


Abstract
Surface-tethered nucleic acids are widely applied in solid-phase assays in which complementary strands must be detected against a complex mixture of other sequences. In response to such needs, numerous methods have been developed for immobilizing nucleic acids on solid supports. Often, detailed analysis of associated chemical transformations and of potential side reactions is difficult to obtain. Combined use of planar and high surface area powder supports allows characterization using surface as well as bulk diagnostic techniques. This approach is followed in the present study in which X-ray photoelectron spectroscopy (XPS), transmission infrared spectroscopy (FTIR) and reactivity titrations are used to investigate siliceous supports modified with an aminosilane precursor followed by a maleimide-bearing crosslinker for attachment of nucleic acids. The supports retain maleimide activity for approximately a day when stored under buffer, but deactivation is accelerated under basic conditions or by incomplete conversion of the precursor aminosilane monolayer. Reactions involving the olefinic bond of the imide as well as its carbonyl groups are observed and analyzed. Attachment of sulfhydryl-terminated oligodeoxyribonucleotides is highly site specific, and immobilized strands exhibit excellent hybridization activity. Quantitative use of XPS for label-free determination of DNA coverage based on calibration against reference materials is also described.
INTRODUCTION
Nucleic acid molecules immobilized on solid supports find widespread applications in genotyping and gene expression analyses (1,2) and in the related area of biosensors (3). These systems also present intriguing fundamental questions, foremost in understanding how surface organization of the nucleic acids impacts their hybridization behavior. On gold-coated surfaces immobilization is most often accomplished through thiolate–Au bonds (4–7). The chemistry of gold supports can be further tailored using alkanethiol molecules to control, e.g. DNA–surface interactions (6). A number of studies have observed that thiolate–Au attachment preserves close to 100% activity of surface-tethered strand ‘probes’ to bind complementary sequences under suitable conditions (6,8–11). In particular, high ionic strengths and sufficiently sparse probe coverages are required to mitigate electrosteric repulsions encountered by hybridizing ‘target’ strands.
Immobilization of nucleic acids on siliceous surfaces such as glass and silica, arguably the most common type of support, is faced with a more complex surface chemistry. The reactive sites on siliceous surfaces are silanol (-Si-OH) groups, which exhibit various reactivities depending on their hydrogen bonding state and connection to the skeletal silica matrix (12). Additional complexity results from the use of organosilanes, such as aminosilanes and mercaptosilanes, as surface modification agents. These reagents can polymerize as well as bind covalently to the support, with a chemistry that is sensitive to presence of water and to catalytic activity, e.g. by amine groups (13,14). Despite these intricacies, the widespread use of siliceous supports continues to motivate improved understanding and protocols for their modification with nucleic acids.
A number of investigators have employed maleimide–thiol conjugation to tether oligodeoxyribonucleotides to siliceous surfaces (15–20). Maleimides are convenient because of their good stability in aqueous environments and their selective and efficient reactivity toward sulfhydryl groups. In a typical approach the solid support is first derivatized with an aminosilane to introduce surface amine groups, followed by reaction with a bifunctional linker molecule containing an amino-reactive site (e.g. succinimide ester or isocyanate) in addition to the maleimide function. Attachment through the amino-reactive moiety leaves the maleimide available for subsequent immobilization of oligonucleotide. Potential side reactions between linker maleimides and silane amines, leading to maleimide deactivation, can be minimized by employing highly amino-reactive functions such as isocyanate to rapidly consume surface amines and prevent subsequent maleimide-amine additions (20).
The present contribution takes a detailed look at the chemistry of siliceous surfaces modified with maleimide groups and their use for immobilization of oligodeoxyribonucleotides. Powder and planar solid supports are employed to allow application of bulk and surface sensitive techniques including infrared spectroscopy (FTIR), titration analysis and X-ray photoelectron spectroscopy (XPS). FTIR and titration analysis are used to track chemical changes in maleimide-activated silica powders when stored under aqueous environments. One outcome from these studies is that decay in maleimide activity with time parallels that in bulk solution; however, faster decrease is observed if remnant silane amine groups are present on the solid support. XPS is used to investigate planar supports modified with oligonucleotides. XPS analysis confirms that, even after five days of immobilization, coupling between native groups on oligodeoxyribonucleotides and maleimide-activated surfaces does not take place. Therefore, attachment of thiol-terminated strands is exclusive through the thiol endgroup. The resultant end-tethered geometry is expected to preserve activity of bound strands toward complementary molecules in solution, as borne out by results of hybridization assays. A method for quantifying DNA coverage from absolute intensity of XPS P 2p emission is also presented.
MATERIALS AND METHODS
Summary of immobilization chemistry
Attachment of thiol-terminated oligodeoxyribonucleotides to siliceous supports was accomplished in three steps (20). In step 1, silica or glass supports were modified with aminopropyl triethoxysilane (APTES; 98% purity; Aldrich). In step 2, the heterobifunctional crosslinker p-maleimidophenyl isocyanate (PMPI, Pierce Biotechnology) was reacted with the support through its isocyanate group to form a urea linkage. In the final third step, thiol-terminated oligonucleotides were immobilized by reaction of the thiol with the PMPI maleimide C=C bond. All oligonucleotides used in these studies were purchased from Qiagen Inc. and were purified by high-performance liquid chromatography (HPLC). In addition to the surface modification steps described above, bismaleimide compounds formed from two PMPI molecules due to reaction between the isocyanate group of PMPI and trace water (21) can attach through maleimide–amine coupling (20,22). This leads to an excess of PMPI residues over those of APTES; however, the surface coverage of active maleimides remains limited by that of APTES residues. A schematic of the attachment chemistry is available as Figure S1 in Supplementary Material.
Preparation of powder supports
Aerosil® 200 fumed silica powder (99.8% amorphous SiO2) from Degussa-Hüls was used to examine steps 1 and 2 of the immobilization scheme, corresponding to the attachment of APTES and PMPI. The large specific surface area (200 m2/g) of this support facilitates application of standard tools such as FTIR and titration analysis for characterizing the chemical state of the surface. Supports bearing only an APTES monolayer will be denoted as silica/APTES, while silica/APTES/PMPI will signify modification with both APTES and PMPI. Preparation of samples followed previously detailed procedures (20) except that, in step 2, the amount of PMPI added to the reaction was fixed at 3 PMPI molecules per nm2 of powder surface present (PMPI concentration ∼40 mM). A lesser amount was used to prepare powders with incomplete conversion of the silica/APTES support.
Characterization of powder supports
FTIR and titration with Ellman's reagent [5,5′-dithio-bis-(2-nitrobenzoic acid); DTNB] were used to monitor chemical changes and activity of powder supports (20). Infrared spectra were collected by averaging 1000 scans at 4 cm−1 resolution. The calibrations used to estimate surface coverage of APTES and PMPI from integrated IR absorbances were:
![]()
![]()
A(ν) is absorbance at wavenumber ν. The numerators are proportional to the amount of APTES (Equation 1) or PMPI (Equation 2), while the denominator is proportional to the total amount of silica in the IR beam and hence to surface area (23). The above equations differ slightly from those reported previously (20) due to tightened integration limits which better isolate infrared absorbances uniquely associated with APTES and silica powder. The PMPI mode used in Equation 2 is affected by hydrolysis of the PMPI residue; therefore, Equation 2 is applicable to freshly made supports only.
Ellman's analysis was performed following the manufacturer's (Pierce Biotechnology) protocol but using mercaptoethanol (MCE) instead of L-cysteine as the titrant. MCE reacts with maleimide groups in a 1:1 stoichiometry; thus, coverage of active maleimide groups on a powder support can be determined by quantifying the number of molecules of MCE that react per area. The number of MCE molecules reacted per area was determined spectrophotometrically by measuring the resultant decrease in bulk MCE concentration using DTNB. DTNB participates in a thiol-exchange reaction with free MCE to release the strongly absorbing species, 2-nitro-5-thiobenzoic acid (extinction coefficient 14150 M−1 cm−1 at 412 nm). The generated absorbance is correlated with MCE concentration according to a calibration curve. Three milligrams of powder were used for each assay.
Preparation of oligodeoxyribonucleotide-modified slides
Glass slides were cleaned for 15 min in a UV-ozone cleaner (Model T10X10/OES, UVOCS Inc.) followed by immersion in deionized (DI) 18.2 MΩ cm water, then toluene and finally anhydrous toluene. Thus cleaned slides were placed in glass bottles containing 2.8 mM solution of APTES in anhydrous toluene for 30 min. Afterwards the slides were successively washed twice in toluene, once with DI water, and once with acetonitrile. The duration of each wash was 10 min.
A pair of APTES-modified slides was used to sandwich a silicone gasket (Grace BioLabs) to create a sealed chamber. The chamber was filled with 2.3 mM PMPI in anhydrous acetonitrile by injection through the silicone gasket with needle and syringe. After a 2 h reaction, the cell was drained and refilled with pure acetonitrile for 10 min, followed by a 10 min wash in SSC1M buffer (0.015 M sodium citrate, 1 M NaCl, pH 7.0). The buffer was drained and the cell was refilled with a 1 μM oligonucleotide probe solution in SSC1M. Immobilization times ranged from 2 to 120 h. Afterwards, slides were washed by refilling/draining the reaction cell four times with pure SSC1M, followed by a fifth refill for 10 min. Only then was the cell disassembled. The slides were washed with DI water (to remove salts), dried under a nitrogen flow, and used directly for characterization or for hybridization assays.
The 20mer probe sequence was d(CGTTGTAAAACGACGGCCAG). Probe P1 included a 3′ modification with a mercaptopropyl –(CH2)3SH moiety. Probe P2, of same sequence as P1, lacked the end modification. P1 was generated from as-received disulfide-terminated precursors by cleaving the disulfide with 200-fold excess of DTT (Pierce Biotechnology) in phosphate buffer (10 mM sodium phosphate, 1 M NaCl, pH 7) for 1 h, followed by purification and buffer switch to SSC1M on PD-10 columns (Amersham Biosciences).
Hybridization assays
For each assay, a set of four 150 μl chambers was created by sandwiching a silicone gasket (Grace BioLabs) between a probe-modified and an unmodified glass slide. Chamber 1 was kept dry as a control to determine the starting probe coverage. Chamber 2 was filled with pure SSC1M buffer for 15 h to monitor the extent of probe loss from the surface. Chamber 3 was filled with 1 μM solution of complementary 20mer ‘TC’ target, d(CTGGCCGTCGTTTTACAACG), in SSC1M for 15 h. Chamber 4 was filled with 1 μM solution of noncomplementary ‘TNC’ target, d(CTAACTGTTACCTCGGTCGG), in SSC1M for 15 h. After 15 h, chambers 3 and 4 were drained and refilled five times with SSC1M buffer to flush out remnant target solution, and then the slide assembly was taken apart. The entire slide was briefly rinsed with chilled ∼4°C DI water to remove any remnant salt solution, followed by immediate drying with a stream of compressed nitrogen. Chilled water was used to decrease the possibility of separation of hybridized duplexes. The dried slides were characterized by XPS to determine coverage of DNA strands (see below). The percentage of probes hybridized to TC targets was estimated from
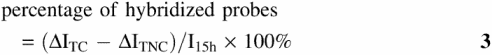
where I15h is P 2p intensity from the probe layer after a 15 h exposure to SSC1M buffer (chamber 2), and ΔITC and ΔITNC are increases relative to this intensity from spots exposed to complementary (chamber 3) and noncomplementary (chamber 4) targets, respectively. Subtraction of ΔITNC in Equation 3 corrects for signal arising from sequence-nonspecific adsorption of targets. By dividing by probe coverage remaining after 15 h of buffer contact, Equation 3 also corrects for probe losses that occur during the hybridization assay.
Characterization of slide supports
XPS measurements were performed on a Physical Electronics PHI 5500 instrument equipped with an Al X-ray monochromatic source (Al Kα line, 1486.6 eV) and a spherical capacitor energy analyzer. Elemental scans were carried out at a pass energy of 58.70 eV and a 45° takeoff angle for carbon (C 1s), silicon (Si 2s and 2p), oxygen (O 1s), phosphorus (P 2p) and nitrogen (N 1s). Typical integration times were 1.5 min for O 1s, 8 min for C 1s, Si 2s and Si 2p, 20 min for N 1s and 80 min for P 2p. Detection limits were ∼0.1% of the total photoelectron intensity. XPS traces were baseline corrected with the program XPSPeak, with baselines modeled as a combination of Shirley and linear functions.
Areal coverages of phosphate atoms were estimated from P 2p intensity using Equation 4, (24)
![]()
where σP is the surface density of P atoms (atoms/area), IP2p is experimentally measured P 2p intensity, θ = 45° is the takeoff angle defined between path of detected photoelectrons and the sample surface, R(θ)P2p is the instrument's response function for the P 2p spectral line and XP2p = 1290 × 10−24 cm2, the differential photoionization cross-section of the P 2p orbital.(25) RP2p (at a binding energy of 133 eV) was interpolated from RSi2p (at 100 eV) and RSi2s (at 151 eV). Values of R at the two silicon lines were measured from fused silica reference slides cleaned in situ in the XPS chamber with a beam of Ar ions until C 1s emission was negligible, and using the relationship R(θ)Si = ISi/ρSi XSi ΛSiS(24). Here, the number density of silicon atoms in fused silica ρSi = 2.2 × 1022 atoms/cm3, the attenuation lengths of photoelectrons in the silica support ΛSi2sS = 3.4 nm and ΛSi2pS = 3.5 nm (26), and the cross-sections XSi2s = 1030 × 10−24 cm2 and XSi2p = 884 × 10−24 cm2 (25). R changed by <3% over the range of interpolation. From the surface density σP of P atoms, oligonucleotide coverage can be obtained by dividing by 19, the number of P atoms per strand. In order to minimize influence of baseline curvature (cf. Figure 6), a consideration important at lower DNA coverages, P 2p intensity was integrated over a 3 eV window centered on the emission maximum rather than over the full emission peak. From analysis of the P 2p peak shape based on samples with high coverage, the full peak area IP2p can be obtained by multiplying the integrated area by 2.60 ± 0.14 (five independent measurements).
Figure 6.

XPS P 2p traces from APTES/PMPI slides reacted for 120 h with P1 (filled circles) or P2 oligodeoxynucleotides (open circles) suspended at 1.0 × 10−6 M concentration in SSC1M buffer.
Equation 4 assumes that IP2p is not attenuated by the presence of an overlayer. For this reason, coverages calculated from Equation 4 represent a lower limit as some attenuation of P 2p emission by the DNA layer itself is expected. The attenuation will be more prominent at higher coverages. A uniform overlayer model (24) was used to estimate the expected extent of attenuation. For 20mer oligonucleotides at a true coverage of 5 × 1012 cm−2, Equation 4 is expected to yield a coverage that is ∼7% lower, for 1 × 1013 cm−2 the discrepancy increases to ∼13%, while for 2 × 1013 cm−2 it is ∼24%. In this study, the surface density of strands remained <1.3 × 1013 cm−2.
RESULTS AND DISCUSSION
Powder supports
The olefinic double bond of maleimides, typically exploited for conjugation of thiol groups to yield thioether linkages, is also reactive toward other nucleophiles. Unprotonated primary and aromatic amines are known to add readily (27–30), and under aqueous environments reaction with hydroxyl anions is a possibility. Such side reactions can deactivate the support toward immobilization of biomolecules. Indeed, one motivation for using PMPI is that its isocyanate is highly amine-reactive, thus facilitating a near complete conversion of amines in the precursor silane film. High initial conversion efficiencies decrease opportunity for subsequent maleimide-amine additions which deactivate the support (20).
Figure 1 plots the percentage of maleimide activity remaining on silica/APTES/PMPI powders when stored under pH 7 buffer for up to one week, determined using Ellman's analysis. When sufficient PMPI was used to ensure full conversion of the APTES precursor film, the decrease in maleimide activity (black squares, solid line) closely paralleled that for maleimide groups in bulk solution (asterisks, dotted line). About 50% activity remained after 20 h of storage (note the logarithmic scale of the x-axis). If, however, the amount of PMPI added was insufficient to fully consume amine groups of the support, the initial decrease in maleimide activity was faster (open circles, dashed line) than in bulk solution. This drop in activity is attributed to deactivation of immobilized PMPI maleimides by reaction, post-immobilization, with remnant APTES amines. For the bulk studies, the bismaleimide bis-maleimidotetraethylene glycol [BM(PEO)4; Pierce Biotechnology] was used in lieu of PMPI since, in aqueous environments, the isocyanate of PMPI degrades to an aromatic amine (21) whose reactivity with maleimide groups could compromise the Ellman assay.
Figure 1.

Remnant fraction of active maleimides on silica/APTES/PMPI supports as a function of storage under 10 mM sodium phosphate, 0.1 M NaCl, pH 7.0. Filled squares and solid line: coverage of 1.1 PMPI/nm2 and 0.9 APTES/nm2, representing a fully converted APTES layer. Open circles and dashed line: coverage of 0.8 PMPI/nm2 and 1.3 APTES/nm2, representing a partially converted APTES layer. Asterisks and dotted line: 0.17 μM solution of BM(PEO)4. The lines are guides to the eye. All points represent an average of two measurements.
Storage of silica/APTES/PMPI supports in air for one week, inside a sealed vial, resulted in 60% retention of activity. Air storage is therefore recommended, if activated supports must be kept for days prior to immobilization of biomolecules. It is suspected, but was not investigated, that retention of activity under air will depend on ambient humidity.
Specifics of the chemical changes accompanying aging of silica/APTES/PMPI supports were investigated by FTIR, using previously reported spectral assignments (20,31). Figure 2 shows absorption spectra from 3000 to 3150 cm−1, covering maleimide and aromatic C–H stretch bands. The maleimide stretch, indicated by arrow, decreases in intensity with storage under pH 7 buffer (Figure 2a). This band is sensitive to the presence of unsaturation on the carbon atoms, and its gradual decrease indicates disappearance of the C=C bond and hence loss of maleimide activity, in agreement with results of Ellman's analysis. Higher pH values accelerate the process, while lower values suppress it (Figure 2b). The pH dependence suggests a chemical mechanism in which addition of HOH across the C=C bond is mediated by hydroxyl anion attack on one of the unsaturated carbons.
Figure 2.

IR absorbance of silica/APTES/PMPI supports in the region of maleimide and aromatic C–H stretching. (a) As a function of immersion time in 10 mM sodium phosphate, 0.1 M NaCl, pH 7.0. Solid line: 0.5 h; dashed line: 4.5 h; dotted line: 168 h. (b) As a function of pH for a 3 h immersion in 10 mM sodium phosphate, 0.1 M NaCl. Solid line: pH 2; dashed line: pH 7; dotted line: pH 12. Arrow in (a) indicates the maleimide C–H band. The structure shown in the inset indicates the corresponding C–H bonds in bold.
Spectral trends between 1350 and 1800 cm−1, where multiple PMPI bands are present, (20) provide evidence of additional chemical changes. Several bands, most notably the symmetric (1778 cm−1) and asymmetric (1717 cm−1) maleimide C=O stretches, and the maleimide symmetric C–N–C stretch (∼1405 cm−1), decrease with storage time (Figure 3a) and at elevated pH (Figure 3b). The attrition of these bands is indicative of opening of the maleimide ring and, as evident from Figure 3b, is facilitated by increased basicity. These observations agree with known hydrolysis of N-alkyl maleimides as a consequence of −OH attack on a carbonyl carbon of the imide 1, producing N-alkylmaleamic acid 2 [Figure 4; (32)].
Figure 3.

IR absorbance of silica/APTES/PMPI powders in the region of carbonyl absorptions. Arrows in (a) indicate maleimide bands whose intensity decreases with (a) immersion time or (b) with elevated pH. Symbols and conditions are as in Figure 2.
Figure 4.

Hydrolysis of N-alkyl maleimides.
Opening of the imide ring is also expected to lead to enhanced amide I and II modes (∼1660 and ∼1560 cm−1, respectively), to carboxylic acid C=O stretching between 1650 and 1680 cm−1 (if hydrogen bonded), and to bands from acid salts -COO− between 1550 to 1650 cm−1 (asymmetric stretch) and between 1330 and 1440 cm−1 (symmetric stretch, broad band) (33). These expectations are consistent with the generally increased absorbance seen in these regions, though complexity of the spectra and band overlap prevent detailed identification of features.
Significantly, should hydrolysis occur at both carbonyl groups, any biomolecules immobilized through the maleimide moiety would be cleaved from the support. Similar outcome would result from hydrolysis of the APTES–PMPI urea linkage. In either scenario, primary amines would be left behind on the solid support. It is therefore significant that primary amine N–H stretch bands were not observed in silica/APTES/PMPI spectra even after seven days of buffer contact. This indicates that hydrolysis was not so extensive as to fully cleave a significant fraction of imide rings or urea bonds.
In summary, titration and spectroscopic analyses show that maleimide-derivatized silica supports lose about half of their activity in a day when stored under pH 7 buffer. Activity can be retained for more than a week when stored under air. Deactivation is accelerated under alkaline conditions, suggesting a mechanism that involves base-catalyzed addition of water to the imide C=C bond. Concomitantly, at a similarly slow rate, hydrolysis at carbonyl sites leads to opening of the maleimide ring.
Immobilization of oligodeoxyribonucleotides
Glass slides, modified with APTES and PMPI, were reacted with probe molecules as described in Methods. Figure 5 shows normalized XPS C 1s traces from APTES, APTES/PMPI, and APTES/PMPI/DNA slides. Addition of PMPI residues is signified by the appearance of a prominent shoulder at higher binding energy, attributed to carbonyl carbons. After DNA attachment, the C 1s trace further broadens on account of diversity of carbon bonding configurations in DNA (34–37).
Figure 5.
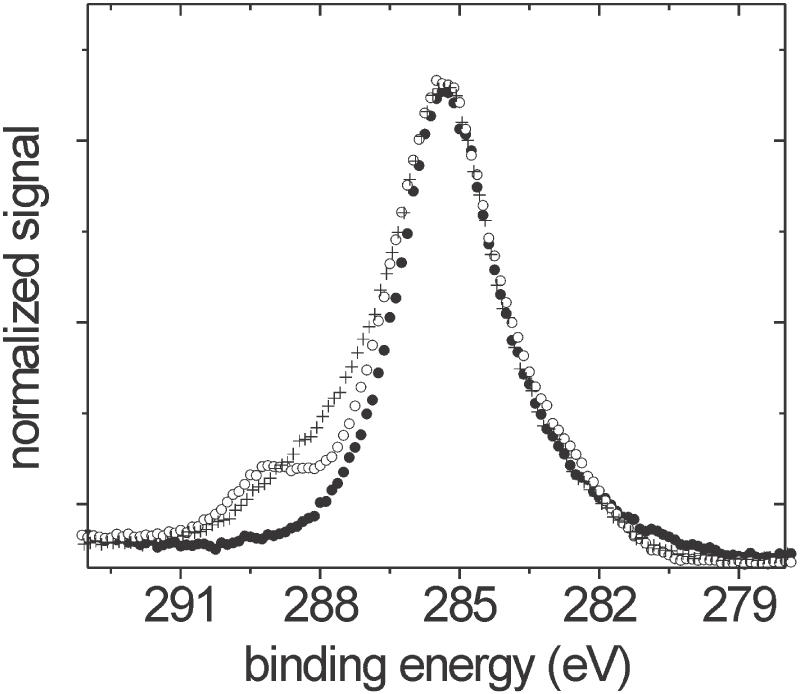
XPS C 1s traces from APTES (filled circles), APTES/PMPI (open circles), and APTES/PMPI/DNA (crosses) modified glass slides. The traces were normalized to facilitate comparison of peak shape.
An important consideration is the possibility of DNA-surface crosslinking at unintended sites along the nucleic acid backbone. Even unreactive moieties, such as aromatic base amines, raise concern if long enough contact times are allowed for the reaction to take place. Such side reactions could lead to multipoint attachment that would be detrimental to the ability of probes to hybridize to target molecules.
Figure 6 depicts P 2p traces measured from maleimide-activated slides after immersion in oligodeoxyribonucleotide solutions in SSC1M buffer for 120 h (5 days). Presence of P 2p emission is indicative of DNA attachment as no other source of phosphorus was used during sample preparation. Within sensitivity limits of the XPS data (∼5 × 1011 strands/cm2), P2 chains, which lack a terminal thiol, do not attach. In contrast, a surface coverage of 2.0 × 1012 cm−2 is found for the P1 oligonucleotides, which contain a terminal thiol. These data convincingly demonstrate that attachment of thiol-terminated oligodeoxyribonucleotides to maleimide-activated supports is highly site-specific, with virtually all strands immobilized in an end-tethered geometry, allaying previous concerns about potential deactivation via side reactions (20). Same conclusions were reached in experiments in which shorter contact times of 2, 5, and 48 h were used. Since maleimide activity will have largely decayed after 120 h (Figure 1), prospects of unintended crosslinking occurring at even longer times are minimal.
These results agree with Adessi et al. (16) who estimated that 88% of thiol-terminated probes were site-specifically immobilized to maleimide supports. In that earlier study site-specificity of attachment was estimated indirectly from extents of hybridization to complementary strands, compared to the direct XPS measurements reported here.
Hybridization of oligodeoxyribonucleotide layers
The ability of probe molecules to bind complementary target strands defines their usefulness for diagnostic assays. Hybridization efficiencies close to 100% have been reported for fairly short (∼20 nt) oligodeoxyribonucleotides self-assembled on Au supports via gold–thiolate bonds, provided the coverage was less than ∼4 × 1012 cm−2 and interactions with the solid support were not too strong (6,8,10,11). On glass supports, with independent determination of probe and target coverage, studies with 20mer to 30mer strands immobilized via a variety of covalent methods find hybridization efficiencies from 9 to 60% at coverages between 1 × 1013 and 2 × 1013 cm−2 (15,18,20,38–41). At lower coverages, in particular below ∼4 × 1012 cm−2 for which close to 100% efficiencies are expected from comparable systems on gold, on siliceous surfaces hybridization efficiencies of between 5 (42) and 100% (41) have been reported. Most values fall between 10 and 50% (20,39–42). The broad diversity of reported behavior reflects the large variety of immobilization protocols used for siliceous supports.
Failure to achieve high extents of hybridization under conditions when full probe activity is expected can serve as a signal for probe deactivation, e.g. through complexation with the support due to multiple covalent bonds or strong physical interactions. Results from the previous section indicate that probe strands attach strictly through one end; therefore, barring excessive physical interactions with the solid support, close to 100% hybridization activity would be expected at lower strand coverages and high ionic strengths. This expectation was tested using four independently prepared samples differing in surface coverage of probe molecules. After preparation, each sample was divided into four regions as described in the Methods section: region 1 was the as-prepared P1 probe layer, region 2 was exposed for 15 h to SSC1M buffer, region 3 was hybridized for 15 h to complementary TC targets, and region 4 was hybridized for 15 h to noncomplementary TNC strands. XPS was used to measure P 2p intensity (IP2p) from each of the four regions. The results are summarized in Figure 7.
Figure 7.

IP2p intensities measured from P1 probe films initially (white bars), after 15 h immersion in SSC1M buffer (black bars), after 15 h hybridization to TC target (lightly hatched bars) and after 15 h hybridization to TNC target (densely hatched bars). The y-axis is proportional to the total (probe + target) coverage of DNA, with an estimated uncertainty due to emission peak integration of ± 10%. The coverage of probe molecules remaining after 15 h immersion in SSC1M buffer, calculated using Equation 4, is listed along the x-axis for each of the four samples studied. The boxes show percentage of probe molecules hybridized to target strands, as calculated from Equation 3.
The difference in IP2p between the ‘initial’ and ‘15 h in buffer’ columns reflects probe removal from the solid support, predominantly attributed to hydrolysis of APTES residues from the glass slide. The loss of probe molecules cannot be attributed to removal of physisorbed strands, as other XPS results show that the washing protocols used do not leave such residue (e.g. see P2 data in Figure 6). Moreover, as mentioned earlier, FTIR analysis of silica/APTES/PMPI powders did not find evidence of significant hydrolysis at other potential sites such as the APTES–PMPI urea bond. The coverage of probe molecules remaining after 15 h, calculated from Equation 4, is shown along the x-axis of Figure 7. Percentages of probe molecules that hybridized to complementary TC targets, calculated from Equation 3, are displayed inside the white boxes.
The results of Figure 7 merit several comments. First, the fraction of probe molecules lost after a 15 h contact with buffer varied somewhat over the four samples studied. These variations likely reflect differences in crosslinking between neighboring APTES residues and between the residues and the solid support, causing variations in layer stability. Second, when the hybridization data are corrected for probe loss, a fairly consistent picture emerges regarding activity of surface-bound strands. For coverages lower than ∼5.5 × 1012 cm−2, hybridization extents close to 80–90% are observed. The data suggest that steric and electrostatic barriers to hybridization are mild at these lower probe coverages, yielding an average hybridization of 89 ± 8%. It is suspected that this number is somewhat underestimated on account of washing the samples prior to XPS measurement (with 4°C water), which may be expected to remove some target strands. Moreover, in correcting for nonspecific adsorption it should be noted that Equation 3 assumes that same-length complementary and noncomplementary target sequences, TC and TNC, produce the same nonspecific signal when same probe coverages and hybridization conditions are used.
For the highest coverage of 9.2 × 1012 cm−2, hybridization to target molecules is clearly suppressed relative to lower probe coverages. These results are consistent with those of Steel et al.(8) in which close to 100% hybridization was observed with 25mer probe films on gold supports and 25mer targets, when probe coverage was <4 × 1012 cm−2. When probe coverage increased to 6.5 × 1012 cm−2, hybridization extents decreased to ∼20%. In the study of Peterson et al. (11) employing a very similar experimental system on gold, hybridization efficiencies fell <80% when probe coverage was in excess of 2 × 1012 cm−2. The present results are in reasonable agreement with these other studies on an entirely different type of solid support. Consistency independent of details of surface chemistry is an important factor in advancing fundamental understanding of surface hybridization reactions.
A previous study that also used APTES/PMPI immobilization found a 40% hybridization efficiency based on an initial probe coverage of 2.2 × 1012 cm−2 and a 30 h hybridization time (20). The reasons for this low efficiency, which were not clarified previously, are attributed to not correcting for probe loss over the time course of hybridization.
CONCLUSIONS
Solid supports activated with maleimide groups provide excellent selectivity toward site-specific immobilization of oligodeoxyribonucleotides, exhibiting no evidence of side reactions with native constituents even after five days of contact under neutral buffer. The absence of undesirable crosslinking reactions is also instrumental in retention of high hybridization activity of immobilized strands. Maleimide-activated supports can be stored under pH 7 buffer for up to a day. Longer times are inadvisable on account of progressive loss of maleimide activity, attributed to base-catalyzed addition of water to the C=C bond of the imide. A challenge faced by applications requiring long hybridization times is limited stability of silane films which are typically used as a first step in modification of siliceous supports. In the present study, up to 40% of probe molecules were lost after 15 h exposure to buffer, presumably due to hydrolysis of APTES silane residues from the solid support. Improved stability should be attainable, e.g. through crosslinking of the surface layer with dendrimer compounds (43).
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
Acknowledgments
ACKNOWLEDGEMENTS
This work was supported by a National Science Foundation CAREER award (DMR-00-93758) and has used shared experimental facilities supported primarily by the MRSEC Program of the NSF (DMR-02-13574) and by the New York State Office of Science, Technology and Academic Research (NYSTAR).
REFERENCES
- 1.Mir K.U. and Southern,E.M. (2000) Sequence variation in genes and genomic DNA: methods for large-scale analysis. Annu. Rev. Genomics Hum. Genet., 1, 329–360. [DOI] [PubMed] [Google Scholar]
- 2.Brown P.O. and Botstein,D. (1999) Exploring the new world of the genome with DNA microarrays. Nature Genet., 21, 33–37. [DOI] [PubMed] [Google Scholar]
- 3.Wang J. (2000) From DNA biosensors to gene chips. Nucleic Acids Res., 28, 3011–3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Okahata Y., Matsunobu,Y., Ijiro,K., Mukae,M., Murakami,A. and Makino,K. (1992) Hybridization of nucleic acids immobilized on a quartz crystal microbalance. J. Am. Chem. Soc., 114, 8299–8300. [Google Scholar]
- 5.Piscevic D., Lawall,R., Veith,M., Liley,M., Okahata,Y. and Knoll,W. (1995) Oligonucleotide hybridization observed by surface plasmon optical techniques. Appl. Surf. Sci., 90, 425–436. [Google Scholar]
- 6.Herne T.M. and Tarlov,M.J. (1997) Characterization of DNA probes immobilized on gold surfaces. J. Am. Chem. Soc., 119, 8916–8920. [Google Scholar]
- 7.Kelley S.O., Boon,E.M., Barton,J.K., Jackson,N.M. and Hill,M.G. (1999) Single-base mismatch detection based on charge transduction through DNA. Nucleic Acids Res., 27, 4830–4837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Steel A.B., Herne,T.M. and Tarlov,M.J. (1998) Electrochemical quantitation of DNA immobilized on gold. Anal. Chem., 70, 4670–4677. [DOI] [PubMed] [Google Scholar]
- 9.Levicky R., Herne,T.M., Tarlov,M.J. and Satija,S.K. (1998) Using self-assembly to control the structure of DNA monolayers on gold: a neutron reflectivity study. J. Am. Chem. Soc., 120, 9787–9792. [Google Scholar]
- 10.Peterson A.W., Heaton,R.J. and Georgiadis,R. (2000) Kinetic control of hybridization in surface immobilized DNA monolayer films. J. Am. Chem. Soc., 122, 7837–7838. [Google Scholar]
- 11.Peterson A.W., Heaton,R.J. and Georgiadis,R.M. (2001) The effect of surface probe density on DNA hybridization. Nucleic Acids Res., 29, 5163–5168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vansant E.F., Van Der Voort,P. and Vrancken,K.C. (1995) Characterization and Chemical Modification of the Silica Surface. Elsevier, New York. [Google Scholar]
- 13.Blitz J., Murthy,R.S.S. and Leyden,D.E. (1987) Ammonia-catalyzed silylation reactions of Cab-O-Sil with methoxymethylsilanes. J. Am. Chem. Soc., 109, 7141–7145. [Google Scholar]
- 14.White L.D. and Tripp,C.P. (2000) An infrared study of the amine-catalyzed reaction of methoxymethylsilanes with silica. J. Colloid Interface Sci., 227, 237–243. [DOI] [PubMed] [Google Scholar]
- 15.Chrisey L.A., Lee,G.U. and O`Ferrall,C.E. (1996) Covalent attachment of synthetic DNA to self-assembled monolayer films. Nucleic Acids Res., 24, 3031–3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adessi C., Matton,G., Ayala,G., Turcatti,G., Mermod,J.-J., Mayer,P. and Kawashima,E. (2000) Solid phase DNA amplification: characterisation of primer attachment and amplification mechanisms. Nucleic Acids Res., 28, e87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Okamoto T., Suzuki,T. and Yamamoto,N. (2000) Microarray fabrication with covalent attachment of DNA using bubble jet technology. Nat. Biotechnol., 18, 438–441. [DOI] [PubMed] [Google Scholar]
- 18.Cavic B.A., McGovern,M.E., Nisman,R. and Thompson,M. (2001) High surface density immobilization of oligonucleotide on silicon. Analyst, 126, 485–490. [DOI] [PubMed] [Google Scholar]
- 19.Oh S.J., Cho,S.J., Kim,C.O. and Park,J.W. (2002) Characteristics of DNA microarrays fabricated on various aminosilane layers. Langmuir, 18, 1764–1769. [Google Scholar]
- 20.Jin L., Horgan,A. and Levicky,R. (2003) Preparation of end-tethered DNA monolayers on siliceous surfaces using heterobifunctional cross-linkers. Langmuir, 19, 6968–6975. [Google Scholar]
- 21.Annunziato M.E., Patel,U.S., Ranade,M. and Palumbo,P.S. (1993) P-maleimidophenyl isocyanate—a novel heterobifunctional linker for hydroxyl to thiol coupling. Bioconjug. Chem., 4, 212–218. [DOI] [PubMed] [Google Scholar]
- 22.Shen G., Horgan,A. and Levicky,R. (2004) Reaction of N-phenyl maleimide with aminosilane monolayers. Colloids Surf. B, Biointerfaces, 35, 59–65. [DOI] [PubMed] [Google Scholar]
- 23.Gorski D., Klemm,E., Fink,P. and Horhold,H.-H. (1988) Investigation of quantitative SiOH determination by silane treatment of disperse silica. J. Colloid Interface Sci., 126, 445–449. [Google Scholar]
- 24.Fadley C.S. (1984) Angle-resolved X-ray photoelectron spectroscopy. Prog. Surf. Sci., 16, 275–388. [Google Scholar]
- 25.Scofield J.H. (1976) Hartree-Slater subshell photoionization cross-sections at 1254 and 1487 eV. J. Electron. Spectrosc., 8, 129–137. [Google Scholar]
- 26.Powell C.J. and Jablonski,A. (2001) NIST Electron Effective-Attenuation-Length Database - Version 1.0. National Institute of Standards and Technology, Gaithersburg, MD. [Google Scholar]
- 27.Crivello J.V. (1973) Polyaspartimides—condensation of aromatic diamines and bismaleimide compounds. J. Polym. Sci., Part A: Polym. Chem., 11, 1185–1200. [Google Scholar]
- 28.Shen Z. and Schlup,J.R. (1998) Mid- and near-infrared spectroscopic investigations of N-phenylmaleimide (NPM)/amine reactions. J. Appl. Polym. Sci., 67, 267–276. [Google Scholar]
- 29.Gambogi J.E. and Blum,F.D. (1992) Molecular mobility of the interface in a model polymer composite: a NMR study. Macromolecules, 25, 4526–4534. [Google Scholar]
- 30.Smyth D.G., Blumenfeld,O.O. and Konigsberg,W. (1964) Reactions of N-ethylmaleimide with peptides and amino acids. Biochem. J., 91, 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Parker S.F., Mason,S.M. and Williams,K.P.J. (1990) Fourier transform Raman and infrared spectroscopy of N-phenylmaleimide and methylene dianiline bismaleimide. Spectrochim. Acta, 46A, 315–321. [Google Scholar]
- 32.Matsui S. and Aida,H. (1978) Hydrolysis of some N-alkylmaleimides. J. Chem. Soc. Perkin Trans.I, 2, 1277–1280. [Google Scholar]
- 33.Socrates G. (1994) Infrared Characteristic Group Frequencies. 2nd edn. John Wiley & Sons Inc., New York. [Google Scholar]
- 34.Barber M. and Clark,D.T. (1970) Molecular core binding energies: carbon and nitrogen 1s levels for cytosine and thymine. Chem. Commun., 24–25. [Google Scholar]
- 35.Barber M. and Clark,D.T. (1970) Molecular core binding energies: carbon and nitrogen 1s levels for adenine. Chem. Commun., 23–24. [Google Scholar]
- 36.Peeling J., Hruska,F.E. and McIntyre,N.S. (1978) ESCA spectra and molecular charge distributions for some pyrimidine and purine bases. Can. J. Chem., 56, 1555–1561. [Google Scholar]
- 37.May C.J., Canavan,H.E. and Castner,D.G. (2004) Quantitative X-ray photoelectron spectroscopy and time-of-flight secondary ion mass spectrometry characterization of the components in DNA. Anal. Chem., 76, 1114–1122. [DOI] [PubMed] [Google Scholar]
- 38.O'Donnell M.J., Tang,K., Koster,H., Smith,C.L. and Cantor,C.R. (1997) High density, covalent attachment of DNA to silicon wafers for analysis by MALDI-TOF mass spectrometry. Anal. Chem., 69, 2438–2443. [DOI] [PubMed] [Google Scholar]
- 39.Rogers Y.-H., Jiang-Baucom,P., Huang,Z.-J., Bogdanov,V., Anderson,S. and Boyce-Jacino,M.T. (1999) Immobilization of oligonucleotides onto a glass support via disulfide bonds: A method for preparation of DNA microarrays. Anal. Biochem., 266, 23–30. [DOI] [PubMed] [Google Scholar]
- 40.Walsh M.K., Wang,X. and Weimer,B.C. (2001) Optimizing the immobilization of single-stranded DNA onto glass beads. J. Biochem. Biophys. Methods, 47, 221–231. [DOI] [PubMed] [Google Scholar]
- 41.Podyminogin M.A., Lukhtanov,E.A. and Reed,M.W. (2001) Attachment of benzaldehyde-modified oligodeoxynucleotide probes to semicarbazide-coated glass. Nucleic Acids Res., 29, 5090–5098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Osborne M.A., Barnes,C.L., Balasubramanian,S. and Klenerman,D. (2001) Probing DNA surface attachment and local environment using single molecule spectroscopy. J. Phys. Chem. B, 105, 3120–3126. [Google Scholar]
- 43.Benters R., Niemeyer,C.M. and Wohrle,D. (2001) Dendrimer-activated solid supports for nucleic acid and protein microarrays. Chembiochem, 2, 686–694. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.