

Abstract
Detection of circulating tumor DNA (ctDNA) after resection of stage II colon cancer may identify patients at the highest risk of recurrence and help inform adjuvant treatment decisions. We used massively parallel sequencing–based assays to evaluate the ability of ctDNA to detect minimal residual disease in 1046 plasma samples from a prospective cohort of 230 patients with resected stage II colon cancer. In patients not treated with adjuvant chemotherapy, ctDNA was detected postoperatively in 14 of 178 (7.9%) patients, 11 (79%) of whom had recurred at a median follow-up of 27 months; recurrence occurred in only 16 (9.8 %) of 164 patients with negative ctDNA [hazard ratio (HR), 18; 95% confidence interval (CI), 7.9 to 40; P < 0.001]. In patients treated with chemotherapy, the presence of ctDNA after completion of chemotherapy was also associated with an inferior recurrence-free survival (HR, 11; 95% CI, 1.8 to 68; P = 0.001). ctDNA detection after stage II colon cancer resection provides direct evidence of residual disease and identifies patients at very high risk of recurrence.
INTRODUCTION
About 1.3 million cases of colorectal cancer are diagnosed annually worldwide (1). In patients with stage II colon cancer (~25% of all colorectal cancer), management after surgical resection remains a clinical dilemma, with about 80% cured by surgery alone (2). The current approach to defining recurrence risk for patients with early-stage colon cancer dates from the original work of C. E. Dukes (3) in the 1930s. For patients with Dukes B cancers [stage II by TNM (tumor-node-metastasis) classification], the risk of recurrence was subsequently refined through the recognition of other clinical and pathological features. Incorporation of these features, such as T4 extension, bowel perforation or obstruction, inadequate nodal sampling, poorly differentiated histology, and lymphovascular invasion (LVI), only modestly affects recurrence risk (4, 5). Deficient mismatch repair (dMMR) status in the tumor defines a low-risk group in which adjuvant chemotherapy is not beneficial (6, 7). Most recently, multiple tissue-based gene signatures have been shown to have prognostic significance, but again with modest hazard ratios (HRs) of 1.4 to 3.7 (8–11).
In practice, adjuvant chemotherapy is more frequently offered to high-risk stage II patients, with the justification that high-risk patients are more likely to derive benefit from treatment. However, an overall survival benefit from adjuvant therapy in patients with stage II colon cancer, including those with high-risk disease based on standard clinicopathologic criteria or gene signatures, remains to be conclusively demonstrated (12–16). The challenge in demonstrating a benefit is in part due to the overall low risk of recurrence in this patient group, requiring very large studies to demonstrate a modest benefit from treatment. Better markers for recurrence risk would allow a high-risk subset to be identified, the selection of which could enrich studies designed to demonstrate adjuvant therapy benefit.
Regardless of whether patients have received adjuvant therapy, early detection of recurrence during follow-up is associated with improved survival in patients with early-stage colorectal cancer (17–20). However, the biomarker now used as the standard of care, carcinoembryonic antigen (CEA), has limited sensitivity and specificity (21, 22). Computed tomography (CT) imaging improves detection of recurrence but is associated with radiation exposure and also has a high rate of false positivity (21).
Sequencing of the DNA from colorectal cancers has identified several genes that are recurrently somatically mutated (23, 24). These tumor-specific DNA mutations can be detected in the cell-free component of peripheral blood [circulating tumor DNA (ctDNA)] in most patients with metastatic disease, allowing for noninvasive molecular characterization of tumors, including genetic changes that are revealed by the selective pressure of targeted therapies (25–28). Additionally, the short half-life of ctDNA (~2 hours) (29) makes ctDNA a useful dynamic marker of tumor bulk, with early decreases in ctDNA amounts reflecting treatment responses that are later confirmed by conventional imaging (30). The possibility that ctDNA could be used to detect micrometastatic disease in patients undergoing surgery with curative intent was suggested in an initial series of 18 patients with advanced colorectal cancer undergoing metastasectomy (29) and also more recently in other solid malignancies such as breast and pancreatic cancers (31, 32).
Here, we report on the results of a prospective correlative biomarker study in stage II colon cancer patients, where the primary aim was to demonstrate that postoperative ctDNA analysis could be used as an indicator of minimal residual disease, thereby identifying patients who would eventually develop recurrent disease detected with conventional radiologic criteria. Secondary aims were to analyze serial samples to explore changes in ctDNA concentration over time, including any impact of adjuvant therapy on ctDNA, and to determine whether persistently detectable ctDNA identified treated patients who would later recur radiologically.
RESULTS
Patient characteristics and postoperative ctDNA status
Patient enrolment and study overview are presented in Fig. 1. Between July 2011 and September 2014, we enrolled 250 patients and collected 1047 plasma samples from 231 eligible patients. We initially subjected the resected primary tumor tissue to targeted massively parallel sequencing using the previously described Safe-SeqS assay (26, 33). At least one somatic mutation was identified in the primary tumor tissue of 230 of 231 (99.6%) eligible cases. For the 230 evaluable patients, we then designed personalized Safe-SeqS assays for the identified mutations to quantify ctDNA in plasma samples. Twenty of these 230 patients (8.7%) harbored the tumor-specific mutation in their plasma (ctDNA-positive) 4 to 10 weeks after surgery, hereafter termed “postoperatively.” Table 1 summarizes the mutations and mutant allele fractions (MAFs) found in the ctDNA-positive cases.
Fig. 1.
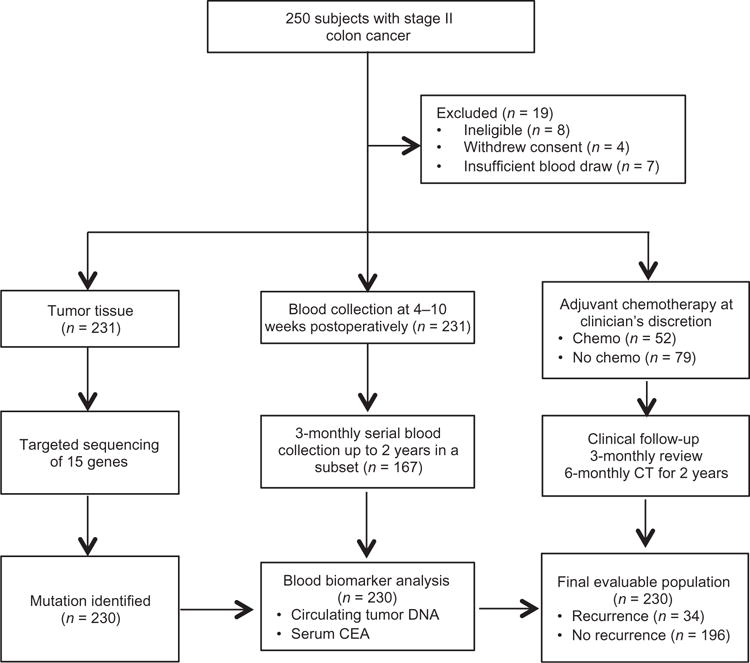
Patient enrolment and sample collection
Table 1.
Mutations and mutant allele fractions detected in postoperative ctDNA-positive cases.
| Mutations | MAF (%) |
|---|---|
| TP53 p.R342X | 1.631 |
| TP53 p.G245D | 0.123 |
| APC p.L878fs | 0.11 |
| TP53 p.R248Q | 0.01 |
| TP53 p.R248Q | 0.006 |
| TP53 p.R248Q | 0.017 |
| APC p.Q1406fs | 0.059 |
| APC p.E1379X | 0.235 |
| KRAS p.G13D | 0.066 |
| TP53 p.R248Q | 0.678 |
| APC p.C1578fs | 0.140 |
| KRAS p.G12D | 0.027 |
| KRAS p.G12V | 0.008 |
| TP53 p.P151fs | 0.003 |
| APC p.S1436fs | 0.046 |
| TP53 p.R282W | 1.774 |
| APC p.D1178fs | 0.007 |
| APC p.E1408fs | 0.046 |
| TP53 p.I254fs | 0.05 |
| APC p.S1010fs | 0.006 |
Baseline patient and pathological characteristics grouped according to postoperative ctDNA status for the 230 evaluable patients are shown in Table 2. The median age was 65 years, and 57% were male. On the basis of pathological characteristics, 16% had poorly differentiated tumors, 17% had T4 disease, 13% had less than 12 lymph nodes (LNs) examined, 19% had LVI, and 18% had dMMR tumors. Of the 230 eligible patients, 52 (23%) patients received adjuvant chemotherapy at their clinician’s discretion. These patients were younger and more likely to have at least one high-risk feature.
Table 2.
Patient and tumor characteristics according to postoperative ctDNA status.
| Characteristics* | All patients (n = 230) | Postoperative ctDNA-positive (n = 20) | Postoperative ctDNA-negative (n = 210) |
|---|---|---|---|
| Age (years) | |||
| Median [interquartile range (IQR)] | 65 (59–73) | 64 (54–68) | 66 (59–74) |
| Range | 23–87 | 43–86 | 23–87 |
| Sex, no. (%) | |||
| Male | 131 (57) | 10 (50) | 121 (58) |
| Female | 99 (43) | 10 (50) | 89 (42) |
| Tumor site, no. (%)† | |||
| Right colon | 103 (45) | 5 (25) | 98 (47) |
| Left colon | 127 (55) | 15 (75) | 112 (53) |
| Differentiation, no. (%) | |||
| Well/moderate | 193 (84) | 18 (90) | 175 (83) |
| Poor | 37 (16) | 2 (10) | 35 (17) |
| T stage, no. (%) | |||
| T3 | 192 (83) | 17 (85) | 175 (83) |
| T4 | 38 (17) | 3 (15) | 35 (17) |
| Lymph node yield, no. (%) | |||
| <12 | 29 (13) | 5 (25) | 24 (11) |
| ≥12 | 201 (87) | 15 (75) | 186 (89) |
| Lymphovascular invasion, no./total no. (%) | |||
| Yes | 41/221 (19) | 6/20 (30) | 35/201 (17) |
| No | 180/221 (81) | 14/20 (70) | 166/201 (83) |
| MMR status, no. (%) | |||
| Proficient | 189 (82) | 19 (95) | 170 (81) |
| Deficient | 41 (18) | 1 (5) | 40 (19) |
| Clinicopathologic risk group, no. (%) | |||
| Low | 140/224 (63) | 10 (50) | 130/204 (64) |
| High | 84/224 (37) | 10 (50) | 74/204 (36) |
| Adjuvant chemotherapy, no. (%)‡ | |||
| Yes | 52 (23) | 6 (30) | 46 (22) |
| No | 178 (77) | 14 (70) | 164 (78) |
There were no significant differences between the groups in any of the characteristics listed in this table.
Right-sided colon cancer was defined as tumors arising from the cecum, ascending, hepatic flexure, or transverse colon; left-sided colon cancer was defined as tumors arising from the splenic flexure, descending, sigmoid, or rectosigmoid colon.
Except for two cases where oxaliplatin-based chemotherapy was used, all other adjuvant treatment was fluorouracil-based chemotherapy.
There was no significant association between postoperative ctDNA status and conventional high-risk clinicopathological factors (Table 1). As of 25 November 2015, median follow-up was 27 months (range, 2 to 52 months). During this period, 34 (14.8%) patients experienced radiologic recurrences, including 27 of 178 (15%) patients not treated with chemotherapy and 7 of 52 (13%) patients treated with adjuvant chemotherapy.
Postoperative ctDNA status and recurrence in patients not treated with adjuvant chemotherapy
To avoid the confounding effect of chemotherapy, we examined the ability of ctDNA analysis at the postoperative time point to predict radiologic recurrence in patients not treated with adjuvant chemotherapy. ctDNA was detected postoperatively in 14 of 178 patients (7.9%) not treated with chemotherapy. Radiologic recurrence was detected during follow-up in 11 of these 14 patients (78.6%). Postoperative ctDNA was negative in the remaining 164 of 178 (92.1%) patients, where disease recurrence was documented in 16 (9.8%) patients. Among all patients not treated with adjuvant chemotherapy, CEA was elevated postoperatively in 2 of 27 cases (7.4%) that recurred and in 5 of 151 cases (3.3%) that did not recur. None of the patients with detectable ctDNA postoperatively had elevated CEA. The relationships between postoperative ctDNA, postoperative CEA, and recurrence status are shown in table S1.
Patients with ctDNA-positive status postoperatively had a markedly reduced recurrence-free survival (RFS) compared to those with a ctDNA-negative status [HR, 18; 95% confidence interval (CI), 7.9 to 40; P = 2.6 × 10−12] (Fig. 2A). Kaplan-Meier estimates of RFS at 3 years were 0% for the ctDNA-positive and 90% for the ctDNA-negative groups. Patients with elevated CEA postoperatively had similar RFS to those with nonelevated CEA (HR, 1.6; 95% CI, 0.30 to 10; P = 0.5) (fig. S1). Of the 40 patients with dMMR tumors, only 1 patient had positive ctDNA postoperatively. This patient did not receive adjuvant therapy and remains recurrence-free at 12 months.
Fig. 2. RFS in patients not treated with adjuvant chemotherapy.
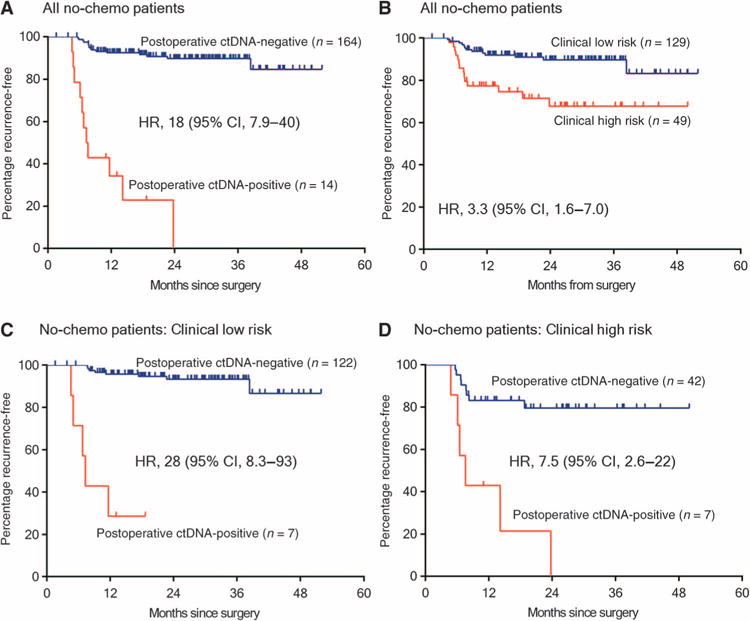
(A) Kaplan-Meier estimates of RFS for all patients not treated with adjuvant chemotherapy, stratified by postoperative ctDNA status. (B) Kaplan-Meier estimates of RFS in the same patients, stratified by clinicopathologic characteristics. (C) Kaplan-Meier estimates of RFS stratified by postoperative ctDNA status in patients with low-risk clinicopathologic characteristics. (D) Kaplan-Meier estimates of RFS stratified by postoperative ctDNA status in patients with high-risk clinicopathologic characteristics. The high-risk group is defined as those having mismatch repair–proficient (pMMR) tumors with at least one of the following poor prognostic features: T4, LN yield <12, LVI, and poor tumor differentiation. The low-risk group is defined as those with no poor prognostic features.
Clinicopathologic variables significantly associated with RFS in univariate analysis were T stage, LN yield, and LVI (Table 3). A borderline association was seen for MMR status (HR, 3.6; 95% CI, 0.86 to 15; P = 0.08). When considering all pathological factors, patients with high clinicopathologic risk had a worse RFS than patients with low clinicopathologic risk (HR, 3.3; 95% CI, 1.6 to 7.0; P = 0.002) (Fig. 2B). Postoperative ctDNA status had a greater impact on RFS than any individual clinicopathological risk factor or any combination of clinicopathological factors. Postoperative ctDNA status added significant prognostic value to patients classified as either low-risk or high-risk on the basis of clinicopathological factors (low-risk: HR, 28; 95% CI, 8.1 to 93; P = 9.2 × 10−8; Fig. 2C; high-risk: HR, 7.5; 95% CI, 2.6 to 22; P = 0.0002; Fig. 2D). After multivariable adjustment, postoperative ctDNA status remained an independent predictor of RFS for patients not treated with chemotherapy (HR, 28; 95% CI, 11 to 68) (Table 3) and for all patients (HR, 14; 95% CI, 6.8 to 28) (Table 3).
Table 3.
Recurrence-free survival analysis by clinicopathological variables and postoperative ctDNA status.
| Variable | Univariate analysis
|
Multivariate analysis
|
||||
|---|---|---|---|---|---|---|
| HR | 95% CI | P | HR | 95% CI | P | |
|
Patients not treated with chemotherapy (n = 178)
| ||||||
| Age, <70 versus ≥70 | 0.92 | 0.43–2.0 | 0.8 | |||
| Sex, male versus female | 1.3 | 0.62–2.8 | 0.5 | |||
| Tumor site, right versus left | 1.5 | 0.69–3.3 | 0.3 | |||
| Tumor differentiation, well/moderate versus poor | 0.39 | 0.09–1.7 | 0.2 | |||
| T stage, T3 versus T4 | 4.0 | 1.7–9.5 | 0.002 | 8.1 | 3.1–21 | <0.001 |
| Lymph node yield, ≥12 versus <12 | 3.1 | 1.3–7.4 | 0.009 | |||
| Lymphovascular invasion, no versus yes | 2.4 | 1.1–5.4 | 0.03 | |||
| MMR status, deficient versus proficient | 3.6 | 0.86–15 | 0.08 | |||
| Clinicopathologic risk group, low versus high | 3.2 | 1.5–6.9 | 0.002 | |||
| Postoperative CEA, normal versus elevated | 1.6 | 0.37–6.8 | 0.5 | |||
| Postoperative ctDNA status, negative versus positive | 18 | 7.9–40 | <0.001 | 28 | 11–68 | <0.001 |
|
All patients (n = 230) | ||||||
| Age, <70 versus ≥70 | 1.0 | 0.50–2.0 | 1.0 | |||
| Sex, male versus female | 1.1 | 0.57–2.2 | 0.7 | |||
| Tumor site, right versus left | 1.1 | 0.55–2.1 | 0.8 | |||
| Tumor differentiation, well/moderate versus poor | 0.32 | 0.08–1.3 | 0.1 | |||
| T stage, T3 versus T4 | 2.4 | 1.2–5.1 | 0.02 | 2.6 | 1.2–5.5 | 0.01 |
| Lymph node yield, ≥12 versus <12 | 2.2 | 0.97–4.8 | 0.06 | |||
| Lymphovascular invasion, no versus yes | 1.9 | 0.92–4.1 | 0.08 | |||
| MMR status, deficient versus proficient | 3.5 | 0.83–14.5 | 0.09 | |||
| Clinicopathologic risk group, low versus high | 2.1 | 1.06–4.2 | 0.03 | |||
| Adjuvant chemotherapy, no versus yes | 0.79 | 0.34–1.8 | 0.6 | |||
| Postoperative CEA, normal versus elevated | 2.8 | 0.98–7.9 | 0.06 | |||
| Postoperative ctDNA status, negative versus positive | 13 | 6.6–27 | <0.001 | 14 | 6.8–28 | <0.001 |
The time-dependent accuracy of postoperative ctDNA in predicting radiologic recurrence at 5, 6, 12, 18, 24, 36, and 40 months, as assessed by time-dependent receiver operating curves (ROC) analysis, is shown in table S2. The sensitivity and specificity of postoperative ctDNA in predicting recurrence at 36 months were 48 and 100%, respectively.
ctDNA dynamics in patients treated with adjuvant chemotherapy
We then assessed the impact of adjuvant chemotherapy on serial ctDNA status during and after completion of chemotherapy. ctDNA was positive in the postoperative period in 6 of 52 (11%) patients who then received at least 3 months of adjuvant chemotherapy. Serial ctDNA concentrations for these patients are shown in Fig. 3 (A to F). The ctDNA status changed from positive to negative during the initial adjuvant treatment phase in five patients (patients A to E). ctDNA became positive again after completion of chemotherapy in two patients (patients A and B), both of whom later recurred radiologically. In the other three patients, ctDNA remained negative after treatment. Two (patients C and D) of these remained recurrence-free at 16 and 34 months. Patient E had radiologic recurrence despite the ctDNA remaining negative, although CEA was elevated at the later time point. ctDNA status for patient F fluctuated over time with no detectable radiologic recurrence at 33 months. Overall, ctDNA positivity immediately after completion of chemotherapy was associated with poorer RFS (HR, 11; 95% CI, 1.8 to 68; P = 0.001) (Fig. 3G). Eight patients did not have blood samples collected after the completion of chemotherapy and were excluded from this analysis.
Fig. 3. ctDNA status before, during, and after adjuvant chemotherapy.
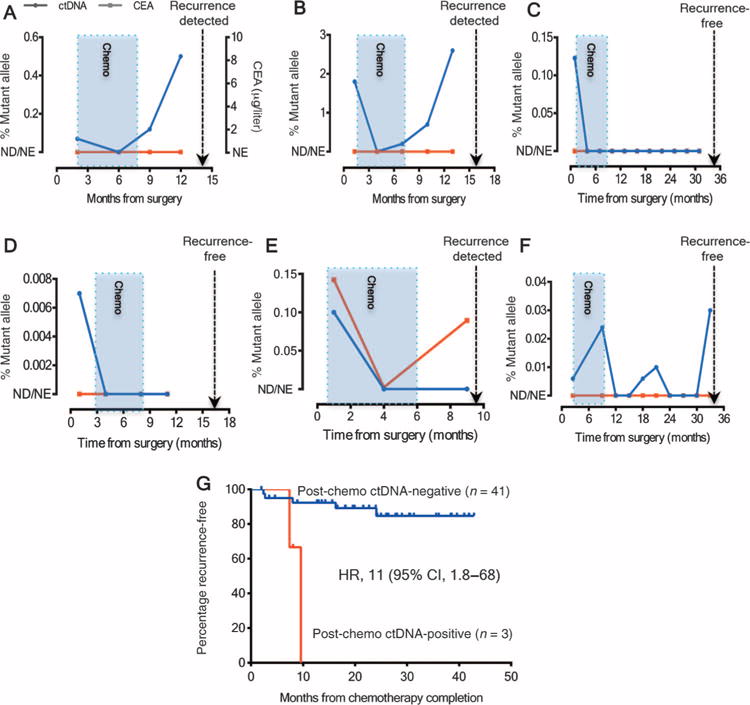
(A to F) ctDNA concentrations (% mutant alleles) for the six patients with positive postoperative ctDNA who subsequently received adjuvant chemotherapy. The blue shaded box indicates the period during which adjuvant chemotherapy was administered. The dotted line indicates the time of radiologic recurrence or last follow-up (if recurrence-free). The ctDNA status of patients represented in (A) and (B) initially became negative, then became positive again at the completion of adjuvant chemotherapy; both patients subsequently suffered a radiologic recurrence. Note that CEAs were not elevated in either patient at any time point. (C to F) Four patients whose ctDNA became negative after completion of chemotherapy. Three of these patients (patients C, D, and F) remained radiologic recurrence-free at their last follow-up visit. (G) Kaplan-Meier estimates of RFS according to post-chemotherapy ctDNA status in patients treated with adjuvant chemotherapy. ND, not detected; NE, not elevated.
Serial ctDNA and CEA measurements during follow-up
We examined the sensitivity of serial ctDNA analysis during the follow-up period to predict subsequent radiologic recurrence. Serial three-monthly ctDNA and CEA values up to radiologic recurrence were available for 27 of 34 patients who had recurred. Samples were not collected at the time of recurrence in seven patients primarily because of individual patients’ decisions. Serial ctDNA up to radiologic recurrence was available from 9 of the 11 patients with positive postoperative ctDNA who were not treated with chemotherapy. Serial ctDNA concentrations (MAFs) up to radiologic recurrence from these patients are illustrated in Fig. 4A. Serial circulating biomarker status and clinical characteristics at radiologic recurrence of patients treated or not treated with adjuvant chemotherapy are detailed in tables S3 and S4, respectively. ctDNA was more frequently positive (23 of 27) than CEA elevation (11 of 27) at the time of radiologic recurrence (85% versus 41%; P = 0.002). The time between ctDNA detection and radiologic recurrence (median, 167 days; IQR, 81 to 279 days) was significantly longer than the time between CEA elevation and radiologic recurrence (median, 61 days; IQR, 0 to 207 days; P = 0.04) (Fig. 4B). The genomic and clinical summaries of all 230 cases included in the final evaluable population are provided in table S5.
Fig. 4. Serial ctDNA status up to radiological recurrence.
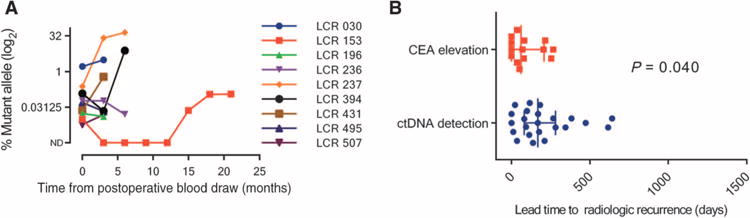
(A) Serial ctDNA measurements up to the time of radiological recurrence for the nine patients who were not treated with adjuvant chemotherapy and who were ctDNA-positive postoperatively. (B) Scatter plot of the lead time to radiological recurrence for ctDNA detection and CEA elevation, with the error bars representing the median and IQR.
DISCUSSION
The decision to treat or not to treat a stage II colon cancer patient with adjuvant chemotherapy remains one of the most challenging areas in colorectal oncology. Currently, up to 40% of stage II patients undergo adjuvant therapy in routine clinical care (34), committing to 6 months of chemotherapy, with the associated risk of potentially serious adverse events and without a method to monitor the impact of adjuvant therapy, for an absolute risk reduction of 3 to 5%. Although multiple clinicopathological markers are now validated and can be combined to define low- and high-risk groups, only a minority of defined high-risk patients will develop recurrence. The benefit of selectively treating these patients with adjuvant therapy also remains to be conclusively proven. Diagnostic approaches that better predict the disease course in this patient population are therefore urgently required.
Here, we have taken a fundamentally different approach to address these issues. We examine postoperative blood samples to identify direct evidence of residual disease in the form of ctDNA that is released into the circulation from cancer cells still present in the patient after surgery but not detectable on imaging. Whereas histopathologic or molecular characteristics of tumors that are associated with a higher recurrence risk indicate a propensity for metastasis, the presence of circulating DNA molecules containing somatic mutations found in an individual’s tumor is a direct indication that occult tumor cells remain after surgery. ctDNA measurements should therefore be considered not as a conventional biomarker of recurrence risk but more like a staging test such as a CT scan. As with a CT scan, which can only demonstrate residual disease when a sufficient tumor bulk is present, ctDNA is not a perfect indicator of residual disease: with a single plasma sample taken during the immediate postoperative period, its sensitivity for predicting recurrence at 36 months is 48%. However, these data demonstrate that the amount of tumor bulk required for ctDNA detection is lower than that for CT scan detection, with ctDNA detection preceding radiologic recurrence in many cases. CT scans have limited specificity, whereas the very high specificity of ctDNA was confirmed in the current study: 97% of patients whose tumors did not recur during the course of the study had negative ctDNA postoperatively, and in the few ctDNA-positive patients without known radiologic recurrence, late recurrence remains a possibility.
Our study has demonstrated that stage II colon cancer patients who were ctDNA-positive postoperatively are at extremely high risk of radiologic recurrence (HR, 18; 95% CI, 7.9 to 40; P = 2.6 × 10−12) when not treated with chemotherapy. This risk is greater than that in patients with stage III colorectal cancer, who are routinely treated with adjuvant therapy. Conversely, patients with negative ctDNA postoperatively were at a low risk of radiologic recurrence (3-year RFS of 90%), not dissimilar to patients with stage I colorectal cancer (35), defining a group where adjuvant therapy is less likely to be helpful. As expected, when we stratified patients into clinicopathological low- and high-risk groups, we found that patients with low risk had a significantly better RFS than patients with high-risk features (P = 0.002). The prognostic impact of postoperative ctDNA status was independent of individual clinicopathological risk features and improved the RFS risk estimates for both patients with clinicopathologic low (HR, 28; 95% CI, 8.1 to 93) and high-risk features (HR, 7.5; 95% CI, 2.6 to 22).
Our study also demonstrated that being ctDNA-positive at the completion of adjuvant chemotherapy treatment predicted a very high risk of radiologic recurrence. Of particular interest are the serial samples from patients who were ctDNA-positive postoperatively, then were treated with chemotherapy on the basis of the clinician’s judgment (in the absence of knowledge of the ctDNA results). These data suggest the possibility that personalized serial measurement of ctDNA could be a real-time marker of adjuvant therapy impact. For example, patients C and D were ctDNA-positive postoperatively, indicating that they had residual disease. Once treated with chemotherapy, their ctDNA became negative and remained so, even after treatment was discontinued. Adjuvant therapy appeared to have an effect on the disease in these patients, and this effect persisted for at least 16 to 34 months (last available follow-up). For patients A and B, the chemotherapy impact was transient in that ctDNA positivity, and later radiologic recurrence, was observed after treatment was completed. In contrast, a substantial fall in ctDNA concentration in the months after surgery was not observed in serial samples from the nine patients with positive postoperative ctDNA who did not receive chemotherapy. Although the finding from this small number of cases is promising, the use of serial ctDNA analysis for providing an early readout of the benefit of adjuvant chemotherapy needs to be validated in a larger cohort of patients.
During surveillance, serial ctDNA measurements appear to be more sensitive than CEA measurement, the current standard-of-care blood test, for predicting radiologic recurrence. Because clinicians were blinded to the ctDNA results but not to the CEA results, a direct comparison between CEA and ctDNA cannot be made. Nevertheless, 85% of patients were ctDNA-positive up to or at the time of radiologic recurrence, whereas CEA was only elevated in 41% of patients. The median lead time from ctDNA detection to radiological recurrence was over 5 months, which might be sufficient to change patient management. Combining radiologic and ctDNA assessments could result in earlier detection of recurrent disease, potentially identifying more patients eligible for curative surgical resection or earlier implementation of systemic therapies. One of the fundamental principles of oncology is that it is easier to cure a small volume of metastatic disease than a large volume (36).
In addition to improving patient selection for adjuvant chemotherapy in the routine clinical practice setting, the inclusion of serial ctDNA analysis as a biomarker in clinical trials is attractive. Enrolling only patients with detectable ctDNA into studies would mean that trial cohorts would be enriched with patients at very high risk of recurrence. This would substantially reduce the sample size required to demonstrate significant differences in outcomes given the very high event rate and would substantially reduce the associated expense of treating and following up many hundreds of patients. Further, not enrolling patients with undetectable ctDNA would mean that very low risk patients who are relatively unlikely to benefit from adjuvant therapy would not be exposed to unnecessary treatment-related risks. If serial ctDNA analysis proves to be a reliable marker of treatment effect, we would also have an early readout of treatment efficacy, meaning that clinical trials could be completed far more efficiently.
Despite the relatively large number of patients in our study, our conclusions are limited by the small number of patients with detectable ctDNA postoperatively. This limits the power to detect statistically significant associations between postoperative ctDNA status and conventional high-risk clinicopathological factors. However, our data suggest that there are no large differences in clinicopathological characteristics between the postoperative ctDNA-positive and ctDNA-negative patients. The relatively small number of patients with detectable ctDNA also makes the study susceptible to inherent biases, which may limit the generalizability of our findings. Notwithstanding this, our finding that postoperative ctDNA is a robust predictor of disease recurrence is consistent with recent reports in other tumor types. In a prospective cohort of early-stage breast cancer patients, ctDNA was detectable 2 to 4 weeks after apparently curative surgery in 7 of 37 (19%) patients and was predictive of early cancer relapse (HR, 25; 95% CI, 4.1 to 130) (31). In a study of early-stage pancreatic cancer, ctDNA was detectable postoperatively in 10 of 20 (50%) patients (32). Patients with detectable ctDNA after surgical resection were more likely to relapse compared with those with undetectable ctDNA (median time to recurrence, 9.9 months versus not reached; P = 0.02). Finally, only one mutation identified in each patient’s tumor tissue was analyzed in our study, yielding a sensitivity of 48% in detecting residual disease and predicting recurrence. The analytical sensitivity of ctDNA detection could potentially be improved by interrogating multiple mutations in a single assay rather than analyzing one mutation per patient, as recently demonstrated by Newman et al. (37).
In summary, this prospective study of stage II colon cancer patients demonstrated that ctDNA analysis of blood samples taken post-operatively defines a population at very high risk of recurrence. This ctDNA measurement is superior to clinicopathological measures currently used to guide adjuvant chemotherapy decisions. In the small number of patients receiving adjuvant chemotherapy, our data highlight the potential utility of biomarkers for tracking adjuvant chemotherapy effectiveness in real time, with implications for modifying or changing therapeutic management before the advent of bulky disease.
MATERIALS AND METHODS
Study design
This prospective multicenter study recruited patients with stage II colon cancer resected with curative intent at 13 Australian hospitals (Australian New Zealand Clinical Trials Registry number ACTRN12612000326897). The overall sample size was driven by the need to include a sufficient number of events, with the expectation that about 35 of 250 unselected patients with stage II colon cancer would experience recurrence in the first three years. The study was approved by the human research ethics committees at each hospital, and all participants provided written informed consent. Eligible patients had a staging CT chest/abdomen/pelvis within 12 weeks of study entry to exclude metastatic disease. Patients with a previous malignancy within the last 5 years were excluded. Blood samples for ctDNA and CEA analysis were collected at 4 to 10 weeks postoperatively, with serial three-monthly blood samples collected for up to 2 years from a subset of patients. The use of adjuvant chemotherapy was at the discretion of the treating clinician, who was blinded to the ctDNA result.
Per-protocol follow-up included three-monthly clinical review and CEA, with six-monthly CT imaging for 2 years. Thereafter, follow-up was according to the participating institutions’ standard of care. Serum CEA was measured by the diagnostic laboratory at each participating site, with CEA concentrations of <5 μg/liter considered normal. All plasma and tumor samples were analyzed at the Ludwig Center at Johns Hopkins.
Pathology
Pathology reports were reviewed for tumor site, LN yield, tumor differentiation, T stage, and LVI. MMR status was assessed by immunohistochemistry for MLH1, MSH2, MSH6, and PMS2 proteins using standard protocols (38). Tumors showing loss of expression in one or more of the proteins were considered MMR-deficient.
We defined a clinicopathologic high-risk group using standard criteria, including pMMR tumors with at least one of the following poor prognostic features: T4, LN yield <12, LVI, and poor tumor differentiation. The low-risk group consisted of patients with dMMR tumors or pMMR tumors with no poor prognostic features.
Identification of somatic mutations in tumor tissue
Formalin-fixed paraffin-embedded (FFPE) tumor sections were macrodissected under a dissecting microscope to ensure a neoplastic cellularity of >30%. DNA was purified with a Qiagen FFPE Kit (Qiagen cat. no. 56494). Polymerase chain reaction (PCR) was used to amplify regions of 15 genes recurrently mutated in colorectal cancer (table S6). Primers were designed and sequencing results were analyzed as previously described (25, 26, 39).
ctDNA analysis
When more than one somatic mutation was identified in a patient’s tumor tissue, the mutation with the highest MAF relative to the MAF in normal control DNA was selected for ctDNA analysis. For each patient, only one mutation identified in the tumor tissue was assessed in the plasma.
Plasma (10 ml) was purified from each patient using the QIAamp Circulating Nucleic Acid kit (Qiagen cat. no. 55114). To distinguish genuine mutations in the samples from artifactual variants arising from sequencing and sample preparation steps, we used Safe-SeqS, an error reduction technology for detection of low-frequency mutations (33). In the Safe-SeqS assay, plasma DNA was aliquoted into 24 wells of a 96-well plate so that an average of 0.5 to 3 ng of DNA was contained in each well. The DNA from each well was then amplified (15 cycles) using primers containing unique identifier sequences (UIDs), which consisted of 14 random bases with an equal probability of A, C, T, and G, to allow for the distinction of each template molecule. The amplified reactions were purified with AMPure XP beads (Beckman Coulter) and eluted in 250 μl of Buffer EB (Qiagen). One percent (2.5 μl) of purified PCR product was then amplified in a second round of PCR with universal primers, as previously described (33). The PCR products were purified with AMPure and sequenced on an Illumina MiSeq instrument.
High-quality sequence reads were selected on the basis of quality scores, which were generated by the Illumina sequencing instrument to indicate the probability that an error was made in base calling. The template-specific portion of the reads was matched to reference sequences. Reads from a common template molecule were then grouped on the basis of the UIDs that were incorporated as molecular barcodes. Artifactual mutations introduced during the sample preparation or sequencing steps were reduced by requiring a mutation to be present in >90% of reads in a UID family in order for that UID to be scored as a “supermutant.” Wells with fewer than 200 UIDs as a result of poor amplification were excluded. DNA from the peripheral blood lymphocytes of healthy individuals was used as a control in each experiment to identify potential false-positive mutations.
Algorithm for classifying ctDNA status
ctDNA was classified as detectable (ctDNA-positive) or undetectable (ctDNA-negative) on the basis of a permutation test that compared the mutation frequency in the sample of interest with the mutation frequencies in controls. First, the MAF, defined as the ratio between the number of supermutants and the number of UIDs for the mutation of interest, was calculated for each well with >200 UIDs. The difference in the distributions of the MAFs between the sample of interest and the controls was then statistically evaluated with the permutation test, using the permTS function of the R package perm (R software version 3.2.3). The one-sided test was used to avoid attributing significance to a ctDNA-negative sample that has fewer supermutants than the associated control. A 0.1 P value was then chosen as the threshold to classify a sample of interest as ctDNA-positive (P < 0.1) or ctDNA-negative. Given the lack of a gold standard, a specificity of at least 0.90 was considered desirable, and a P value equal to 0.1 yielded 0.90 specificity when performing leave-one-out cross-validation on the controls.
The controls with the highest MAF (upper tail of the MAF distribution among controls) would inevitably be classified as positive by this permutation test in a leave-one-out cross-validation performed over all controls. The actual specificity realized in our experiments with patients is therefore expected to be higher than the 90% estimated from the control samples. Among patients not treated with chemotherapy, there were only 3 cases out of 178 with ctDNA detection and no recurrence, yielding a specificity of ~98% [false-positive rate (FPR), 2%]. Furthermore, the classification algorithm is based on testing differences in MAF distributions. It is therefore expected that a false positive is more likely in cases with low ctDNA concentrations, such as those shown in Fig. 3 (D and F).
The availability of multiple samples (n) from the same patients increases the probability of observing at least one false positive. Specifically, this probability is given by 1-P0, where P0 is the probability of 0 successes in a binomial with parameters n and p, with p being the FPR. At the same time, with multiple time points, the probability that a false positive will occur more than once in the same patient is 1-P0-P1, which is smaller than an FPR of 2% when n < 11. Thus, assessing multiple sequential samples from the same patient can increase the overall specificity.
Statistical analysis
A preplanned analysis was conducted when 32 (90%) of the predicted events had occurred. The primary outcome measure was RFS assessed by standard radiologic criteria. RFS was measured from date of surgery to documented first radiologic recurrence or death as a result of colorectal cancer and was censored at last follow-up or non–colorectal cancer–related death. Multivariate analysis was performed with manual backward stepwise Cox regression modeling considering T stage, postoperative ctDNA status, LN yield, LVI, and MMR status. T stage and postoperative ctDNA were the only independent variables included in the final model used to estimate the probability of RFS over time. Data on LVI were not available from nine cases. We used multiple imputation (Stata version 12.1) to handle subjects with missing data. We used a previously described time-dependent ROC analysis (40) performed with the R package survivalROC (version 1.0.3) to assess the time-dependent accuracy of postoperative ctDNA in predicting recurrence at 5, 6, 12, 18, 24, 36, and 40 months. We compared two groups using the χ2 or Fisher’s exact test for categorical variables and the Mann-Whitney (rank-sum) test for continuous variables. Statistical analysis was performed using Stata version 12.1 (StataCorp LP) and GraphPad Prism version 6.07 (GraphPad Software Inc.), where P values <0.05 were considered significant.
Supplementary Material
Fig. S1. Recurrence-free survival in patients not treated with adjuvant chemotherapy stratified by postoperative CEA status.
Table S1. Relationship between postoperative ctDNA, postoperative CEA, and recurrence status for patients not treated with chemotherapy.
Table S2. Time-dependent predictive accuracy of postoperative ctDNA for recurrence at 5, 6, 12, 18, 24, 36, and 40 months.
Table S3. Serial circulating biomarker status and clinical characteristics of patients not treated with adjuvant chemotherapy who experienced recurrence.
Table S4. Serial circulating biomarker status and clinical characteristics of chemotherapy-treated patients who experienced recurrence.
Table S5. Genomic and clinical summary for all patients included in the final evaluable population.
Table S6. Panel of 15 genes used for identification of somatic mutations in tumor tissue.
Acknowledgments
We thank all participating hospitals (Western Health, Royal Melbourne Hospital, Eastern Health, Alfred Hospital, Austin Health, Cabrini Health, Border Medical Oncology, Canberra Hospital, South West Oncology, Queen Elizabeth Hospital, Flinders Centre for Innovation in Cancer, Barwon Health, and Royal Women’s and Brisbane Hospital) for patient enrollment and sample collection; J. Ptak, J. Schaeffer, L. Dobbyn, and C. Blair for expert technical assistance; N. Turner, P. Robertson, M. Chapman, A. Woolley, and S. Carroll for administrative assistance; and Victorian Cancer Biobank for sample processing.
Funding: This study is supported by the Virginia and D.K. Ludwig Fund for Cancer Research, the Conrad N. Hilton Foundation, the Sol Goldman Sequencing Facility at Johns Hopkins, NIH grants (CA57345, R37-CA43460, U01-CA152753, and P30-CA006973), Victorian Cancer Agency Translational Research Grant, and Victorian Cancer Agency Clinical Research Fellowship (to J.T.).
Footnotes
SUPPLEMENTARY MATERIALS
www.sciencetranslationalmedicine.org/cgi/content/full/8/346/346ra92/DC1
Author contributions: J.T., B.V., and P.G. designed the study; B.V., Y.W., S.S., I.K., N.S., K.W.K., and N.P. performed massively parallel sequencing and bioinformatics analyses; C.T. and L.L. were responsible for developing the algorithm for classifying ctDNA status; M.C. performed pathology assessment of the tumor tissue; H.-L.W., S.K., I.S., R.W., M.S., B.T., J.D., I.J., A.H., T.H., and T.J.P. contributed to patient recruitment; J.T., Y.W., C.T., L.L., M.T., R.L.S., L.A.D., B.V., and P.G. analyzed and interpreted the data; all authors contributed to the writing and review of the manuscript.
Competing interests: K.W.K., N.P, L.A.D., and B.V. are founders of PapGene and Personal Genome Diagnostics and members of the Scientific Advisory Boards of Morphotek and Sysmex-Inostics. I.K. is an employee of PapGene. These companies and others have licensed patent applications on genetic technologies from Johns Hopkins, some of which result in royalty payments to K.W.K., N.P., L.A.D., B.V., and I.K. The terms of these arrangements are being managed by Johns Hopkins University in accordance with its conflict of interest policies.
Data and materials availability: DNA sequence data on individual patients have been deposited in the European Genome-phenome Archive (accession no. EGAS00001001839).
REFERENCES AND NOTES
- 1.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–E386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 2.Edge S, Byrd DR, Compton CC, Fritz AG, Greene FL, Trotti A. AJCC Cancer Staging Manual. Springer; New York: 2010. p. 648. [Google Scholar]
- 3.Dukes CE. The classification of cancer of the rectum. J Pathol Bacteriol. 1932;35:323–332. [Google Scholar]
- 4.Quah H-M, Chou JF, Gonen M, Shia J, Schrag D, Landmann RG, Guillem JG, Paty PB, Temple LK, Wong WD, Weiser MR. Identification of patients with high-risk stage II colon cancer for adjuvant therapy. Dis Colon Rectum. 2008;51:503–507. doi: 10.1007/s10350-008-9246-z. [DOI] [PubMed] [Google Scholar]
- 5.Niedzwiecki D, Bertagnolli MM, Warren RS, Compton CC, Kemeny NE, Benson AB, III, Eckhardt SG, Alberts S, Porjosh GN, Kerr DJ, Fields A, Rougier P, Pipas JM, Schwartz JH, Atkins J, O’Rourke M, Perry MC, Goldberg RM, Mayer RJ, Colacchio TA. Documenting the natural history of patients with resected stage II adenocarcinoma of the colon after random assignment to adjuvant treatment with edrecolomab or observation: Results from CALGB 9581. J Clin Oncol. 2011;29:3146–3152. doi: 10.1200/JCO.2010.32.5357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ribic CM, Sargent DJ, Moore MJ, Thibodeau SN, French AJ, Goldberg RM, Hamilton SR, Laurent-Puig P, Gryfe R, Shepherd LE, Tu D, Redston M, Gallinger S. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med. 2003;349:247–257. doi: 10.1056/NEJMoa022289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sargent DJ, Marsoni S, Monges G, Thibodeau SN, Labianca R, Hamilton SR, French AJ, Kabat B, Foster NR, Torri V, Ribic C, Grothey A, Moore M, Zaniboni A, Seitz JF, Sinicrope F, Gallinger S. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J Clin Oncol. 2010;28:3219–3226. doi: 10.1200/JCO.2009.27.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kopetz S, Tabernero J, Rosenberg R, Jiang Z-Q, Moreno V, Bachleitner-Hofmann T, Lanza G, Stork-Sloots L, Maru D, Simon I, Capellà G, Salazar R. Genomic classifier ColoPrint predicts recurrence in stage II colorectal cancer patients more accurately than clinical factors. Oncologist. 2015;20:127–133. doi: 10.1634/theoncologist.2014-0325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Venook AP, Niedzwiecki D, Lopatin M, Ye X, Lee M, Friedman PN, Frankel W, Clark-Langone K, Millward C, Shak S, Goldberg RM, Mahmoud NN, Warren RS, Schilsky RL, Bertagnolli MM. Biologic determinants of tumor recurrence in stage II colon cancer: Validation study of the 12-gene recurrence score in Cancer and Leukemia Group B (CALGB) 9581. J Clin Oncol. 2013;31:1775–1781. doi: 10.1200/JCO.2012.45.1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gray RG, Quirke P, Handley K, Lopatin M, Magill L, Baehner FL, Beaumont C, Clark-Langone KM, Yoshizawa CN, Lee M, Watson D, Shak S, Kerr DJ. Validation study of a quantitative multigene reverse transcriptase–polymerase chain reaction assay for assessment of recurrence risk in patients with stage II colon cancer. J Clin Oncol. 2011;29:4611–4619. doi: 10.1200/JCO.2010.32.8732. [DOI] [PubMed] [Google Scholar]
- 11.Zhang JX, Song W, Chen ZH, Wei JH, Liao YJ, Lei J, Hu M, Chen GZ, Liao B, Lu J, Zhao HW, Chen W, He YL, Wang HY, Xie D, Luo JH. Prognostic and predictive value of a microRNA signature in stage II colon cancer: A microRNA expression analysis. Lancet Oncol. 2013;14:1295–1306. doi: 10.1016/S1470-2045(13)70491-1. [DOI] [PubMed] [Google Scholar]
- 12.O’Connor ES, Greenblatt DY, LoConte NK, Gangnon RE, Liou J-I, Heise CP, Smith MA. Adjuvant chemotherapy for stage II colon cancer with poor prognostic features. J Clin Oncol. 2011;29:3381–3388. doi: 10.1200/JCO.2010.34.3426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Figueredo A, Charette ML, Maroun J, Brouwers MC, Zuraw L. Adjuvant therapy for stage II colon cancer: A systematic review from the Cancer Care Ontario Program in evidence-based care’s gastrointestinal cancer disease site group. J Clin Oncol. 2004;22:3395–3407. doi: 10.1200/JCO.2004.03.087. [DOI] [PubMed] [Google Scholar]
- 14.Mamounas E, Wieand S, Wolmark N, Bear HD, Atkins JN, Song K, Jones J, Rockette H. Comparative efficacy of adjuvant chemotherapy in patients with Dukes’ B versus Dukes’ C colon cancer: Results from four National Surgical Adjuvant Breast and Bowel Project adjuvant studies (C-01, C-02, C-03, and C-04) J Clin Oncol. 1999;17:1349–1355. doi: 10.1200/JCO.1999.17.5.1349. [DOI] [PubMed] [Google Scholar]
- 15.QUASAR Collaborative Group. Gray R, Barnwell J, McConkey C, Hills RK, Williams NS, Kerr DJ. Adjuvant chemotherapy versus observation in patients with colorectal cancer: A randomised study. Lancet. 2007;370:2020–2029. doi: 10.1016/S0140-6736(07)61866-2. [DOI] [PubMed] [Google Scholar]
- 16.André T, de Gramont A, Vernerey D, Chibaudel B, Bonnetain F, Tijeras-Raballand A, Scriva A, Hickish T, Tabernero J, Van Laethem JL, Banzi M, Maartense E, Shmueli E, Carlsson GU, Scheithauer W, Papamichael D, Möehler M, Landolfi S, Demetter P, Colote S, Tournigand C, Louvet C, Duval A, Fléjou J-F, de Gramont A. Adjuvant fluorouracil, leucovorin, and oxaliplatin in stage II to III colon cancer: Updated 10-year survival and outcomes according to BRAF mutation and mismatch repair status of the MOSAIC study. J Clin Oncol. 2015;33:4176–4187. doi: 10.1200/JCO.2015.63.4238. [DOI] [PubMed] [Google Scholar]
- 17.Jeffery GM, Hickey BE, Hider P. Follow-up strategies for patients treated for non-metastatic colorectal cancer. Cochrane Database Syst Rev. 2002:CD002200. doi: 10.1002/14651858.CD002200. [DOI] [PubMed] [Google Scholar]
- 18.Renehan AG, Egger M, Saunders MP, O’Dwyer ST. Impact on survival of intensive follow up after curative resection for colorectal cancer: Systematic review and meta-analysis of randomised trials. BMJ. 2002;324:813. doi: 10.1136/bmj.324.7341.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Figueredo A, Rumble RB, Maroun J, Earle CC, Cummings B, McLeod R, Zuraw L, Zwaal C. Gastrointestinal Cancer Disease Site Group of Cancer Care Ontario’s Program in Evidence-based Care, Follow-up of patients with curatively resected colorectal cancer: A practice guideline. BMC Cancer. 2003;3:26. doi: 10.1186/1471-2407-3-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pita-Fernàndez S, Alhayek-Aí M, González-Martín C, López-Calviño B, Seoane-Pillado T, Pértega-Díaz S. Intensive follow-up strategies improve outcomes in nonmetastatic colorectal cancer patients after curative surgery: A systematic review and meta-analysis. Ann Oncol. 2015;26:644–656. doi: 10.1093/annonc/mdu543. [DOI] [PubMed] [Google Scholar]
- 21.Chao M, Gibbs P. Caution is required before recommending routine carcinoembryonic antigen and imaging follow-up for patients with early-stage colon cancer. J Clin Oncol. 2009;27:e279–e280. doi: 10.1200/JCO.2009.25.6156. [DOI] [PubMed] [Google Scholar]
- 22.Benson AB, III, Desch CE, Flynn PJ, Krause C, Loprinzi CL, Minsky BD, Petrelli NJ, Pfister DG, Smith TJ, Somerfield MR. American Society of Clinical Oncology, 2000 update of American Society of Clinical Oncology colorectal cancer surveillance guidelines. J Clin Oncol. 2000;18:3586–3588. doi: 10.1200/JCO.2000.18.20.3586. [DOI] [PubMed] [Google Scholar]
- 23.Wood LD, Parsons DW, Jones S, Lin J, Sjöblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, Silliman N, Szabo S, Dezso Z, Ustyanksky V, Nikolskaya T, Nikolsky Y, Karchin R, Wilson PA, Kaminker JS, Zhang Z, Croshaw R, Willis J, Dawson D, Shipitsin M, Willson JKV, Sukumar S, Polyak K, Park BH, Pethiyagoda CL, Pant PVK, Ballinger DG, Sparks AB, Hartigan J, Smith DR, Suh E, Papadopoulos N, Buckhaults P, Markowitz SD, Parmigiani G, Kinzler KW, Velculescu VE, Vogelstein B. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 24.Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Diaz LA, Jr, Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, Allen B, Bozic I, Reiter JG, Nowak MA, Kinzler KW, Oliner KS, Vogelstein B. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012;486:537–540. doi: 10.1038/nature11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, Bartlett BR, Wang H, Luber B, Alani RM, Antonarakis ES, Azad NS, Bardelli A, Brem H, Cameron JL, Lee CC, Fecher LA, Gallia GL, Gibbs P, Le D, Giuntoli RL, Goggins M, Hogarty MD, Holdhoff M, Hong SM, Jiao Y, Juhl HH, Kim JJ, Siravegna G, Laheru DA, Lauricella C, Lim M, Lipson EJ, Marie SKN, Netto GJ, Oliner KS, Olivi A, Olsson L, Riggins GJ, Sartore-Bianchi A, Schmidt K, Shih IM, Oba-Shinjo SM, Siena S, Theodorescu D, Tie J, Harkins TT, Veronese S, Wang TL, Weingart JD, Wolfgang CL, Wood LD, Xing D, Hruban RH, Wu J, Allen PJ, Schmidt CM, Choti MA, Velculescu VE, Kinzler KW, Vogelstein B, Papadopoulos N, Diaz LA., Jr Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6:224ra24. doi: 10.1126/scitranslmed.3007094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, Valtorta E, Schiavo R, Buscarino M, Siravegna G, Bencardino K, Cercek A, Chen CT, Veronese S, Zanon C, Sartore-Bianchi A, Gambacorta M, Gallicchio M, Vakiani E, Boscaro V, Medico E, Weiser M, Siena S, Di Nicolantonio F, Solit D, Bardelli A. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486:532–536. doi: 10.1038/nature11156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morelli MP, Overman MJ, Dasari A, Kazmi SMA, Mazard T, Vilar E, Morris VK, Lee MS, Herron D, Eng C, Morris J, Kee BK, Janku F, Deaton FL, Garrett C, Maru D, Diehl F, Angenendt P, Kopetz S. Characterizing the patterns of clonal selection in circulating tumor DNA from patients with colorectal cancer refractory to anti-EGFR treatment. Ann Oncol. 2015;26:731–736. doi: 10.1093/annonc/mdv005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, Thornton K, Agrawal N, Sokoll L, Szabo SA, Kinzler KW, Vogelstein B, Diaz LA., Jr Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008;14:985–990. doi: 10.1038/nm.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tie J, Kinde I, Wang Y, Wong HL, Roebert J, Christie M, Tacey M, Wong R, Singh M, Karapetis CS, Desai J, Tran B, Strausberg RL, Diaz LA, Jr, Papadopoulos N, Kinzler KW, Vogelstein B, Gibbs P. Circulating tumor DNA as an early marker of therapeutic response in patients with metastatic colorectal cancer. Ann Oncol. 2015;26:1715–1722. doi: 10.1093/annonc/mdv177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garcia-Murillas I, Schiavon G, Weigelt B, Ng C, Hrebien S, Cutts RJ, Cheang M, Osin P, Nerurkar A, Kozarewa I, Garrido JA, Dowsett M, Reis-Filho JS, Smith IE, Turner NC. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci Transl Med. 2015;7:302ra133. doi: 10.1126/scitranslmed.aab0021. [DOI] [PubMed] [Google Scholar]
- 32.Sausen M, Phallen J, Adleff V, Jones S, Leary RJ, Barrett MT, Anagnostou V, Parpart-Li S, Murphy D, Kay Li Q, Hruban CA, Scharpf R, White JR, O’Dwyer PJ, Allen PJ, Eshleman JR, Thompson CB, Klimstra DS, Linehan DC, Maitra A, Hruban RH, Diaz LA, Jr, Von Hoff DD, Johansen JS, Drebin JA, Velculescu VE. Clinical implications of genomic alterations in the tumour and circulation of pancreatic cancer patients. Nat Commun. 2015;6:7686. doi: 10.1038/ncomms8686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kinde I, Wu J, Papadopoulos N, Kinzler KW, Vogelstein B. Detection and quantification of rare mutations with massively parallel sequencing. Proc Natl Acad Sci USA. 2011;108:9530–9535. doi: 10.1073/pnas.1105422108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wirtzfeld DA, Mikula L, Gryfe R, Ravani P, Dicks EL, Parfrey P, Gallinger S, Pollett WG. Concordance with clinical practice guidelines for adjuvant chemotherapy in patients with stage I–III colon cancer: Experience in 2 Canadian provinces. Can J Surg. 2009;52:92–97. [PMC free article] [PubMed] [Google Scholar]
- 35.O’Connell JB, Maggard MA, Ko CY. Colon cancer survival rates with the new American Joint Committee on Cancer sixth edition staging. J Natl Cancer Inst. 2004;96:1420–1425. doi: 10.1093/jnci/djh275. [DOI] [PubMed] [Google Scholar]
- 36.Bozic I, Reiter JG, Allen B, Antal T, Chatterjee K, Shah P, Moon YS, Yaqubie A, Kelly N, Le DT, Lipson EJ, Chapman PB, Diaz LA, Jr, Vogelstein B, Nowak MA. Evolutionary dynamics of cancer in response to targeted combination therapy. Elife. 2013;2:e00747. doi: 10.7554/eLife.00747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Newman AM, Lovejoy AF, Klass DM, Kurtz DM, Chabon JJ, Scherer F, Stehr H, Liu CL, Bratman SV, Say C, Zhou L, Carter JN, West RB, Sledge GW, Jr, Shrager JB, Loo BW, Jr, Neal JW, Wakelee HA, Diehn M, Alizadeh AA. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat Biotechnol. 2016;34:547–555. doi: 10.1038/nbt.3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shia J. Immunohistochemistry versus microsatellite instability testing for screening colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome. Part I. The utility of immunohistochemistry. J Mol Diagn. 2008;10:293–300. doi: 10.2353/jmoldx.2008.080031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kinde I, Bettegowda C, Wang Y, Wu J, Agrawal N, Shih IM, Kurman R, Dao F, Levine DA, Giuntoli R, Roden R, Eshleman JR, Carvalho JP, Marie SKN, Papadopoulos N, Kinzler KW, Vogelstein B, Diaz LA., Jr Evaluation of DNA from the Papanicolaou test to detect ovarian and endometrial cancers. Sci Transl Med. 2013;5:167ra4. doi: 10.1126/scitranslmed.3004952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heagerty PJ, Lumley T, Pepe MS. Time-dependent ROC curves for censored survival data and a diagnostic marker. Biometrics. 2000;56:337–344. doi: 10.1111/j.0006-341x.2000.00337.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Recurrence-free survival in patients not treated with adjuvant chemotherapy stratified by postoperative CEA status.
Table S1. Relationship between postoperative ctDNA, postoperative CEA, and recurrence status for patients not treated with chemotherapy.
Table S2. Time-dependent predictive accuracy of postoperative ctDNA for recurrence at 5, 6, 12, 18, 24, 36, and 40 months.
Table S3. Serial circulating biomarker status and clinical characteristics of patients not treated with adjuvant chemotherapy who experienced recurrence.
Table S4. Serial circulating biomarker status and clinical characteristics of chemotherapy-treated patients who experienced recurrence.
Table S5. Genomic and clinical summary for all patients included in the final evaluable population.
Table S6. Panel of 15 genes used for identification of somatic mutations in tumor tissue.