with T. b. brucei and checked for parasitemia on day 14. One group, taken as the control, was treated with a single dose of 40 mg/kg Berenil on day 21. From days 22 to 25 the control group was treated with the vehicle DMSO in PBS. The other group of mice was treated with 1 at a dose of 20 mg/kg/day on days 21–25. Following a 2-day hiatus, treatment with 1 was resumed on days 28–32. Parasitemia was checked from both groups via examination of tail blood on days 14, 28, 35, 39, and 43. The results are reported in Figure 2 and Table S-4.
Figure 2.

Mice without parasitemia in a mouse model of CNS infection with T. b. brucei
On day 28, 2 days after the 1st regimen of treatment was completed, 3/5 mice treated with 1 showed no detectable parasitemia. Following the 2nd round of treatment, this number increased to 4/5 mice showing no detectable parasitemia on day 35. When checked on day 39 however, all mice treated with 1 showed a full relapse of parasites in the blood. The control group comparatively showed no detectable levels of parasitemia when checked on days 28, 35, and 39 in all 5 mice, and only 2 mice showing a relapse on day 43. Since recrudescence was observed even in animals where the bloodstream infection was cleared by 1, we interpret this result to suggest that compound 1 does not have sufficient antiparasitic activity in the brain to cure Stage 2 HAT. This may be attributed in part to the high PPB (>99.9%) and rapid clearance (rat hepatocyte Clint=24.9 µL/min/106 cells) of 1.
Target Identification
Though the potent activity of 1 was uncovered from a set of compounds selected on the basis of their likely kinase inhibition activity, we noted that the structure of 1 was quite similar to a class of previously reported T. brucei methionyl-tRNA synthetase (MetRS) inhibitors (such as compound 1312, Figure 3).16 T. brucei has a single MetRS and is necessary for cell growth as previously reported using RNA interference.16 We wished to determine whether 1 inhibited T brucei MetRS.
Figure 3.

Structures of previously published T. brucei MetRS inhibitors (131216, 143317, and Met-SA118)
Biochemical assessment
A functional assay for the T. brucei MetRS enzyme based on ATP-depletion was previously described.19 Compound 1 was determined to be a highly potent inhibitor, with an IC50 below the lower detection limit of the assay (<25 nM) (Figure 4). The positive control compound, Met-SA118 Figure 3, is also a potent inhibitor with an IC50 of <25 nM.
Figure 4.

T. brucei MetRS inhibition assay. IC50s of both the positive control, Met-SA1, and compound 1 are <25 nM (the lower detection limit of the assay as described in the results and discussion). Error bars represent standard error of the mean of three replicates. Pentamidine, a negative control, had no T. brucei MetRS inhibition up to 10 µM (results not shown).
Potency shift in resistant parasites
To further test if compound 1 is targeting the T. brucei MetRS, a T. brucei growth inhibition cell assay was conducted with a strain that is resistant to MetRS inhibitors. The resistant strain was generated by serial in vitro passage of wild-type T. brucei with the MetRS inhibitor 1433 (Figure 3) and was shown to have ~35-fold upregulated expression of MetRS mRNA.18 The EC50 of 1 and 1433 are ~107-fold and ~48-fold less potent against the 1433-resistant strain compared to the wild-type (Figure 5 and Table 8), consistent with the hypothesis that 1 acts by inhibiting the MetRS enzyme. Pentamidine does not inhibit T. brucei MetRS (data not shown) and does not show a difference in EC50 values between the 1433-resistant and wild-type strains.
Figure 5.

Wild-type and 1433-resistant T. brucei growth inhibition assays. Results from two independent assays, as indicated with the 1 and 2. Error bars represent standard error of the mean of three replicates.
Table 8.
Wild-type and 1433-resistant T. brucei growth inhibition assays. EC50s represent averages and standard error of the mean of two independent assays.
| Strain | |||
|---|---|---|---|
| Compound | WT EC50 (nM) | 1433 resistant EC50 (nM) | 1433 resistant EC50 / WT EC50 |
| Pentamidine | 1.5 ± 0.2 | 1.1 ± 0.1 | 0.73 |
| 1433 | 98 ± 1 | 4700 ± 230 | 48 |
| 1 | 0.13 ± 0.01 | 14 ± 1 | 108 |
Structural biology
Even though crystals of TbruMetRS•Met would disintegrate after ~20 seconds in contact with a solution of 1, it was possible to replace the bound methionine with 1 in the binding site using short soaking times.20 The structure in complex with 1 was determined at 2.6 Å resolution and deposited in the Protein Data Bank with PDB identifier 5TQU. Data collection and refinement statistics are shown in Table S5 (supporting information) and the electron density map for the compound is shown in Figure 6A. Compound 1 binds to TbruMetRS in a manner similar to previously-reported compound-bound structures.20, 21 Characteristically, two pockets are involved in binding of the inhibitor (Figure 6B): the enlarged methionine pocket (EMP), formed mainly by hydrophobic residues including the ones engaged in methionine binding, and the “auxiliary pocket” (AP), not present in the methionine bound state.20 The EMP, formed mainly as a result of the movement of TbruMetRS residues Val473, Trp474 and Phe522, is occupied by the 4,6-dichloroindolyl-ring moiety which also interacts with residues Ile248 and Tyr481. The AP is occupied by the benzimidazole moiety which interacts with His289, Gly290 and Val473, while establishing a hydrogen bond with Asp287 through one of the nitrogens of the benzimidazole ring with a distance of 3.0 Å. The flexible linker of 1 forms hydrophobic interactions with the aromatic group of residue Tyr250 and a hydrogen bond through the exocyclic amine of the benzimidazole moiety with the catalytic Asp287 with a distance of 2.8 Å (Figure 6C). In a novel interaction pattern for TbruMetRS inhibitors, the OH group in the hydroxyethyl substituent of the 4,6-dichloroindolyl-ring moiety makes a hydrogen bond with residue Asp287 with a distance of 2.6 Å. As a result, Asp287 plays a central role in establishing polar contacts with the three different regions of 1: the linker, the benzimidazole moiety and the OH group in the hydroxyethyl moiety. Furthermore, at least one water molecule is part of what seems to be a crucial hydrogen bonding network involving Asp287 and 1 (Figure 6C).
Figure 6.

Binding of 1 to TbruMetRS. (A) The TbruMetRS•1 structure (PDB: 5TQU) with the difference electron density map calculated by omitting the inhibitor, contoured at the 3σ level (positive density in grey, negative density in red). (B) General features of the 1 binding mode. The position of the methionine sulfur atom in the TbruMetRS•methionine structure (PDB: 4EG1)20 is shown with a yellow cross. The protein surface and the two pockets, EMP and AP, where the inhibitor is bound are shown. (C) Hydrogen bond network in the TbruMetRS•1 structure. The label for residue Asp287 is highlighted with a rectangle. The hydroxyethyl substituent of the 4,6-dichloroindolyl-ring moiety establishes an extra hydrogen bond with residue Asp287. (D) Chemical structures of compound 1 and the previously reported TbruMetRS inhibitor 1331. (E) Superposition of compounds 1 and 1331 (PDB: 4EG7)20 bound to TbruMetRS. (F) Hydrogen bond interactions between inhibitor and enzyme in the TbruMetRS•1331 structure.
When comparing the new structure to previously reported TbruMetRS complexes with inhibitors bound, the overall binding mode of 1 is quite similar. Taking the TbruMetRS•1331 structure (Figure 6D) as a representative of previously determined inhibitor bound structures, it appears that the two ring systems of each inhibitor occupy similar positions. For instance, the two chlorine atoms are only 0.8 and 0.3 Å (Cl-4 and Cl-6, respectively) apart in the two structures (Figure 6E) and the linkers follow a similar path. The key difference is the extra interaction of the hydroxyl group of 1 with Asp287 (compare Figures 6C and 6f), representing one additional hydrogen bond between the inhibitor and the target enzyme.
CONCLUSIONS
In summary, we report the structure-activity relationships for the various regions of 1, and described its activity in a mouse model of CNS infection of T. brucei. In addition, the compound’s mechanism of action via T. brucei MetRS was conclusively elucidated by a combination of biochemical, cellular, and crystallographic techniques. Despite the excellent in vitro anti-parasitic potency of 1, the lack of CNS activity in the late-stage model of HAT necessitates further optimization of CNS permeability and pharmacokinetics; this work is ongoing.
Supplementary Material
Scheme 5. Acyclic eastern half synthesis.

Reagents and conditions: (a) Boc2O, CHCl3, 0 °C→rt, o.n.; (b) 20, toluene, µW, 150 °C, 6 hrs; (c) NaH, DMF, CH3I, rt, 1 hr; (d) TFA, CH2Cl2, rt, o.n.; (e) H2O, µW, 150 °C, 1 hr
Scheme 6. 1 cyclohexyl linker replacement synthesis.

Reagents and conditions: (a) Boc2O, CH2Cl2, 0 °C→rt, o.n.; (b) 20, toluene, µW, 150 °C, 6 hrs; (c) TFA, CH2Cl2, rt, o.n.
Acknowledgments
Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award numbers R56AI099476 (to MPP), R01AI114685 (to MPP and MN), R01AI084004 (to WGJH) and R01AI097177 (to FSB and RMR). We acknowledge the support of a Fulbright Fellowship to XB-A. Funding from Northeastern University is also acknowledged. We wish to thank Erkang Fan (University of Washington) for providing compounds 1433 and Met-SA1. We thank Christophe L. M. Verlinde for his contributions to the structure validation process. We thank Stewart Turley and Robert Steinfeldt for providing support for the X-ray data collection and computing environment at the Biomolecular Structure Center of the University of Washington. Crystallography performed in support of the work benefitted from remote access to resources at the Stanford Synchrotron Radiation Lightsource supported by the U.S. Department of Energy Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515 and by the National Institutes of Health (P41GM103393).
Footnotes
Experimental details for the pharmacokinetics experiments, chemical syntheses, X-ray crystallography, and in vitro and in vivo biological methods are available in the Supporting Information. In addition, annotation of compounds with NEU registry numbers and SMILES strings are provided. This information is available free of charge via the Internet at http://pubs.acs.org/.
REFERENCES
- 1.Jacobs RT, Nare B, Wring SA, Orr MD, Chen D, Sligar JM, Jenks MX, Noe RA, Bowling TS, Mercer LT, Rewerts C, Gaukel E, Owens J, Parham R, Randolph R, Beaudet B, Bacchi CJ, Yarlett N, Plattner JJ, Freund Y, Ding C, Akama T, Zhang YK, Brun R, Kaiser M, Scandale I, Don R. SCYX-7158, an Orally-Active Benzoxaborole for the Treatment of Stage 2 Human African Trypanosomiasis. PLoS Negl Trop Dis. 2011;5:e1151. doi: 10.1371/journal.pntd.0001151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Torreele E, Bourdin Trunz B, Tweats D, Kaiser M, Brun R, Mazué G, Bray MA, Pécoul B. Fexinidazole - A New Oral Nitroimidazole Drug Candidate Entering Clinical Development for the Treatment of Sleeping Sickness. PLoS Negl Trop Dis. 2010;4:e923. doi: 10.1371/journal.pntd.0000923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hay M, Thomas DW, Craighead JL, Economides C, Rosenthal J. Clinical development success rates for investigational drugs. Nat Biotech. 2014;32:40–51. doi: 10.1038/nbt.2786. [DOI] [PubMed] [Google Scholar]
- 4.Diaz R, Luengo-Arratta SA, Seixas JD, Amata E, Devine W, Cordon-Obras C, Rojas-Barros DI, Jimenez E, Ortega F, Crouch S, Colmenarejo G, Fiandor JM, Martin JJ, Berlanga M, Gonzalez S, Manzano P, Navarro M, Pollastri MP. Identification and characterization of hundreds of potent and selective inhibitors of Trypanosoma brucei growth from a kinase-targeted library screening campaign. PLoS Negl Trop Dis. 2014;8:e3253. doi: 10.1371/journal.pntd.0003253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mansueto M, Frey W, Laschat S. Ionic Liquid Crystals Derived from Amino Acids. Chemistry - A European Journal. 2013;19:16058–16065. doi: 10.1002/chem.201302319. [DOI] [PubMed] [Google Scholar]
- 6.Loison S, Cottet M, Orcel H, Adihou H, Rahmeh R, Lamarque L, Trinquet E, Kellenberger E, Hibert M, Durroux T, Mouillac B, Bonnet D. Selective Fluorescent Nonpeptidic Antagonists For Vasopressin V2 GPCR: Application To Ligand Screening and Oligomerization Assays. Journal of medicinal chemistry. 2012;55:8588–8602. doi: 10.1021/jm3006146. [DOI] [PubMed] [Google Scholar]
- 7.Attenburrow J, Cameron AFB, Chapman JH, Evans RM, Hems BA, Jansen ABA, Walker T. 194. A synthesis of vitamin a from cyclohexanone. Journal of the Chemical Society (Resumed) 1952:1094–1111. [Google Scholar]
- 8.Montero A, Goya P, Jagerovic N, Callado LF, Meana JJ, Girón Ro, Goicoechea C, Martin MI. Guanidinium and aminoimidazolinium derivatives of N-(4-piperidyl)propanamides as potential ligands for µ opioid and I2-imidazoline receptors: synthesis and pharmacological screening. Bioorganic & Medicinal Chemistry. 2002;10:1009–1018. doi: 10.1016/s0968-0896(01)00356-x. [DOI] [PubMed] [Google Scholar]
- 9.New OM, Dolphin D. Design and Synthesis of Novel Phenothiazinium Photosensitiser Derivatives. European Journal of Organic Chemistry. 2009;2009:2675–2686. [Google Scholar]
- 10.Schmuck C, Dudaczek J. Ion Pairing Between the Chain Ends Induces Folding of a Flexible Zwitterion in Methanol. European Journal of Organic Chemistry. 2007;2007:3326–3330. [Google Scholar]
- 11.Marugan JJ, Zheng W, Motabar O, Southall N, Goldin E, Westbroek W, Stubblefield BK, Sidransky E, Aungst RA, Lea WA, Simeonov A, Leister W, Austin CP. Evaluation of Quinazoline Analogues as Glucocerebrosidase Inhibitors with Chaperone Activity. Journal of medicinal chemistry. 2011;54:1033–1058. doi: 10.1021/jm1008902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cortez NA, Aguirre G, Parra-Hake M, Somanathan R. New heterogenized C2-symmetric bis(sulfonamide)-cyclohexane-1,2-diamine-RhIIICp* complexes and their application in the asymmetric transfer hydrogenation (ATH) of ketones in water. Tetrahedron Letters. 2009;50:2228–2231. [Google Scholar]
- 13.Beena Joshi S, Kumar N, Kidwai S, Singh R, Rawat DS. Synthesis and Antitubercular Activity Evaluation of Novel Unsymmetrical Cyclohexane-1,2-diamine Derivatives. Archiv der Pharmazie. 2012;345:896–901. doi: 10.1002/ardp.201200219. [DOI] [PubMed] [Google Scholar]
- 14.Wager TT, Hou X, Verhoest PR, Villalobos A. Moving beyond rules: The development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem. Neurosci. 2010;1:435–449. doi: 10.1021/cn100008c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shultz MD, Cheung AK, Kirby CA, Firestone B, Fan J, Chen CH, Chen Z, Chin DN, Dipietro L, Fazal A, Feng Y, Fortin PD, Gould T, Lagu B, Lei H, Lenoir F, Majumdar D, Ochala E, Palermo MG, Pham L, Pu M, Smith T, Stams T, Tomlinson RC, Toure BB, Visser M, Wang RM, Waters NJ, Shao W. Identification of NVP-TNKS656: The Use of Structure-Efficiency Relationships To Generate a Highly Potent, Selective, and Orally Active Tankyrase Inhibitor. Journal of medicinal chemistry. 2013;56:6495–6511. doi: 10.1021/jm400807n. [DOI] [PubMed] [Google Scholar]
- 16.Shibata S, Gillespie JR, Kelley AM, Napuli AJ, Zhang Z, Kovzun KV, Pefley RM, Lam J, Zucker FH, Van Voorhis WC, Merritt EA, Hol WG, Verlinde CL, Fan E, Buckner FS. Selective inhibitors of methionyl-tRNA synthetase have potent activity against Trypanosoma brucei Infection in Mice. Antimicrob Agents Chemother. 2011;55:1982–1989. doi: 10.1128/AAC.01796-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shibata S, Gillespie JR, Ranade RM, Koh CY, Kim JE, Laydbak JU, Zucker FH, Hol WGJ, Verlinde CLMJ, Buckner FS, Fan E. Urea-Based Inhibitors of Trypanosoma brucei Methionyl-tRNA Synthetase: Selectivity and in Vivo Characterization. Journal of Medicinal Chemistry. 2012;55:6342–6351. doi: 10.1021/jm300303e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ranade RM, Zhang Z, Gillespie JR, Shibata S, Verlinde CLMJ, Hol WGJ, Fan E, Buckner FS. Inhibitors of Methionyl-tRNA Synthetase Have Potent Activity against Giardia intestinalis Trophozoites. Antimicrobial Agents and Chemotherapy. 2015;59:7128–7131. doi: 10.1128/AAC.01573-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pedro-Rosa L, Buckner FS, Ranade RM, Eberhart C, Madoux F, Gillespie JR, Koh CY, Brown S, Lohse J, Verlinde CL, Fan E, Bannister T, Scampavia L, Hol WG, Spicer T, Hodder P. Identification of potent inhibitors of the Trypanosoma brucei methionyl-tRNA synthetase via high-throughput orthogonal screening. J Biomol Screen. 2015;20:122–130. doi: 10.1177/1087057114548832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koh Cho Y, Kim Jessica E, Shibata S, Ranade Ranae M, Yu M, Liu J, Gillespie JR, Buckner Frederick S, Verlinde Christophe LMJ, Fan E, Hol Wim GJ. Distinct States of Methionyl-tRNA Synthetase Indicate Inhibitor Binding by Conformational Selection. Structure. 2012;20:1681–1691. doi: 10.1016/j.str.2012.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koh CY, Kim JE, Wetzel AB, de van der Schueren WJ, Shibata S, Ranade RM, Liu J, Zhang Z, Gillespie JR, Buckner FS, Verlinde CLMJ, Fan E, Hol WGJ. Structures of Trypanosoma brucei Methionyl-tRNA Synthetase with Urea-Based Inhibitors Provide Guidance for Drug Design against Sleeping Sickness. PLoS Neglected Tropical Diseases. 2014;8:e2775. doi: 10.1371/journal.pntd.0002775. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.