

Abstract
Identifying evolutionarily conserved blocks in orthologous genomic sequences is an effective way to detect regulatory elements. In this study, with the aim of elucidating the architecture of the regulatory network, we systematically estimated the degree of conservation of the upstream sequences of 3,750 human–mouse orthologue pairs along 8-kb stretches. We found that the genes with high upstream conservation are predominantly transcription factor (TF) genes. In particular, developmental process-related TF genes showed significantly higher conservation of the upstream sequences than other TF genes. Such extreme upstream conservation of the developmental process-related TF genes suggests that the regulatory networks involved with developmental processes have been evolutionarily well conserved in both human and mouse lineages.
Keywords: cis-element, development, noncoding, ZFHX1B, Hirschsprung disease
Cross-species genome-wide comparison of noncoding orthologous sequences has been demonstrated to be effective for identifying regulatory sequences for Saccharomyces species (1, 2). For higher eukaryotes, orthologous noncoding sequence comparison has been successfully applied to human and mouse sequences (3–5). These results can contribute to the elucidation of the architecture of regulatory networks.
However, because comprehensive knowledge regarding regulatory networks remains to be elucidated, particularly for higher eukaryotes, direct comparison of their regulatory networks is still difficult. Thus, in the present study, with the aim of elucidating the features of regulatory networks that are characteristic of higher eukaryotes, we systematically estimated the degree of the sequence conservation upstream of human–mouse orthologous genes and categorized the gene function according to the Gene Ontology (GO) Consortium (6).
In higher eukaryotes, the regulatory sequences are located in a wider range outside the coding sequences than in yeast. However, to date, 85% of mouse regulatory sequences have been estimated to be located within 2 kb from the promoter, and most promoters reside immediately upstream of the transcription start site (7), both of which play major roles in gene expression control. Thus, between humans and mice, we can expect that the degree of orthologous upstream sequence conservation in the kilobase range could reflect the evolutionary conservation of features related to gene expression control.
In the present study, we examined the upstream sequences of 3,750 human–mouse orthologous gene pairs and constructed a global alignment of the 8-kb upstream sequences for each of the orthologous gene pairs based on their local alignments. To identify human–mouse orthologous genes, we focused on genes that have been assigned an identical official gene symbol (www.gene.ucl.ac.uk/nomenclature/) between humans and mice, because these kinds of genes are annotated not only on the basis of sequence homology but also on evidence from functional and physiological experiments.
We report here that the genes with high upstream conservation are predominantly transcription factor (TF) genes. Furthermore, we show that the developmental process-related TF genes have significantly higher conservation of the upstream sequences than other TF genes.
Materials and Methods
Orthologue Identification and Upstream Sequence Collection. We searched the human and mouse Reference Sequence (RefSeq) (8) annotations from the National Center for Biotechnology Information (ftp://ftp.ncbi.nih.gov/refseq/LocusLink/LL_tmpl) for genes whose human and mouse official gene symbols were identical (9,207 gene pairs, as of February 2, 2004). Next, we selected only the nuclear protein-coding genes (7,408 genes). For these genes, we collected the corresponding genomic sequences, i.e., the RefSeq contig entries, according to the contig feature descriptions in the RefSeq annotations. We surveyed the entire annotation of every contig to check whether there were any genic sequences within the 9-kb stretch upstream of the first coding site for each of the genes collected. Then, we excised the 8-kb genomic sequence immediately upstream of the coding start site for every gene that did not contain any descriptions of genic regions within its 9-kb upstream stretch. We set a 1-kb margin to decrease the frequency of cases in which the excised 8-kb sequences overlapped with promoter regions or 3′ regulatory sequences of adjacent genes. For genes having alternative coding start sites, we always used the most 5′ coding start site according to the annotation.
Genomic Global Alignment. Initially, we made local nucleotide alignments of every human–mouse orthologue pair of genomic sequences by using blast 2 sequences (9). To appropriately align the short conserved regulatory sequences in the noncoding regions, we reduced the mismatch penalty to –2 and shortened the word size to 7. We processed the resultant set of alignments by using the program realigner, which we developed to obtain genomic global alignments based on the results from blast 2 sequences. First, we selected the local alignments by using the following set of criteria: hit length >7 bps, identity of 70% or higher, and hit strand in the same direction. For these local alignments, realigner performed the following two steps: (i) when two local alignments overlapped, the program removed the alignment with the lower bit score and retained the other and (ii) when two local alignments were not syntenic, the alignment with the lower bit score was removed and the other was retained. realigner performed steps i and ii in decreasing order of the bit score for each local alignment of each sequence pair. In these steps, if the bit scores to be compared were equal, then the longer hit-stretch and then the more downstream alignments had the higher priority. Finally, the numbers of identical sites for every local alignment were summed for each orthologue pair.
Simulation Analysis. We generated 10,000 pairs of 8-kb random sequences. Each pair of 8-kb sequences was generated so that its frequencies of A, T, G, C, and N became proportional to the observed average counts of all of the examined human and mouse 8-kb sequences, respectively. These 10,000 random sequence pairs were then processed in the same way as described above.
Validation of Genomic Alignment Procedures by the Eukaryotic Promoter Database (EPD) Data Set. We downloaded the EPD (10) data set (Release 77_1, February 2004) from ftp://ftp.epd.unil.ch/pub/databases/epd/77_1. We first retrieved every human EPD entry that was assigned an official gene symbol. Then, we selected every entry that matched the official gene symbol of any of the genes in our analysis (345 EPD entries). Because each EPD entry had a 60-bp promoter region sequence (spanning from 50 bp upstream to 10 bp downstream of the experimentally validated transcription start site), we further selected every EPD entry whose entire 60-bp sequence had an exact match with a consecutive 60-bp stretch of the corresponding human 8-kb upstream sequence (266 EPD entries). Then, we checked whether each of these 266 EPD sequences overlapped with or was covered by the conserved human upstream sequence of the corresponding alignments.
Identification of TF Genes by GO Annotation. We regarded a given gene that was assigned the GO term “transcription factor activity” as a TF gene. This retrieval procedure identified 305 TF genes (Ntf). We adopted this tight retrieval strategy so as not to overestimate the number of TF genes and to thereby make the statistical test sufficiently stringent.
Categorization of Genes by GO Annotation. We first searched the GO annotation for controlled terms that included “development,” “metabolism,” “biosynthesis,” “cell cycle,” and “signal transduction.” We then checked every retrieved term manually to confirm that it was relevant to each corresponding category. We finally confirmed 347 developmental process-related genes (Ndev).
Statistical Analysis. We counted the total number of genes that were assigned any of the GO terms in the categories of molecular function or biological process for either the mouse or human annotation (Ntotal = 2,883). Assuming a binomial distribution of ptf = Ntf/Ntotal, we calculated the cumulative probability, p, of observing T or more TF genes in the top n genes as follows, unless specified otherwise:
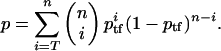 |
Retrieval of SNP Information for the ZFHX1B Gene. We searched the RefSeq contig annotations for every description of variation linked to the SNP Database (11) (www.ncbi.nlm.nih.gov/SNP) within the range of the 8-kb upstream stretch of the ZFHX1B gene. We also confirmed the variation information according to the H-Invitational Database (12) (www.h-invitational.jp).
Results
Alignment of the 8-kb Upstream Sequences of the Human–Mouse Orthologue Pairs. We identified 9,207 genes whose human and mouse official gene symbols were identical. We then selected only the nuclear protein-coding genes, which amounted to 7,408 genes. We regarded these gene pairs as orthologues. Among these, we were able to collect 9-kb genomic upstream nucleotide sequences without any described genic regions for 3,750 orthologous gene pairs. Then, we excised 8-kb stretches upstream of the translation start sites. We set a 1-kb margin to decrease the frequency of cases in which the excised 8-kb sequences overlapped with promoter regions or 3′ regulatory sequences of adjacent genes. For all of the 3,750 pairs of human and mouse genes, the accessions of the contig entries used are shown in Table 3, which is published as supporting information on the PNAS web site, together with the positions of the excised sequences. Finally, we were able to make a global pairwise alignment for each of the 3,750 orthologue pairs by using realigner.
Simulation Analysis. To estimate the level of the number of identical sites obtained by chance in the 8-kb alignments, we conducted a simulation study in which 10,000 pairs of randomly generated 8-kb sequences were processed in the same way as described above (Fig. 1). Because 10 of the 10,000 random sequence pairs exceeded 287 as the number of identical sites, we regarded any orthologous genomic global alignment whose number of identical sites was below this value as not significant at the P < 0.001 level and only used the orthologues above this threshold for further analyses (3,055 of 3,750 orthologue pairs).
Fig. 1.

Bar graph showing the frequencies of the 3,750 human–mouse orthologue pairs relative to the number of identical sites along the 8-kb upstream sequences. The area of each bar corresponds to each relative frequency. The line graph shows the relative frequency of the result of the simulation study in which 10,000 randomly generated 8-kb sequence pairs were processed in the same way as the human–mouse orthologue alignments.
Validation for Conserved Sequences Reflecting Regulatory Regions. To confirm that the conserved upstream sequences obtained by the alignment procedures were actually able to reflect the major regulatory regions, we focused on the experimentally validated promoter regions that were registered in the EPD. We could assign 266 entries in the EPD to the human upstream sequences under analysis. As a result, 80.0% of the 266 EPD sequences were covered by conserved sequences in the alignments over at least a 10-bp stretch, and, for 40.0% of the 266 entries, the entire 60-bp EPD promoter region was completely covered in the conserved sequences. These results indicate that our genomic alignment procedures are effective for detecting the regulatory regions.
Repetitive Sequences of Highly Upstream-Conserved Genes. Within all of the upstream sequences of the top 1–10 upstream-conserved orthologue pairs, we found very few transposable element (TE) insertions (i.e., 0.42% of the human and 0.56% of the mouse 8-kb upstream sequences) by using repeatmasker (http://ftp.genome.washington.edu/RM/RepeatMasker.html), although TE insertions have been reported to occupy ≈45% of the human (13) and 37.5% of the mouse (7) genome sequences. This result indicates that the highly conserved upstream sequences that we identified have been under strong selection against TE insertions.
High Degree of Upstream Conservation for TF Genes. We have listed the top 30 upstream-conserved orthologues in Table 1 (see also Fig. 2 and the list of all of the 3,055 genes under analysis shown in Table 4, which is published as supporting information on the PNAS web site). Notably, we found that nine of the 10 most upstream-conserved orthologues were TF genes (P < 2 × 10–8). Within the top 30 upstream-conserved genes, 19 were also TF genes (P < 5 × 10–12) (Table 1). The high occurrence of TF genes also continued for the top 200 upstream-conserved genes, i.e., 62 of the top 200 genes encoded TFs (P < 5 × 10–15), although TF genes occupy only 10.0% (305/3,055) of the genes analyzed.
Table 1. The top 30 upstream-conserved orthologous genes.
| Rank | Official gene symbol | No. of identical sites | Gene name |
|---|---|---|---|
| 1 | ZFHX1B | 6,000 | Zinc finger homeobox 1b |
| 2 | HOXC9 | 5,455 | Homeobox C9 |
| 3 | FOXP2 | 5,402 | Forkhead box P2 |
| 4 | LHX2 | 4,912 | LIM homeobox 2 |
| 5 | NR4A3 | 4,873 | Nuclear receptor subfamily 4, group A, member 3 |
| 6 | OTX2 | 4,601 | Orthodenticle homolog 2 (Drosophila) |
| 7 | PITX2 | 4,536 | Paired-like homeodomain transcription factor 2 |
| 8 | NR4A2 | 4,413 | Nuclear receptor subfamily 4, group A, member 2 |
| 9 | INHBA | 4,400 | Inhibin, beta A (activin A, activin AB alpha polypeptide) |
| 10 | SIX1 | 4,398 | Sine oculis homeobox homolog 1 (Drosophila) |
| 11 | NTNG2 | 4,393 | Netrin G2 |
| 12 | PAX6 | 4,362 | Paired box gene 6 (aniridia, keratitis) |
| 13 | SP8 | 4,235 | Sp8 transcription factor |
| 14 | BAI3 | 4,178 | Brain-specific angiogenesis inhibitor 3 |
| 15 | MLLT10 | 4,110 | Myeloid/lymphoid or mixed-lineage leukemia (trithorax homolog, Drosophila); translocated to 10 |
| 16 | EYA1 | 4,069 | Eyes absent homolog 1 (Drosophila) |
| 17 | OTP | 4,055 | Orthopedia homolog (Drosophila) |
| 18 | DNAJB5 | 3,995 | DnaJ (Hsp40) homolog, subfamily B, member 5 |
| 19 | PROX1 | 3,932 | Prospero-related homeobox 1 |
| 20 | MEF2C | 3,931 | MADS box transcription enhancer factor 2, polypeptide C (myocyte enhancer factor 2C) |
| 21 | ELAVL2 | 3,860 | ELAV (embryonic lethal, abnormal vision, Drosophila)-like 2 (Hu antigen B) |
| 22 | HOXD4 | 3,857 | Homeobox D4 |
| 23 | NR2F1 | 3,841 | Nuclear receptor subfamily 2, group F, member 1 |
| 24 | PAX2 | 3,838 | Paired box gene 2 |
| 25 | DLL1 | 3,809 | delta-like 1 (Drosophila) |
| 26 | HOXD3 | 3,802 | Homeobox D3 |
| 27 | PCDH7 | 3,793 | BH-protocadherin (brain-heart) |
| 28 | NRXN3 | 3,767 | Neurexin 3 |
| 29 | CDK6 | 3,763 | Cyclin-dependent kinase 6 |
| 30 | LDB1 | 3,753 | LIM domain binding 1 |
Orthologous genes are listed in the decreasing order of the number of identical sites within their 8-kb upstream sequences. Nineteen of the 30 genes are assigned “transcription factor activity,” according to the GO annotation (shown in bold).
Fig. 2.

Schematic diagrams showing the alignments of the top three upstream-conserved human–mouse orthologues along the 8-kb sequences immediately upstream of the translation start sites. Each conserved block is color-coded according to the percent identity. The alignments are drawn in proportion to the physical location of the genomic sequences.
Relationship Between the Degree of Upstream Sequence Conservation and Gene Function. To further elucidate the relationship between the gene function and the degree of upstream sequence conservation, we subcategorized the genes by their function, based on the GO annotation, into the following six categories: (i) metabolism, (ii) biosynthesis, (iii) cell cycle, (iv) signal transduction, (v) developmental process, and (vi) TFs. As a result, we found that the TF genes and developmental process-related genes showed the highest degrees of upstream sequence conservation within the six categories (Fig. 3). In more detail, 61 developmental process-related genes appeared within the top-200 upstream-conserved genes (P < 5 × 10–12), although such genes occupy only 11.4% (347/3,055) of the genes in the present analysis.
Fig. 3.

The relative frequency for each of the six functional categories is shown, along with ranks that represent the degree of upstream sequence conservation. For instance, rank 200 includes the top 1–200 upstream-conserved genes, and the points on each line graph represent the relative frequency within the corresponding rank for the corresponding functional category. The genes under analysis were subdivided into six functional categories according to the GO annotation.
In contrast to the TF genes and developmental process-related genes, genes involved in metabolism and biosynthesis showed less upstream sequence conservation. These categories of genes belonged more frequently to the lower ranks of upstream sequence conservation. An intermediate degree of upstream conservation was shown for cell cycle- and signal transduction-related genes, which were distributed almost evenly throughout all of the ranks concerning the degree of upstream conservation (Fig. 3). Although a considerable number of cell cycle-related genes contribute to common processes among eukaryotes, the degree of upstream sequence conservation remained much less than that of the developmental process-related genes and TF genes. Our results clearly show that the TF genes have a remarkably high degree of conservation in the upstream regions that are considered to play major regulatory roles.
Higher Conservation of Upstream Regions of Developmental Process-Related TF Genes. We further focused on the genes that were involved in both TF activity and developmental processes. These developmental process-related TF genes gave an even higher degree of upstream sequence conservation. Notably, >27% of the developmental process-related TF genes appeared in the top 200 upstream-conserved rank. They also appeared more frequently in the top three ranks (i.e., the top 1–800 upstream-conserved genes) than the TF genes that are not involved in developmental processes (P < 1 × 10–6; rank-sum test) and non-TF genes that are involved in developmental processes (P < 1 × 10–15; rank-sum test) (Fig. 4). We showed that developmental process-related TF genes have a much higher degree of upstream sequence conservation.
Fig. 4.

The relative frequency for developmental process-related TF genes is shown, along with ranks that represent the degree of upstream sequence conservation, together with the relative frequencies of TF genes that are not involved in the developmental process and the developmental process-related non-TF genes.
Candidates for Regulatory SNPs in the 8-kb Upstream-Conserved Sequence of the ZFHX1B Gene. According to the RefSeq contig annotation, we found that 11 SNPs were located within the 8-kb stretch upstream of the human ZFHX1B gene, which was the most upstream-conserved orthologue between humans and mice. Of these, six were within the human-mouse conserved sequences that we identified (Table 2). We showed the physical locations of all of the conserved sequences (96,873 sequence blocks) that we identified in the alignments of the 3,055 human–mouse orthologous 8-kb upstream sequence pairs in Table 5, which is published as supporting information on the PNAS web site.
Table 2. Candidates for regulatory SNPs within 8-kb upstream of the ZFHX1B gene.
| dbSNP accession no. | Locations in RefSeq contig (accession no. NT_005058.14) | Positions relative to coding start site, bps upstream |
|---|---|---|
| 3755092 | 13887422 | 497 |
| 3806477 | 13888534 | 1,609 |
| 3806475* | 13888795 | 1,870 |
| 1465531 | 13889161 | 2,236 |
| 5835003* | 13889221 | 2,296 |
| 3835062 | 13890430-13890431 | 3,505-3,506 |
| 1427300 | 13891454 | 4,529 |
| 10593682* | 13893217-13893219 | 6,292-6,294 |
| 10601407* | 13893220-13893222 | 6,295-6,297 |
| 7602487* | 13893446 | 6,521 |
| 7603150* | 13894021 | 7,096 |
Note that the ZFHX1B gene is encoded in its minus strand in its RefSeq contig sequence.
SNP Database (dbSNP) accessions for which the SNP entry is located within the human—mouse conserved sequence block that we identified.
Discussion
High Upstream Conservation for Development-Related TF Genes. In the present study, we have shown that the genes with high upstream sequence conservation are predominantly TF genes. More interestingly, we have also elucidated that developmental process-related TF genes have an even higher degree of upstream sequence conservation.
Because our resultant upstream conserved sequences could reflect regulatory sequences, these TF genes may have a larger number of regulatory sequences and may form highly connected regulatory networks between TF genes, such as those found in yeast by using genome-wide experiments (14). Because such highly connected biological networks have been reported to be preferentially retained evolutionarily (15), the potentially high connectivity of the regulatory networks that are constituted of TF genes (in particular, developmental process-related TF genes) might contribute to the evolutionary stasis.
Therefore, the surplus degree of upstream conservation we found for the developmental process-related TF genes can illustrate the evolutionary constraint that has been considered to be inherent in the program of ontogeny, or so-called developmental constraint (16, 17).
Pleiotropism of ZFHX1B Could Explain the Requirement of Its Strict Regulation. We found that the most upstream-conserved human–mouse orthologous gene, ZFHX1B, had 26 conserved sequence blocks and showed 75% nucleotide identity on average along the 8-kb upstream sequence. The ZFHX1B gene encodes a TF, Smad-interacting protein 1 (SIP1) (18), and mutations in the exons of this gene have been reported to cause a form of Hirschsprung disease (19, 20), the most common congenital malformation (≈1 in 5,000 live births). Besides Hirschsprung disease, mutations in one allele of the ZFHX1B gene have been shown to cause a wide spectrum of congenital anomalies (19, 20), such as microcephaly and malformations of the brain (cerebral atrophy or callosal agenesis), heart (patent ductus arteriosus), face morphology, and genitourinary organs (21), along with complex developmental disorders, such as mental retardation, delayed motor development, and epilepsy (19–21). These findings indicate that haploinsufficiency of the ZFHX1B gene can have a variety of influences on multiple organs in multiple phases. This wide breadth of pleiotropism of the ZFHX1B gene (20) should require strict temporal and spatial control of its expression, which could account for the large number of upstream regulatory sequences.
Candidates for Regulatory SNPs for Further Examination. Although all of the mutations in ZFHX1B reported to cause disease phenotypes have been within the exons, it would also be meaningful to examine nucleotide changes in the highly conserved noncoding upstream sequences of ZFHX1B that we identified, because partial dysregulation of this gene could cause milder forms of those clinical features. Alternatively, such mutations could be genetic factors relevant to other complex diseases, because of its wide breadth of pleiotropism.
We found 11 SNPs located within the 8-kb stretch upstream of ZFHX1B, six of which were included in the conserved sequences (Table 2). These six SNPs could be candidates for regulatory SNPs. Because we identified 96,873 conserved sequence blocks in the 3,055 pairs of human–mouse orthologous 8-kb upstream sequences (shown in Table 5), these evolutionarily conserved sequences may be good targets for identifying novel mutations that are related to diseases or disease susceptibilities, and they may also be useful as targets for identifying regulatory sequences in high-throughput experimental analyses.
In summary, the present study has demonstrated the possible molecular basis of evolutionary constraint related to the regulatory program of ontogeny as the surplus degree of upstream sequence conservation for developmental process-related TF genes. By means of cross-species comparison of genome sequences and the utilization of functional annotations, we have demonstrated the gene expression regulatory features characteristic of TF genes and development-related genes, which await further experimental evidence on a genome-wide scale.
Supplementary Material
Acknowledgments
We thank K. Hotta and K. Sumiyama for fruitful discussions.
Author contributions: H.I. performed research; and T.G. and H.I. designed research.
Abbreviations: TF, transcription factor; GO, gene ontology; EPD, Eukaryotic Promoter Database.
References
- 1.Kellis, M., Patterson, N., Endrizzi, M., Birren, B. & Lander, E. S. (2003) Nature 423, 241–254. [DOI] [PubMed] [Google Scholar]
- 2.Cliften, P., Sudarsanam, P., Desikan, A., Fulton, L., Fulton, B., Majors, J., Waterston, R., Cohen, B. A. & Johnston, M. (2003) Science 301, 71–76. [DOI] [PubMed] [Google Scholar]
- 3.Wasserman, W. W., Palumbo, M., Thompson, W., Fickett, J. W. & Lawrence, C. E. (2000) Nat. Genet. 26, 225–228. [DOI] [PubMed] [Google Scholar]
- 4.Blanchette, M. & Tompa, M. (2002) Genome Res. 12, 739–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pennacchio, L. A. & Rubin, E. M. (2001) Nat. Rev. Genet. 2, 100–109. [DOI] [PubMed] [Google Scholar]
- 6.Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., Davis, A. P., Dolinski, K., Dwight, S. S., Eppig, J. T., et al. (2000) Nat. Genet. 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Waterston, R. H., Lindblad-Toh, K., Birney, E., Rogers, J., Abril, J. F., Agarwal, P., Agarwala, R., Ainscough, R., Alexandersson, M., An, P., et al. (2002) Nature 420, 520–562. [DOI] [PubMed] [Google Scholar]
- 8.Pruitt, K. D. & Maglott, D. R. (2001) Nucleic Acids Res. 29, 137–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tatusova, T. A. & Madden, T. L. (1999) FEMS Microbiol. Lett. 174, 247–250. [DOI] [PubMed] [Google Scholar]
- 10.Schmid, C. D., Praz, V., Delorenzi, M., Périer, R. & Bucher, P. (2004) Nucleic Acids Res. 32, D82–D85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sherry, S. T., Ward, M. & Sirotkin, K. (1999) Genome Res. 9, 677–679. [PubMed] [Google Scholar]
- 12.Imanishi, T., Itoh, T., Suzuki, Y., O'Donovan, C., Fukuchi, S., Koyanagi, K. O., Barrero, R. A., Tamura, T., Yamaguchi-Kabata1, Y., Tanino, M., et al. (2004) PloS Biol. 2, 1–20. [Google Scholar]
- 13.Lander, E. S., Linton, L. M., Birren, B., Nusbaum, C., Zody, M. C., Baldwin, J., Devon, K., Dewar, K., Doyle, M., FitzHugh, W., et al. (2001) Nature 409, 860–921. [DOI] [PubMed] [Google Scholar]
- 14.Lee, T. I., Rinaldi, N. J., Robert, F., Odom, D. T., Bar-Joseph, Z., Gerber, G. K., Hannett, N. M., Harbison, C. T., Thompson, C. M., Simon, I., et al. (2002) Science 298, 799–804. [DOI] [PubMed] [Google Scholar]
- 15.Wuchty, S., Oltvai, Z. N. & Barabási, A.-L. (2003) Nat. Genet. 35, 176–179. [DOI] [PubMed] [Google Scholar]
- 16.Gould, S. J. & Lewontin, R. C. (1979) Proc. R. Soc. London Ser. B 205, 581–598. [DOI] [PubMed] [Google Scholar]
- 17.Maynard-Smith, J., Burian, R., Kauffman, S., Alberch, P., Campbell, J., Goodwin, B., Lande, R., Raup, D. & Wolpert, L. (1985) Q. Rev. Biol. 60, 265–287. [Google Scholar]
- 18.Verschueren, K., Remacle, J. E., Collart, C., Kraft, H., Baker, B. S., Tylzanowski, P., Nelles, L., Wuytens, G., Su, M. T., Bodmer, R., et al. (1999) J. Biol. Chem. 274, 20489–20498. [DOI] [PubMed] [Google Scholar]
- 19.Wakamatsu, N., Yamada, Y., Yamada, K., Ono, T., Nomura, N., Taniguchi, H., Kitoh, H., Mutoh, N., Yamanaka, T., Mushiake, K., et al. (2001) Nat. Genet. 27, 369–370. [DOI] [PubMed] [Google Scholar]
- 20.Yamada, K., Yamada, Y., Nomura, N., Miura, K., Wakako, R., Hayakawa, C., Matsumoto, A., Kumagai, T., Yoshimura, I., Miyazaki, S., et al. (2001) Am. J. Hum. Genet. 69, 1178–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mowat, D. R., Wilson, M. J. & Goossens, M. (2003) J. Med. Genet. 40, 305–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}