

Abstract
Background
The availability of the peach genome sequence has fostered relevant research in peach and related Prunus species enabling the identification of genes underlying important horticultural traits as well as the development of advanced tools for genetic and genomic analyses. The first release of the peach genome (Peach v1.0) represented a high-quality WGS (Whole Genome Shotgun) chromosome-scale assembly with high contiguity (contig L50 214.2 kb), large portions of mapped sequences (96%) and high base accuracy (99.96%). The aim of this work was to improve the quality of the first assembly by increasing the portion of mapped and oriented sequences, correcting misassemblies and improving the contiguity and base accuracy using high-throughput linkage mapping and deep resequencing approaches.
Results
Four linkage maps with 3,576 molecular markers were used to improve the portion of mapped and oriented sequences (from 96.0% and 85.6% of Peach v1.0 to 99.2% and 98.2% of v2.0, respectively) and enabled a more detailed identification of discernible misassemblies (10.4 Mb in total). The deep resequencing approach fixed 859 homozygous SNPs (Single Nucleotide Polymorphisms) and 1347 homozygous indels. Moreover, the assembled NGS contigs enabled the closing of 212 gaps with an improvement in the contig L50 of 19.2%.
Conclusions
The improved high quality peach genome assembly (Peach v2.0) represents a valuable tool for the analysis of the genetic diversity, domestication, and as a vehicle for genetic improvement of peach and related Prunus species. Moreover, the important phylogenetic position of peach and the absence of recent whole genome duplication (WGD) events make peach a pivotal species for comparative genomics studies aiming at elucidating plant speciation and diversification processes.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-017-3606-9) contains supplementary material, which is available to authorized users.
Keywords: Prunus persica, WGS assembly, SNPs, SSRs, Linkage mapping, NGS resequencing, Gap patching, Recombination rates, Centromeric regions
Background
The WGS (Whole Genome Shotgun) approach for sequencing complex eukaryotic genomes [1, 2] has contributed to the assembling many genomes of non-model and crop species. Poplar [3] and grape [4] were the first plant genomes sequenced with this approach. The advantages of WGS sequencing, as compared to the BAC by BAC (BAC, Bacterial Artificial Chromosome) approach [5–7] are the speed of sequencing and the reduced cost. However, a weakness of the WGS sequencing approach is it tends to produce a more fragmented assembly with reduced contiguity, also coupled with the risk of large-scale misassemblies. This is especially true for complex eukaryotic genomes and the assembly process can be confounded with recent duplication events (either segmental or Whole Genome Duplication, WGD), large regions of expanded repeats (up to 85% of the genome in species such as corn and wheat [8, 9]), and residual heterozygosity. The result is the production of a fragmented sequence with poor contiguity metrics such as the N50 (number of DNA stretches that contain half of the genome) and the L50 (the shortest sequence length at 50% of the genome). The potential issues are greater if a highly heterozygous individual is chosen as reference [10, 11]. However, in the BAC by BAC approach, the use of local sequence information (i.e. the single BAC clone) mitigates the risk of large-scale misassembly. The advent of Next Generation Sequencing (NGS) technologies has exacerbated WGS assembly drawbacks, typically producing a more fragmented assembly. NGS produces shorter sequence reads compared to the Sanger method, making genome assembly more difficult and requiring the development of a range of dedicated bioinformatics tools and novel alignment algorithms [12]. A de novo short-read NGS assembly needs high genome coverage, mainly to overcome the reduced overlap length and improve the contiguity of the resulting assembly [13]. Henson et al. [14] calculated that by increasing the reads length from 50 bp to 1000 bp, the contig L50 value of the human genome can theoretically increase from 3 kb to about 9,000 kb. Moreover, the lack of a chromosome-scale assembly, combined with putative misassemblies (usually undetected in non-anchored WGS genomes) precludes extensive use in evolutionary and comparative genomics studies, as well as Genome Wide Association Studies (GWAS). The availability of a chromosome-scale assembly is, therefore, crucial to maximally leverage the advantages of the WGS sequences. The coupling of WGS genome assembly with highly saturated and high resolution molecular genetic maps has been proposed to cope with the lack of chromosome-scale WGS genomes [15–17]. Genetic maps enable the reconstruction of a chromosome-scale sequence by positioning WGS scaffolds in their correct order and orientation to arrange them in long stretches of DNA, representing the individual chromosomes, called “pseudomolecules” or “pseudochromosomes”. Inconsistencies between the position of markers on the map and in the assembly can highlight putative misassembled sequences that can be further resolved by breaking the chimeric scaffolds and rearranging the broken pieces in their correct order and orientation. Fragmented genome assemblies, such as those obtained with short reads NGS data, need dense genetic maps for a large fraction of the assembled sequence to be anchored on chromosomes.
The availability of WGS genome assemblies in many species combined with NGS platforms has fueled variant discovery through alignment of resequenced reads of different accessions to the reference genome. Millions of Single Nucleotide Polymorphisms (SNPs) and small insertions/deletions (indels) dispersed throughout the genome have been discovered in different species [18–20]. This discovery, coupled with high-throughput genotyping technologies, such as SNP arrays [21–26] and genotyping by sequencing (GBS) [27–30], has accelerated the construction of high-resolution genetic maps, enabling map-sequence integration of WGS scaffolds in highly fragmented de novo NGS assemblies. Medium and high-throughput genotyping tools have been developed in Prunus for peach [31] and cherry [32] and in other Rosaceae species such as apple [21, 33] and strawberry [22].
To overcome the limitations of short read sequencing technologies (such as Illumina) third generation methodologies, based on single molecule sequencing, have been recently released such as the one of Pacific Bioscience [34] and Moleculo [35]. These methodologies, are capable of obtaining much longer reads, up to 50 kb [36], with the trade-off of an increase in error rates of 13-15% [37, 38] in comparison to 0.2–0.8% in Illumina short reads [38]. To solve this problem, several strategies have been proposed such as the integration of long reads with more accurate NGS reads. This hybrid sequencing strategy has been recently used in pineapple [39, 40] apple [41] and Arabidopsis thaliana Ler [42].
The peach [Prunus persica (L.) Batsch] genome sequence was obtained by the International Peach Genome Initiative (IPGI [20]) and is an 8.5-fold WGS high quality draft sequence [43] with long contiguity, high base accuracy, and a large portion of sequences mapped on chromosomes. Sequences were obtained using the Sanger methodology and a complete homozygous reference accession, the ‘Lovell’ double haploid PLov2-2n. The Prunus reference map (TxE [44–46]) was used to anchor the first release of the peach genome (Peach v1.0) obtaining eight pseudomolecules representing the eight Prunus chromosomes. Approximately half of the markers were placed by genotyping only six seedlings using the BIN mapping strategy [47, 48]. Thus, even if the BIN mapped markers were useful to assign scaffolds to chromosomes and check scaffold integrity, in most cases they did not provide sufficient information for anchoring the scaffolds on chromosomes. Moreover, the TxE map is an interspecific map and was obtained with a limited number of individuals (88) resulting in a reduced recombination frequency in some regions [49, 50], providing only a rough estimation of the recombination frequency at a short physical distance. The anchoring markers (i.e. those having sequence information associated) are unevenly distributed along the linkage groups leaving portions of the genome uncovered. Later analyses of the peach genome revealed that 4% of the sequence of the Peach v1.0 (with ten major scaffolds larger than 300 kb) was not included in the pseudomolecules; with ten scaffolds (7% of the total sequence) anchored with unknown orientation and a large number of mapped scaffolds had no markers on their ends (>500 kb), making it difficult to detect putative chimerism [20].
In this work, we describe the efforts aimed at improving the peach chromosome-scale build (Peach v1.0 [20]) using a set of linkage maps and resequencing the reference accession (‘Lovell’ double haploid). Two mapping strategies were used: i) a targeted approach where markers (Simple Sequence Repeats, SSRs, and SNPs) were targeted in specific regions of the peach genome (i.e. in map gaps, unmapped, not oriented scaffolds, and uncovered scaffold ends) and mapped in two already available linkage maps [45, 51]; ii) a whole genome approach that made use of the IPSC 9 K SNP array v1 [31] to genotype a large set of progeny [50, 52, 53]. Moreover, Illumina NGS resequencing of the reference accession at high coverage was performed to correct sequencing errors (false SNPs and indels) and to close a number of gaps in the Peach v1.0 assembly increasing the contiguity of the final peach genome.
Methods
Plant material, DNA extraction and quality test
Four biparental mapping populations were used to refine the peach genome (Peach v1.0):
-
i)
67 seedlings of the ‘Texas’ x ‘Earligold’ F2 population (TxE [46]), an interspecific cross between almond and peach, maintained at the experimental station of CREA-FRU in Rome, Italy (latitude: from 41°47'43.72"N to 41°47'46.75"N; longitude: from 12°33'48.78"E to12°33'52.58"E);
-
ii)
242 seedlings of the peach selection IF7310828 x Ferganensis BC1 population (PxF [52]) maintained at the experimental station of CREA-FRU;
-
iii)
305 seedlings of the ‘Contender’ x ‘Ambra’ F2 population (CxA [51]) maintained in a farm belonging to the Municipality of Castel San Pietro (Bologna, Emilia Romagna, Italy) leased to ASTRA (latitude: from 44u24944.180 N to: 44u24930.080 N; longitude: from 11u35947.210E, to: 11u3692.000E);
-
iv)
62 seedlings of the Maria Dolce x SD81 F1 cross (MDxSD) maintained at the experimental station of CREA-FRU.
Young leaves were collected from each seedling and lyophilized. DNA was extracted with the DNeasy Plant Mini Kit (QIAGEN), quantified with the NanoDrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) and with the PicoGreen® Assay (Thermo Fisher Scientific) for samples genotyped on the IPSC 9 K SNP array. For Sequenom analysis, DNA was extracted from the seedlings of the CxA progeny after Mercado et al. [54].
Target SSRs identification and mapping in TxE progeny
Target SSRs were selected from among the 63,145 identified within the Peach v1.0 genome sequence [55] to increase marker density in order to leverage in large unanchored scaffolds, and correct order/orientation of anchored scaffolds. Sequences of ~600 bp flanking both sides of the repeated motif were first blasted against Peach v1.0, using the BLAST facility available on Phytozome [56, 57], and only non-repetitive regions were selected to design specific primer pairs by the Primer 3 software [58]. Only single locus SSR markers were used for further analysis. The selected SSR primers were first tested on the BIN set and parents of the TxE progeny. Only the most physically distant polymorphic markers in each target region were genotyped on the whole TxE progeny. To check for scaffold integrity, some SSRs were developed in the distal region of uncovered scaffold ends. In the presence of suspected chimeric regions, further markers spaced approximately every 100 kb were identified, developed, and mapped to restrict the size of misassembly containing regions. Primer sequences and features are listed in Additional file 1: Table S1.
All PCR reactions were carried out in a 10 μL volume with a final concentration of 1x PCR buffer, 1.5 mM MgCl2, 200 μM of each dNTP and 0.1 μM of each primer, 10 ng genomic DNA, and 0.5U of Platinum®Taq DNA Polymerase (Invitrogen TermoFisher). The amplification profile was: one cycle at 94 °C for 5 min, followed by 10 touchdown cycles, with a decrement of the annealing step temperature of 0.5 °C/cycle, starting with a denaturation at 94 °C for 30 s, an annealing step five degrees above the primer-specific annealing temperature (Ta °C, reported in Additional file 1: Table S1) for 30 s, and an elongation at 72 °C for 30 s, followed by 25 cycles at 94 °C for 30 s, Ta °C for 30 s, 72 °C for 30 s, and a final elongation of 30 min at 72 °C. PCR products were then separated on a 3% high-resolution agarose gel (MetaPhor™Agarose, Lonza) in TBE 1x with a voltage of ∼ 5 V/cm and stained with the GelRed™ (Biotium). Markers that could not be easily scored on agarose gel were separated by capillary electrophoresis on a CEQ 8000 Genetic Analyzer (Beckmann Coulter).
SSRs genotyped in the whole TxE progeny were integrated with the previous TxE dataset [45] and mapped using the software MAPMAKER [59], grouping them at a LOD score higher than 5. They were located by using the TRY and RIPPLE commands. After mapping, the ERROR DETECTION command of MAPMAKER was used and putative double recombinants were manually checked.
SNP identification and mapping in the CxA progeny
The F1 parent of the CxA progeny was resequenced with an Illumina platform (Project SRA0532230, Accession # SRX150230 [20]). The CLC Genomics Workbench 5.5 (CLC Bio, Aarhus, Denmark) was used for read alignment and SNP calling. Only reads aligning to a single location with at least 92% identity over at least 90% of their length were considered. For SNP calling, variants were retained when: i) the coverage ranged between 0.5 and 2 X of the average coverage (computed excluding zero coverage regions); ii) minor allele frequency > 30%; iii) the polymorphic nucleotide Phred-scaled quality score ≥ 20 and the average quality ≥ 15 for the 11 bp surrounding the putative SNP.
SNPs were manually selected based on their distribution on the peach genome. SNPs surrounded by repetitive sequences and/or located within a stretch of bases identical to that of the SNP itself (i.e. a short stretch of A in an A/G SNP) were avoided. The surrounding sequences (about 150 bp/side) were obtained from the peach Gbrowse available on the IGA website [60] and blasted against the v1.0 peach genome at GDR [61] to verify their uniqueness. Only unique SNP-surrounding sequences were used to design the assays (a locus specific primer pair and a single-base extension primer or probe for each SNP), and combine them in multiplex reactions (hereafter called iPlex) by the software Mass ARRAY Design 3.1.
All locus-specific PCR primers and probes were blasted against the peach genome to further verify their specificity. Only the SNPs that passed all these quality checks were retained for further analyses.
A total of ten iPlex were designed to attain the optimal genome coverage, including two iPlex (steps 9 and 10) with SNPs selected in specific uncovered regions, or where SNPs previously tested were not useful. Information about the primers used for the genotyping is listed in Additional file 1: Table S2.
Genotyping in the CxA progeny was performed using iPLEX Gold technology [62] and Mass ARRAY high-throughput DNA analysis mass spectrometry (Sequenom, Inc) at the Centre for Applied Biomedical Research (CRBA) of Bologna.
The Sequenom data for each SNP were first verified by checking the heterozygosity of the CxA F1 parent and its consistency with the two grandparents ‘Contender’ and ‘Ambra’. All the SNP data were integrated with the dataset of 31 SSR markers genotyped following Eduardo et al. [51], and then analyzed by JoinMap 3.0 software [63] with the default parameters and the Kosambi [64] mapping function. Linkage groups were established at LOD value (independent LOD score) higher than 10.
All these SNPs were also tested on the BIN set of the TxE mapping progeny.
IPSC 9 K SNP array genotyping and mapping
The IPSC 9 K SNP array [31] was used to genotype 242 individuals of the PxF and 62 of the MDxSD progenies, using the Illumina Infinium II design probes, and the dual color channel assay (Infinium HD Assay Ultra, Illumina). SNP genotypes were scored with the Genotyping Module of the Genome Studio Data Analysis software (Illumina, Inc.). SNPs with a GeneTrain score ≥0.4 and less than 10% missing data were retained. Allele segregation was also checked and SNPs showing unexpected segregations (as, for example, with parents homozygous for the same allele) or unexpected genotype classes (with respect to the parental genotypes), were inspected using Genome Studio. If possible they were re-clustered using the “define cluster” function. Those still showing missing or unexpected classes were discarded.
Linkage analysis and map construction were performed with JoinMap 4.1 [65] using the CP and BC1 population types for PxF and MDxSD, respectively. Linkage groups were established at LOD value (independent LOD score) higher than 10 as described above. The Multipoint Maximum Likelihood mapping algorithm was used with the default parameters. Kosambi units [64] were used; for the CP population type, Haldane map distances were manually converted in Kosambi units using the formula provided in JoinMap 4.1 manual.
Map-sequence integration
The original raw version of the Peach v1.0 assembly, post filtering organelle, repetitive, and small scaffolds (< 1 kb) sequences, was used for the new map-sequence integration. Markers were placed on the WGS scaffolds using two methods as reported in Verde et al. [20]. SSR and SNP markers having primer sequences (i.e. the MASSARRAY developed markers) were placed using three successive rounds of electronic PCR (e-PCR [66]) with N = 0, N = 1 and N = 3. Markers with a known sequence, including RFLP (Restriction Fragment Length Polymorphism) and SNP markers or SSRs whose primers had not been found with the three rounds of ePCR, were placed with BLASTN. The additional breaks were made in regions of low BAC/Fosmid coverage and the broken sequences reordered according to the new maps. The mapped WGS scaffolds were joined as described in Verde et al [20] to form 8 pseudomolecules (Pp01 to Pp08). Each map join is denoted by 10.000 N bps.
In this work, to avoid confusion between v1.0 and v2.0 releases, scaffolds composing the Peach v1.0 assembly were named and are hereafter referred to as “Scaffold_##”. The 40 WGS scaffolds included in the 8 v1.0 pseudomolecules were named as “Scf_##”. The WGS scaffolds composing the v2.0 pseudomolecules (Pp01 to Pp08) were named as “Super_##”.
Lovell DH resequencing
Resequencing of the ‘Lovell’ double haploid (PLov2-2n) was performed using the MiSeq Illumina platform. Paired-end reads (43x 2x250 bp, 600 bp insert size and 21x 2x250 3 kb and 6 kb insert size Additional file 1: Table S3) were assembled with the AbySS software [67] after quality checking and filtering. The resulting contigs were used to patch gaps in the Peach v1.0 assembly after the new breaks and joins described above were applied (hereafter referred to as “modified v1.0 assembly”). Contigs were aligned to the repeat masked modified v1.0 assembly using BLAT [68]. Contigs whose ends aligned to either side of a gap, with at least 1/3 of the contig length anchoring to the edges of the gap at ≥95% identity, were used to patch the gap. Sequence and quality scores were then integrated into the v1.0 modified assembly. Finally, homozygous SNPs and indels were corrected using ~43x Illumina reads. Reads were aligned using BWA [69] and variants (SNPs and indels) called using the standard GATK pipeline [70] including base quality score recalibration, indel realignment, and duplicate removal.
Physical vs genetic distance comparison and identification of centromeric regions
MareyMaps were obtained, for each mapping progeny, by plotting the genetic positions of molecular markers (in centimorgans, cM) against their physical position on the Peach v2.0 (in Megabase pairs, Mb) [71]. Cumulative recombination curves for each chromosome were estimated using the cubic spline interpolation method with default parameters and the cross-validation type present in the MareyMap package. The recombination value per position was obtained calculating the slope per markers and their curves were plotted for each chromosome.
In order to identify the putative centromeric region of each chromosome, regions displaying the lowest recombination rate, as highlighted by the MareyMaps, were manually checked on the Peach v2.0 using the JBrowse available on Phytozome [57] for the absence of transcripts and the abundance of repeated elements associated with centromeric and pericentromeric regions. Sequences retrieved from Neumann et al. [72], representing a catalog of plant repeated elements associated with centromeric regions, were aligned with BLASTN [57] to the peach genome assembly. Sequences aligning within the putative peach centromeric regions (1 Mb of sequence around the predicted centromere), with at least 63% of identity and an e-value greater than 4 x 10−16, were retained.
Recombination frequency was compared among all progenies by multiple comparison statistics implemented in PAST 2.12 [73]. For each linkage map used in this study (TxE, CxA, and PxF), recombination rate was estimated at individual whole-chromosome scale as the ratio between genetic (cM) and physical (Mb) distances. One-way analysis of variance (ANOVA) was applied with Tukey’s pairwise post-hoc test. The Levene’s test for homoscedasticity and the Shapiro-Wilk test for normal distribution were also applied to check the assumptions for the applicability of the ANOVA. In the case of violation, the non-parametric Kruskal-Wallis test was applied instead, with the Mann-Whitney pairwise post-hoc comparison and the Bonferroni correction.
Results and Discussion
Four linkage maps were used to improve the peach genome. TxE and CxA were already available [45, 51] and were enriched using a targeted approach. Another map was the result of a de-novo mapping of the PxF progeny [52] obtained using the IPSC 9 K SNP array [31] and tripling the mapping progeny size. Finally, the MDxSD map, also obtained with the IPSC 9 K SNP array, was used to specifically address some inconsistencies at the top of linkage group 6 (LG6).
Targeted SSR identification and mapping in TxE
A total of 111 SSRs (RPPG set) were identified in the Peach v1.0 assembly and primers were designed and then tested in the TxE BIN set (Additional file 1: Table S1). Twenty markers were developed on the major unmapped scaffolds, 14 were individuated within the randomly oriented scaffolds and 77 were identified within the 24 uncovered scaffold ends. Thirty-eight out of the total were monomorphic in TxE and 73 were BIN mapped (65.8% of polymorphism; Additional file 1: Table S1). Out of 56 microsatellites falling within genic regions, 40 (71.4%) were polymorphic. Three polymorphic markers (RPPG14-003, RPPG16-002, RPPG5-005) were mapped in TxE, and found in successive analysis to have their primer pair on the same flanking side of the microsatellite region. These three markers were retained and reclassified as indel markers. These incidental length polymorphisms in the TxE interspecific progeny reflect the different genomic structure of the closely related almond and peach species. The same length variation was observed in six out of the seven Knox genes whose fragment size indicated differences between the two parents ranging from 2 bp to 20 bp in length [74]. A much higher level of polymorphism (89.2%) with RFLPs in the TxE progeny had been already observed (Dettori, unpublished results) in comparison to that (28.4%) of the intraspecific PxF progeny [52]. Thirty-two well-spaced SSRs, out of the 73 polymorphic ones, were mapped in the whole TxE progeny to resolve orientation, misassembly, or ordering discrepancies.
In the preliminary steps of the peach genome assembly 54 SSRs [47, 48, 75] targeting specific regions had been mapped in the whole progeny (Additional file 1: Table S4). In addition, seven markers (SNPs and indels) targeting the peach KNOX genes [74] had also been included. The final map (Additional file 2: Figure S1, Additional file 1: Table S4) is composed of 655 markers and covers 511.3 cM. Three hundred and twenty-nine markers, having sequence information associated, were used for the Peach v2.0 map-sequence integration. They cover 472.7 cM (92.3% of the total genetic distance in TxE) and 220 Mb (97.5% of the v2.0 pseudomolecule length, Table 1). Only four gaps larger than 10 cM are present. In addition, 449 ROSCos BIN mapped markers [76] and four SNPs from the Prunus-Malus consensus sequence [77] were included in this study and integrated with the WGS scaffolds. Furthermore, 53 SNPs from the CxA F1 parent and 41 SSRs isolated in this study (RPPG set) were also BIN mapped. In total 1,224 TxE markers were integrated within the assembly, 895 BIN mapped (348 used in Peach v1.0 and 547 added in this study), and 329 mapped in the whole progeny (Table 1, Additional file 1: Table S4).
Table 1.
Anchoring statistics of the Peach v2.0 assembly
| Chromosome (LG) Pseudomolecule | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mapping progeny | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | Total | |
| Number of markers integrateda | TxE | 256 (186) | 143 (101) | 155 (113) | 120 (83) | 123 (90) | 159 (121) | 143 (107) | 125 (94) | 1,224 (895) |
| PxF | 269 | 207 | 292 | 408 | 153 | 219 | 224 | 202 | 1,974 | |
| CxA | 29 | 24 | 36 | 42 | 12 | 29 | 25 | 28 | 225 | |
| Total | 554 | 374 | 483 | 570 | 288 | 407 | 392 | 355 | 3,423 | |
| Number of scaffolds anchored | TxE | 10 | 9 | 10 | 7 | 5 | 8 | 4 | 4 | 57 |
| PxF | 9 | 8 | 8 | 7 | 5 | 7 | 4 | 3 | 51 | |
| CxA | 9 | 8 | 9 | 6 | 3 | 9 | 4 | 4 | 52 | |
| Total | 10 | 9 | 10 | 7 | 5 | 9 | 4 | 4 | 58 | |
| Genetic distances covered (cM) | TxE | 77.5 | 42.7 | 44.1 | 51.7 | 47.6 | 81 | 70.6 | 57.5 | 472.7 |
| PxF | 117.4 | 70.9 | 69.9 | 69.3 | 62.1 | 81.1 | 67.3 | 67.6 | 605.6 | |
| PxF F1 | 139.8 | 88.1 | 71.9 | 76.7 | 59.1 | 91 | 63.6 | 68.4 | 658.6 | |
| PxF recurrent | 53.7 | 31 | -- | 44.2 | 27.6 | 45.9 | 19.6 | 10.4 | 232.4 | |
| CxA | 98 | 36.3 | 67.7 | 64.5 | 36.5 | 79.9 | 62.3 | 64.4 | 509.6 | |
| Physical distance in bp covered with the integrated markers and (%) of Peach v2.0 pseudomolecules | TxE | 47,190,243 (98.6) | 29,794,491 (98.0) | 27,174,422 (99.3) | 24,974,520 (96.6) | 17,300,580 (93.5) | 30,384,999 (98.8) | 21,009,142 (93.8) | 22,199,033 (98.3) | 220,027,430 (97.5) |
| PxF | 46,854,330 (97.9) | 29,975,524 (98.6) | 26,162,111 (95.6) | 25,167,755 (97.4) | 17,989,526 (97.3) | 29,985,579 (97.5) | 22,201,468 (99.2) | 20,421,932 (90.5) | 218,758,225 (96.9) | |
| PxF F1 | 46,854,330 (97.9) | 29,652,167 (97.5) | 26,162,111 (95.6) | 25,167,755 (97.4) | 17,989,526 (97.3) | 29,209,364 (94.9) | 20,322,548 (90.8) | 20,087,434 (89.0) | 215,445,235 (95.5) | |
| PxF recurrent | 39,665,700 (82.9) | 22,115,897 (72.7) | -- | 19,074,112 (73.8) | 5,684,854 (30.7) | 26,868,520 (87.3) | 3,306,229 (14.8) | 3,031,158 (13.4) | 119,746,470 (53.1) | |
| CxA | 45,955,086 (96) | 19,656,032 (64.6) | 27,246,203 (99.6) | 25,053,083 (96.9) | 7,348,324 (39.7) | 29,848,481 (97.0) | 21,600,273 (96.5) | 22,073,557 (97.8) | 198,781,039 (88.1) | |
| Total No of bases anchored (bp) | 47,851,208 | 30,405,870 | 27,368,013 | 25,843,236 | 18,496,696 | 30,767,194 | 22,388,614 | 22,573,980 | 225,694,811 | |
For each map and for each chromosome the number of markers, number of anchored scaffolds, genetic and physical distance covered with the integrated markers and the total number of anchored bases are reported
aIn brackets the BIN mapped markers in TxE
Targeted SNP identification and mapping in CxA
From the resequencing of the CxA F1 parent, 265 SNPs have been developed and included in a total of ten iPLEX assays, with an average number of 26.5 SNP each iPlex. Among the tested SNPs, 194 were useful for mapping while 71 were not: of these, 49 were monomorphic (38 showing only one allele and 11 being heterozygous in the whole progeny), 12 showed only two over three expected genotypes and ten presented more than 25% of missing data. The latter group also included five markers with no amplification in the whole progeny, possibly due to assay failure.
The fraction of scorable polymorphic SNPs ranged from 52.2% to 86.2% in different iPLEX assays, with an average success rate of 73.2% (Additional file 1: Table S5). The relatively negative result on iPLex 9 and 10 is likely due to forcing the iPlex design to develop markers in target regions.
The observed SNP calling efficiency was evaluated by comparing our results with those obtained by Verde et al. [20], which used more stringent parameters for SNP calling. This a posteriori analysis showed that 40.8% of the non-polymorphic SNPs were false positives (29 markers out of 71) but at the same time five true SNPs over the 194 mapped ones (7%) would have been lost using the more stringent conditions (false negatives). Finally, with the more stringent parameters, the total efficiency of SNP design would have increased from 73.2% to 80.1% (Additional file 1: Table S5).
The CxA map was first obtained with 31 SSRs on 169 F2 progeny [51]. To improve the chromosome-scale assembly the number of progeny was increased to 305. The additional individuals were genotyped with SSR markers, adding 194 targeted polymorphic SNPs to the map. Fifteen of these SNPs were already linked on LG4 to a candidate gene controlling maturity date in peach [78]. Another set of twelve SNPs on LG5 surrounding the nectarine G locus had been described [79]. A total of 20 SNPs were identified on unmapped scaffolds, ten on the randomly oriented scaffolds and 15 in putative chimeric regions. Fifty-three of these SNPs were also BIN mapped in TxE, as already described in the previous paragraph. The final CxA map (Additional file 2: Figure S1; Additional file 1: Table S6) includes 225 markers (SSRs and SNPs) corresponding to 211 unique genetic positions, covering 509.6 cM with only four gaps larger than 10 cM. All the pseudomolecules are almost completely covered (198.8 Mb, 88.1% of the v2.0 pseudomolecule length, Table 1), with the exception of Pp02 lacking the bottom portion (about 10 Mb, ~35% of the total length) and Pp05 missing the upper portion (about 11 Mb, ~60% of the total length).
IPSC 9 K SNP array mapping in PxF
The 242 trees of the PxF progeny were genotyped with the IPSC 9 K SNP array [31]. Out of the 8,144 placed on the array, a total of 3,399 polymorphic SNPs (41.7%) were identified; 1,669 SNPs were informative for the F1 parent (segregating in a 1:1 ratio), 641 were informative for both parents (1:2:1 ratio) and 1,089 were informative for the recurrent parent (1:1 ratio). Of the three types of segregation, only the first two were used for the map-sequence integration because the unequal recombination frequencies between the two parents of the cross inhibited efficient joining of their genetic information. In fact, the recurrent parent (IF7310828) displays a marked reduction in recombination frequency in comparison to that of the F1 parent (1.941 vs 3.057 cM/Mb, on average, Table 2, Additional file 1: Tables S7, S8, S9). As a consequence, the map-sequence integration was unreliable due to the heavily skewed order of markers in regions with strong differences in recombination frequencies between the two parents. Moreover, the distribution of the informative SNPs for the recurrent parent was uneven across the genome resulting in a fragmented linkage map with 12 groups. In fact, five groups were split in two groups each (LG2, 5, 6, 7 and 8), one was completely missing (LG3) resulting in a reduced genome coverage (119.7 Mb, 53.1% of the Peach v2.0 pseudomolecule length, Table 1, Additional file 1: Table S9). The inspection of the IPSC 9 K SNP array markers segregating in the recurrent parent highlighted that they did not give additional information for integration except for two contiguous minor scaffolds (Super_23 and Super_456, 1.5 Mb in total) found in the upper part of the Pp01. Seven markers located in these two scaffolds informative for the recurrent parent, were selected and integrated into the map. The presence of bridge markers in that region (about 10 Mb) segregating in a 1:2:1 ratio and the low number of 1:1 markers informative for the F1 parent (5 out of 52) enabled a reliable integration. When a single SNP was mapped in a linkage group that differed from the expected placement from the array information, and no further evidence of misassembly was observed in the same genetic region (i.e. SNP not located at the scaffold terminals), this marker was deemed as putatively duplicated (from putative paralogous genes) and excluded.
Table 2.
Genetic/physical ratio (cM/Mb) for each map and each chromosome
| LG-Ppa | TxE | PxF | PxF F1 | PxF recurrent | CxA | WxB | DvsS |
|---|---|---|---|---|---|---|---|
| 1 | 1.642 | 2.506 | 2.984 | 1.354 | 2.133 | 2.261 | 1.858 |
| 2 | 1.433 | 2.365 | 2.971 | 1.402 | 1.847 | 2.141 | 1.435 |
| 3 | 1.623 | 2.672 | 2.748 | -- | 2.485 | 2.340 | 2.174 |
| 4 | 2.070 | 2.754 | 3.048 | 2.317 | 2.575 | 2.544 | 2.384 |
| 5 | 2.751 | 3.452 | 3.285 | 4.855 | 4.967 | 3.777 | 2.672 |
| 6 | 2.666 | 2.705 | 3.115 | 1.708 | 2.677 | 2.393 | 1.770 |
| 7 | 3.360 | 3.031 | 3.130 | 5.928 | 2.884 | 3.003 | 1.930 |
| 8 | 2.590 | 3.310 | 3.405 | 3.431 | 2.918 | 2.775 | 1.812 |
| Total | 2.148 | 2.768 | 3.057 | 1.941 | 2.564 | 2.553 | 1.954 |
a LG Linkage Group, Pp Pseudomolecule
After filtering, we mapped 1,974 SNPs in total (1,566 segregating in a 1:1 ratio informative for the F1 parent, 401 segregating in a 1:2:1 ratio and 7 informative for the recurrent parent) corresponding to 567 unique genetic loci. The map (Additional file 2: Figure S1, Additional file 1: Table S7) covers 605.6 cM (corresponding to 218.8 Mb, 96.9% of the v2.0 pseudomolecule length, Table 1) with only one gap larger than 10 cM.
Integration of unmapped scaffolds in Peach v2.0 pseudomolecules
One hundred and ninety-four scaffolds (8.7 Mb in total, 4% of the total assembly size), had not been included in the eight peach v1.0 pseudomolecules. To anchor the larger unmapped scaffolds (> 300 kb) in Peach v1.0, we first used the TxE map. Twenty SSRs (Additional file 1: Table S1) were targeted in the unmapped portion of the peach genome and 17 polymorphic ones (11 fully mapped) enabled anchoring of the ten major scaffolds (v1.0 Scaffold_9 to Scaffold_18) and fixing the orientation for two of them (v1.0 Scaffold_9 and Scaffold_10, 2.1 Mb and 851 kb, respectively; Additional file 2: Figure S1, Additional file 1: Tables S3 and S9). The same was done using the CxA map (Additional file 1: Table S2). This map (Additional file 2: Figure S1, Additional file 1: Table S6), in addition to being from an intraspecific cross, was obtained with a large mapping progeny (305 plants) providing a fine estimation of the recombination frequencies even at a small scale (about 100 kb). With this approach, we were able to confirm the anchoring of the ten major scaffolds (>300 Kb) and anchoring of an extra minor scaffold (v1.0 Scaffold_36, 23 kb in size). The high resolution of the CxA map also enabled the ordering of two contiguous small scaffolds (v1.0 Scaffold_12 and Scaffold_16) on pseudomolecule 2, that in TxE were unordered and unoriented, and fixing the orientation of five scaffolds (v1.0 Scaffold_9 Scaffold_10, Scaffold_11, Scaffold 12 and Scaffold_15, 4.9 Mb in total). Only 19 markers of the IPSC 9 K SNP array were located in the unmapped portion of the genome and six of them were polymorphic in PxF. The position of five of the ten major scaffolds previously mentioned (v1.0 Scaffold_10, Scaffold_12 Scaffold_13, Scaffold_14, and Scaffold_17) was confirmed using these markers.
Together, these analyses positioned 11 unmapped scaffolds on the v2.0 pseudomolecules (Additional file 1: Table S10). They cover 7.2 Mb of sequence (3.2% of the total assembly); five of them were also orientable (4.9 Mb, 2.2% of the total assembly; Additional file 1: Table S10).
Ordering and orientation of Peach v1.0 randomly oriented sequences
Ten mapped scaffolds (15.8 Mb) of the Peach v1.0 genome were placed with random orientation due to the lack of recombination among markers or because they were anchored with only one marker. To resolve the orientation of these scaffolds, previously BIN mapped markers, located by the ends of the non-oriented scaffolds were mapped in the whole TxE progeny. For scaffolds where no BIN mapped markers were available, 14 SSR primer pairs (Additional file 1: Table S1) were designed towards the end, tested on the TxE BIN set and the most physically distant polymorphic markers (8 SSRs) were mapped in the whole progeny. In this way, five integrated scaffolds (v2.0 Super_23, Super_25, and Super_10 on Pp01, Super_ 20 on Pp02 and Super_19 on Pp05, Additional file 2: Figure S1, Additional file 1: Tables S3 and S9) representing 10.4 Mb of sequence were oriented along the pseudomolecules. Moreover, the attempt to orient a scaffold on Pp02 (v2.0 Super_20) revealed that it was incorrectly placed along the pseudomolecule due to a mismapped marker (CPDCT044) in TxE. This scaffold was correctly positioned at the top of the Pp02. The higher genetic resolution of CxA map (Additional file 2: Figure S1, Additional file 1: Tables S5 and S9) confirmed the position of Super_20 at the top of Pp02 and the correct orientation of a major scaffold at the bottom of Pp011 (v2.0 Super_10). It also rectified the orientation of two wrongly oriented scaffolds due to mapping artifacts in TxE: one on top of Pp07 (v2.0 Super_11, 4.8 Mb) and the other in the middle portion of Pp08 (v2.0 Super_15, 2.9 Mb). The high density and resolution of the PxF map further confirmed the orientation of the above-mentioned scaffolds and enabled the correct orientation of three other scaffolds in Peach v1.0 (v2.0 Super_451 on Pp03 and Super_26 and Super_29 on Pp04; 2.9 Mb). In a region of Pp03 (12–17.6 Mb), indicating high recombination frequency suppression, five scaffolds (v2.0 Super_451, Super_18, Super_27, Super_31, Super_32) were ordered with low probability in TxE. The higher resolution of CxA and PxF helped to resolve uncertainties (i.e. the position of Super_18 embedded between Super_31/Super_32 and Super_451/Super_27). However, the orientation of Super_31 and Super_32 was indeterminate in v2.0 since the corresponding markers cosegregated in all maps. Moreover, it was not possible to have certainty of the order of Super_451 and Super_27 located in the same region (Pp3, 15.3–17.6 Mb). In fact, markers on those scaffolds were ordered in TxE with low probability (alternative positions to the accepted one were only slightly less likely, with a difference in log-likelihood of 0.37, i.e. 2.34 folds less likely). In CxA and in PxF only one of these two scaffolds was anchored in each map (Super_451 in PxF and Super_27 in CxA) giving no additional information on their order. For these scaffolds, the Peach v1.0 order, based on TxE, was retained. However, recently published maps [80, 81] obtained using the IPSC 9 K SNP array [31] have enabled verification of their order and orientation. In particular in the MxR_01 map [80] two SNPs (SNP_IGA_336437 and SNP_IGA_339719) mapping on Super_27 at 39.5 and 41.1 cM, respectively (Peach v2.0 position at 17,026,649 and 17,569,078 nt, respectively) and other four (SNP_IGA_326457, SNP_IGA_328528, SNP_IGA_331373 and SNP_IGA_333074) mapping on Super_451 at 42.8, 45.9, 49.2 and 50.7 cM, respectively (Peach v2.0 position 15,586,851, 15,899,181, 16,311,538 and 16,634,203 nt, respectively) revealed that the order established in Peach v2.0 is incorrect and will be inverted in a future release. This is also confirmed by the DvsS map [81] in which two SNPs (SNP_IGA_338615 at 17,411,354 nt on Super_27 and SNP_IGA_325296 at 15,442,995 nt on Super_451) were mapped at 31 and 32 cM, respectively. The map obtained by Sánchez et al. [80] confirmed the orientation of Super_451 established only by PxF map and v2.0 orientation of Super_27 was shown to be correct, though it was anchored by only one marker in TxE and CxA.
Together, all the ten scaffolds randomly oriented in Peach v1.0 and two wrongly placed (summing up 23.6 Mb of sequence, 10.4% of the Peach v2.0 total length) were correctly oriented in Peach v2.0 pseudomolecules. Currently, only six minor scaffolds (Super_31, Super_32, Super_34, Super_36, Super 35, and Super_54, Additional file 1: Table S10) summing up 2.4 Mb are randomly oriented in this release. Moreover, only a known mis-order (Super_27 and Super_451) and a random order (Super_31 and Super_32) in the central part of chromosome 3 are still present in Peach v2.0 (Additional file 1: Table S10). These will be rectified in a future release of the peach genome.
Scaffold ends checking and correction of misassembled sequences
Within the 40 scaffolds composing the Peach v1.0 pseudomolecules, 24 terminals were not covered with molecular markers for at least 500 kb of their length, 13 having an uncovered portion larger than 1 Mb with the largest one being of 3.1 Mb. These regions are potential sites of misassembled sequence. To check scaffold consistency, SSR and SNP markers were developed in the distal part of these uncovered regions. Markers mapped in TxE and CxA maps (48 SSRs and 15 SNPs, respectively) helped to reveal five out of these 24 uncovered scaffold ends as sites of misassembly: three in pseudomolecule 4, one in pseudomolecule 3 and one in pseudomolecule 7. On pseudomolecule 4, there were two scaffolds bearing two different chimeric regions, resulting in six pieces in total that needed to be relocated on different chromosomes. For this purpose, the two most distant polymorphic markers in each chimeric region were mapped in the whole TxE progeny in order to locate and orient the new broken scaffolds within the peach pseudomolecules. A particular case occurred in the v1.0 integrated scaffolds Scf_450 and Scf_451 located on pseudomolecule 3 and pseudomolecule 7, respectively. They originated from a chimeric scaffold that had been broken in a wrong position in the Peach v1.0 assembly due to insufficient marker coverage within the putative chimeric region (about 1.2 Mb gap). In v2.0, with the help of the new mapping data, we refined the breakpoint, so that 385 kb from v1.0 Scf_450 in pseudomolecule 3 were re-joined to the formerly broken Scf_451 to form the new Super_452 on v2.0 Pp07 (Additional file 1: Table S10).
The whole genome mapping approach of the PxF map confirmed all of the chimeric scaffolds and enabled identification of two other cases of misassembly on the top of v1.0 pseudomolecule 6. In this region two scaffolds (Scf_26 and Scf_457, 14.8 Mb total sequence, Additional file 1: Table S10) were chimeric and needed to be broken in one point each. Three of the broken portions, 4.6 Mb of sequence (Super_447, Super_464 and Super_446, 602 kb, 3.3 Mb and 709 kb, respectively), had to be rearranged within the same chromosomal region (Additional file 2: Figure S1, Additional file 1: Tables S6, S7, S8, S9). To support the rearrangements at the top of v1.0 pseudomolecule 6, we used additional information from the MDxSD map. In this map, LG6 is composed of 153 SNP markers for a total of 27 single genetic positions covering a genetic distance of 54.9 cM with an average of 2.03 cM between markers and a major gap of 6.5 cM (Additional file 2: Figure S1, Additional file 1: Table S11). The total physical distance covered amounts to 29,6 Mb (96.1% of Pp06 length). Twenty-one MDxSD markers mapped in the 4,6 Mb region (spanning 9.7 cM) confirm the order of the three broken scaffolds highlighted by PxF and the orientation of two of them (Super_447 and Super_464). Without high resolution and high-density linkage maps highlighting discrepancies in this 4.6 Mb region (41 markers in PxF and 21 in MDxSD), this problem could not have been solved since the TxE and CxA maps have low marker density in that region. Scaffold ends were also checked using additional information from recently published linkage maps [80–82]. In the current assembly, only 3 scaffold ends larger than 500 kb are still not covered with markers, all of them lying in highly repeated centromeric regions, with the largest being a 594 kb stretch of sequence (Additional file 1: Table S10).
In total, 10.4 Mb (4.6% of the total assembly) of sequence from Peach v1.0, were relocated in their correct chromosomal positions with the correct orientation.
Base accuracy and contiguity improvement through reference accession resequencing
The ‘Lovell’ double haploid (PLov2-2n) was resequenced from 5 libraries (Additional file 1: Table S3), producing 73,881,213 paired-end reads, corresponding to 43-fold paired end fragment sequence coverage (2x250, 600 bp insert size) and 21-fold mate pair coverage (2x250, 3 kb and 6 kb insert size). This set of reads was assembled using ABySS (v1.3.6) [67], producing 30,131 scaffolds greater than 500 bp for a total of 180.7 Mb of sequence. Scaffolds produced in the ABySS assembly were broken into contigs, and a total of 206 contigs representing 3.2 Mb of sequence were used to close 212 Peach v1.0 gaps, with a gain of 25,199 kb (Table 4). The overall contiguity was improved, with the total number of contigs in Peach v2.0 decreased by 7.5% dropping from 2,730 to 2,525 and the contig L50 increased by 19.2% (from 214.2 kb to 255.4 Kb). Finally, 859 homozygous SNPs and 1,347 indels were corrected using ~43x paired end fragment Illumina reads (Table 3).
Table 4.
Summary statistics of the Peach v2.0 chromosome-scale assembly statistics and its comparison with the v1.0
| Peach v2.0 | Peach v1.0 | |
|---|---|---|
| Number of scaffolds | 191 | 202 |
| Number of contigs | 2,525 | 2,730 |
| Scaffold sequence | 227.4 Mb | 227.3 Mb |
| Mapped scaffold sequence | 225.7 Mb (99.2%) | 218.4 Mb (96%) |
| Oriented scaffold seqeuence | 223.3 Mb (98.2%) | 194.6 Mb (85.6%) |
| Contig sequence | 224.6 Mb | 224.6 Mb |
| Scaffold N/L50 | 4/27.4 Mb | 4/26.8 Mb |
| Contig N/L50 | 250/255.4 kb | 294/214.2 kb |
| Number of scaffolds > 50 KB | 11 | 21 |
| % main genome in scaffolds > 50 kb | 99.4% | 99.4% |
Table 3.
Summary of gap patching and indel and SNP correction
| Pseudomolecules | No. of contigs | No. of gaps closed | Gap bases patched | Initial contig length | Post gap-patching contig length | Bases gained | Indels corrected | SNP corrected |
|---|---|---|---|---|---|---|---|---|
| Pp01 | 36 | 36 | 6,820 | 47,412,656 | 47,417,444 | 4,788 | 269 | 143 |
| Pp02 | 27 | 27 | 6,884 | 29,982,897 | 29,985,295 | 2,398 | 185 | 117 |
| Pp03 | 29 | 30 | 4,831 | 27,022,361 | 27,022,947 | 586 | 167 | 128 |
| Pp04 | 24 | 24 | 3,895 | 25,545,546 | 25,549,276 | 3,730 | 133 | 132 |
| Pp05 | 19 | 21 | 10,824 | 18,291,031 | 18,295,669 | 4,638 | 110 | 58 |
| Pp06 | 25 | 26 | 9,635 | 30,419,305 | 30,423,361 | 4,056 | 197 | 117 |
| Pp07 | 29 | 30 | 7,501 | 22,049,797 | 22,053,146 | 3,349 | 157 | 75 |
| Pp08 | 17 | 18 | 3,485 | 22,391,144 | 22,392,798 | 1,654 | 129 | 89 |
| Totals: | 206 | 212 | 53,875 | 223,114,737 | 223,139,936 | 25,199 | 1,347 | 859 |
Chromosome-scale assembly and comparison to other published genomes
In the new map-sequence integration, a total of 3,423 markers (Table 1) from three linkage maps were used in the integration of the original raw peach assembly obtained prior to the build of Peach v1.0 pseudomolecules: 1,224 markers were integrated in TxE (895 of them BIN mapped), 1974 in PxF and 225 in CxA (Table 1). The improved TxE map (Additional file 2: Figure S1, Additional file 1: Table S4) enabled the anchoring of 57 scaffolds (225,7 Mb; 99.2% of the peach genome) and orienting of 40 scaffolds (207.5 Mb; 91.3% of the peach genome; Additional file 1: Table S10). The CxA map (Additional file 2: Figure S1, Additional file 1: Table S6) anchored 52 scaffolds (211.1 Mb; 92.8% of the assembled sequences) and oriented 33 (178.1 Mb; 78.3% of the assembled sequences; Additional file 1: Table S10). The PxF map (Additional file 2: Figure S1, Additional file 1: Table S7) anchored 51 scaffolds (220.8 Mb; 97.1% of the assembled sequences) and oriented 44 (215.6 Mb: 94.8% of the assembled sequences; Additional file 1: Table S10). Finally, 153 SNPs mapped in MDxSD on LG 6 were instrumental in confirming some faults at the top of Pp06 (Additional file 1: Table S11). Important amendments were made in the v2.0 assembly including the portion of mapped and oriented sequences and the identification and correction of discernible misassemblies. A visual comparison between the two versions of the peach genome (v1.0 vs v2.0) is shown with the MareyMap plot (Fig. 1). High-quality assemblies are those in which the plots are characterized by a monotonically increasing function. Both Peach v1.0 and v2.0 show a general increasing tendency, however, in few v1.0 regions the function decreases highlighting this release faults (misordered, misoriented or local misassembled scaffolds such as in Pp01, Pp06, Pp07). Furthermore, differences in the physical length between the two assemblies, observed in all but Pp05 pseudomolecule, reflect the inclusion of previously unmapped scaffolds and the correction of inter-pseudomolecule misassemblies (Additional file 2: Figure S1 and Additional file 1: Table S10).
Fig. 1.
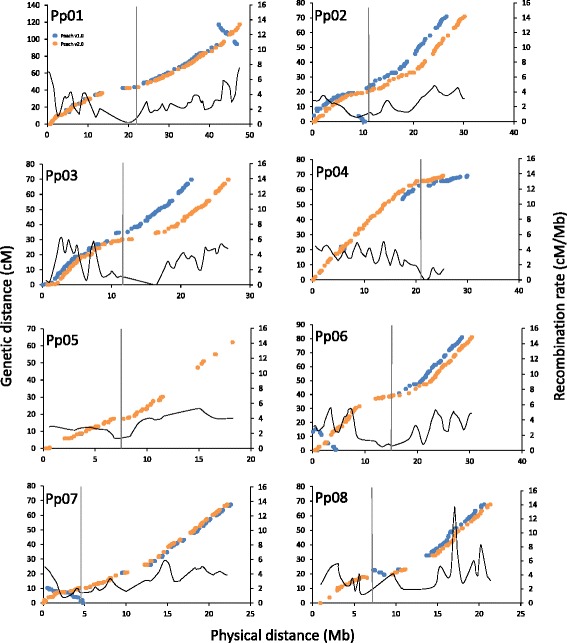
Plots of genetic-by-physical distances (MareyMap). Comparison of v1.0 and v2.0 physical distances (Mb, in the horizontal axis) and PxF genetic distances (cM, in the vertical axis). Dots represent the mapped markers. The vertical bars indicate the putative position of the centromere. The solid line represents the recombination rate plotted along the 8 pseudomolecules
The total number of major WGS scaffolds (> 1 kb) after filtering and scaffold breaking and prior to pseudomolecule construction was 241, spanning 226,911,381 bp with a scaffold N50/L50 of 10/7.3 Mb (Additional file 1: Table S12). The final chromosome-scale assembly is arranged in 191 stretches of non-contiguous sequences (8 pseudomolecules and 183 scaffolds) spanning 227,411,381 bp with a contig length of 224,638,928 bp and a scaffold contig coverage of 98.8% (Additional file 1: Table S113). Fifty-eight scaffolds spanning 225.7 Mb of sequences were integrated into the 8 Peach v2.0 pseudomolecules corresponding to 99.2% of the total sequence (Table 4, Additional file 1: Table S10). A comparison between the statistics of the two chromosome-scale assemblies, Peach v1.0 and Peach v2.0 is also shown in Table 4. Fifty-two scaffolds, summing up 223.3 Mb (98.2%), are correctly oriented in the new assembly (Additional file 1: Table S10). The unmapped portion of the genome comprises 183 scaffolds with a length of 1.7 Mb (0.8%) containing only 62 predicted genes [57]. Additional file 1: Table S10 resumes the integration of Peach v2.0 assembly, reporting the scaffolds mapped in the 8 pseudomolecules, their position in Peach v1.0 and Peach v2.0, the orientation information in all the maps and the size of uncovered scaffold ends.
Table 5 reports a comparison among some of the major plant genome assemblies released to date. The table was adapted and updated from Verde et al.[20] with newly published genomes and the updated genome releases (soybean, poplar, Brachypodium, Sorghum, Physcomitrella) available on Phytozome [57]. Among the WGS assemblies, Peach v2.0 displays one of the largest portions of sequences mapped on chromosomes (99.2%). The scaffold N50/L50 (10/7.3 Mb), prior to pseudomolecules build, shows the best values among those available. Chromosome-scale N50/L50 (4/27.8 Mb) is comparable with other chromosome-scale assemblies. If we exclude the finished genomes (rice and Arabidopsis) and those almost finished (Brachipodium and Sorghum), the peach genome shows one of the best contig N50/L50 (250/255.4 kb), including the recent WGS pineapple genome [39] (contig L50 126.5 kb) obtained combining the third generation single molecule long reads (PACBIO and Moleculo) with the NGS short reads (Illumina and 454). The long read sequencing in pineapple was instrumental in increasing the scaffold L50 metric from 91 to 640 kb and the contig L50 from 6.5 to 126.5 kb [39]. PACbio long reads were also useful to improve the assembly of the A. thaliana Ler genome [42]; the integration of 17x PACbio long-reads with the Illumina short reads improved the scaffold L50 that increased from 4.1 Mb to 12.8 Mb. The long read single molecule sequencing technologies are playing an increasingly important role in sequencing projects. In fact, the bioinformatics efforts of the assembling procedure are assisted and empowered by the availability of sequence stretches entirely spanning the repetitive regions, which are a major contributor to gaps present in assemblies.
Table 5.
Comparison of the peach genome to other published plant genomes
| Genome release [Reference] | Coverage | Assembled scaffold sequence Mb | Mapped sequences Mb (%) | N50 | L50 Mb | N50 | L50 Mb | Contig N50 | Contig L50 kb | Sequencing methods |
|---|---|---|---|---|---|---|---|---|---|---|
| Scaffold WGSa | Scaffold Chrb | |||||||||
| Peach (Prunus persica) v2.0 [20] | 8.47x | 227.4 | 225.7 (99.2) | 10 | 7.3 | 4 | 27.4 | 250 | 255.4 | Sanger (WGS) |
| Apple (Malus x domestica) [10] | 16.9x | 598.3 | 528.3 (88.3) | 80 | 2 | -- | -- | 16171 | 13.4 | Sanger, 454 (WGS) |
| Arabidopsis thaliana c [7] | -- | 119.7 | 119.7 (100) | -- | -- | 3 | 23.5 | -- | -- | Sanger, (BAC by BAC) |
| Rice (Oryza sativa)d [5] | -- | 382.2 | 382.2 (100) | -- | -- | 6 | 30.8 | -- | -- | Sanger, (BAC by BAC) |
| Soybean (Glycine max)e v2.0 [88] | 8.04x | 955.4f | 932.5 (97.6) | -- | -- | 10 | 48.6f | 1548f | 182.8f | Sanger (WGS) |
| Poplar (Populus trichocarpa)e v3.0 [3] | 9.44xf | 423f | 388 (91.7) | -- | -- | 8f | 19.5f | 206f | 552.8f | Sanger (WGS) |
| Grape (Vitis vinifera)e [4] | 8.4x | 467.5 | 290.2 (62.1) | -- | 2.1 | 14 | 13.9 | 2012 | 66.4 | Sanger (WGS) |
| Papaya (Carica papaya)e [89] | <3x | 271.7 | 235 (86.5) | -- | -- | 74 | 1.3 | 7109 | 10.6 | Sanger (WGS) |
| Brachypodium distachyon v3.1 [90] | 9.4x | 271.9 | 271.1 (99.8) | -- | -- | 3 | 59.3 | -- | 22000f | Sanger (WGS) |
| Sorghum bicolor e v3.1 [91] | 8.5x | 697.6 | 625.6 (89.7) | -- | -- | 6 | 62.4 | -- | 1200f | Sanger (WGS) |
| Selaginella moellendorffii [92] | 7x | 212.6 | -- (--) | 38 | 1.7 | -- | -- | 515 | 119.8 | Sanger (WGS) |
| Physcomitrella patens e, v3.3 [93] | 8.92x | 466.7 | -- (--) | 86 | 1.7 | 12f | 17.4f | 312f | 464.9f | Sanger (WGS) |
| Tomato (Solanum lycopersicon) [94] | 25x | 781.3 | 759.9 (97.3) | 52 | 4.5 | 6 | 64.8 | 3641 | 55.7 | Sanger, 454, Solid, Illumina (WGS) |
| Banana (Musa paradisiaca) [95] | 20.5x | 472.2 | 331.8 (70.3) | 65 | 1.3 | 8 | 28.6 | 2113 | 43.1 | Sanger, 454 (WGS) |
| Citrus (Citrus x clementina) [96] | 6.97x | 309.9 | 288.6 (93.1) | -- | 6.8 | -- | 31.4 | -- | 115.9 | Sanger (WGS) |
| Watermelon (Citrullus lanatum) [97] | 108.64x | 353.5 | 330 (93.4) | 42 | 2.4 | -- | -- | 3315 | 26.4 | Illumina (WGS) |
| Amborella trichopoda [98] | 30x | 706 | -- | 50 | 4.9 | -- | -- | 6448 | 29.4 | Sanger, 454, Illumina (WGS) |
| Medicago truncatula [99] | -- | 328.9 | 297.1 (90.3) | 53 | 1.27 | 4 | 38.9 | -- | -- | Sanger, 454, Illumina (WGS, BAC by BAC) |
| Melon (Cucumis melo) [100] | 13.52x | 361.4 | 316.3 (87.5) | 26 | 4.68 | 6g | 17.7g | -- | 18.2 | Sanger, 454 (WGS) |
| Coffee (Coffea canephora) [101] | 30x | 568.6 | 364.1 (64.0) | 108 | 1.3 | 5g | 32g | 2290 | 51.1 | Sanger, 454, Illumina (WGS) |
| Cotton (Gossipium raimondii) [102] | 103.6x | 775.2 | 567.2 (73.2) | 95 | 2.3 | -- | -- | 4918 | 44.9 | Illumina (WGS) |
| Pineapple (Ananas comosus) [39] | 410x | 381.9 | 315.8 (82.7) | -- | 0.64 | 13g | 11.8 | -- | 126.5 | PACbio, Illumina, 454, Moleculo, (WGS) |
aN50/L50 statistics of the WGS assembly prior to pseudomolecule build
bN50/L50 statistics of the chromosome-scale assembly
c Arabidopsis assembly, obtained using BAC by BAC approach, represents the golden standard for plant genome. Statistics were calculated from TAIR10 release. (http://www.ncbi.nlm.nih.gov/mapview/stats/BuildStats.cgi?taxid=3702&build=9&ver=2)
dRice assembly, obtained using BAC by BAC approach, represents the golden standard for plant genome. Statistics were calculated from IRGSP Releases Build 4.0 (http://rgp.dna.affrc.go.jp/IRGSP/Build4/build4.html)
eData retrieved from Schmutz et al. [88]; they recalculated the original statistics to better match chromosome-scale assemblies
fData from recent releases retrieved from Phytozome
gData were recalculated based on the original statistics reported in the paper
We compared the statistics of the current peach release with the standards established by Chain et al. [43]. According to these standards, the Peach v2.0 assembly can be classified as “Improved High-Quality Draft” since actions have been taken in assigning almost the whole sequence to chromosomes, in solving discernible misassemblies, filling gaps to reduce the number of contigs and correcting base errors. Moreover, the annotation of the release (Peach v2.1a) has greatly improved gene completeness using a large amount of RNA-seq data, as well as the annotated repeats which include low copy repeats and a complete set of Helitron transposons. The number of gene models in v2.1a decreased to 26,873 (was 27,853 in v1.0) resulting also in less fragmented gene models. The average number of transcripts per gene model increased to 1.75 from 1.03 (47,089 transcripts in v2.1a vs 28,689 in v1.0). The annotation improvements, not described in this work, are briefly reported on Phytozome [57], GDR [61] and IGA [60] websites. For all these features the improved peach release (v2.0 assembly and v2.1a annotation) can be further classified according to Chain et al. [43] as an “Annotation-Directed Improvement”, making it a useful tool for genome comparison and evolutionary studies, including gene studies such as alternative splicing analysis and metabolic pathway reconstruction.
Physical vs genetic distance comparison and identification of centromeric regions
The availability of the TxE, CxA and PxF linkage maps covering most of the peach genome enabled a chromosome-scale comparison of the recombination frequencies along the genome. In Fig. 1, Additional file 3: Figure S2, Additional file 4: Figure S3, and Additional file 5: Figure S4, MareyMaps are plotted together with a function describing the genetic/physical ratio (cM/Mb). Average physical/genetic distance ratios for each individual chromosome and cross were calculated (Table 2) to be 2.148 cM/Mb in the interspecific cross TxE and 2.564 cM/Mb and 2.768 cM/Mb in the intraspecific crosses CxA and PxF, respectively.
The MareyMap plots (Fig. 1, Additional file 3: Figure S2, Additional file 4: Figure S3 and Additional file 5: Figure S4), constructed for the three different mapping progenies, indicates the expected monotonic increase along each chromosome except for a flat region where a marked suppression of recombination can be observed in each pseudomolecule. The survey of these regions on Peach v2.0 JBrowse [57] revealed the almost complete absence of predicted genes and the abundance of repetitive elements. This concurrent evidence suggest that these are likely the centromeric regions (pointed out as a vertical bar in Fig. 1, Additional file 3: Figure S2, Additional file 4: Figure S3, and Additional file 5: Figure S4) which are known to be mainly composed of interspersed tandem repeats and retrotransposons. Despite the centromeres have highly conserved function during cell division, their DNA sequences are not conserved [83]. In particular, in plants, the centromeric satellite DNA repeats are species-specific. Recent studies suggest that these sequences underwent a rapid evolution revealing no sequence similarity among species diverged more than 50 Mya [84]. Other important components of the centromeric regions are represented by the retrotransposons directly involved in the centromere evolution and function [72]. A BLAST analysis against the peach genome was performed using a set [72] of 335 centromeric retrotransposon sequences belonging to 33 different plant species. In particular, eight plant centromere retrotransposons indicated similarity with the peach putative centromeric regions: three from Medicago truncatula (AC131249.44, AC147471.14, CT010572.8), two from Pinus taeda (AC241271.1, AC241322.1), one from Picea glauca (AF229251.1), one from Beta vulgaris (AJ539424.1), one from Vitis vinifera (AM426079.1). These results strongly support that these regions are the peach centromeres. The accuracy of the Peachv2.0 assembly and the Peach v2.1a annotation enabled the positioning of the centromeric regions, which are generally very difficult to assemble due to their highly repetitive sequence composition. Linkage maps are of little use to assemble fragmented centromeric regions due to the suppression of recombination. As an example, the putative peach centromeric region in Pp03 (spanning Super_31 and Super_32, 12-13.2 Mb) indicated no recombination in the nearly 1000 meiosis analyzed in the three different mapping progenies, leaving the related scaffolds unoriented and unordered. The identification of putative centromeric regions for all of the eight Peach v2.0 chromosomes attests the completeness of the current peach genome assembly.
To verify whether there were significant differences in the recombination frequencies among the three maps, an analysis of variance was performed. Since the PxF map was obtained by map data of the two parents of the cross (the F1 and the recurrent IF7310828), the individual maps of the two parents were also considered in the analysis. Both the ANOVA and the non-parametric Kruskal-Wallis statistics were applied showing no significant difference among the five maps (F = 0.9026 with p = 0.4733; Hc = 7.329 with p = 0.195). However, due to the low coverage (referring to local rather than global recombination rates) of some linkage groups (Table 1) chromosomes with coverage lower than 50% of the total length (G3, G5, G7, G8 in PxF recurrent map and G5 in CxA) were excluded thus revealing significant differences with the ANOVA test (F = 8.16; p = 0.00014). Moreover, to enlarge the comparison panel, two recently published maps, obtained with the IPSC 9 K SNP array, were included: the peach intraspecific ‘NJ Weeping’ x ‘Bounty’ F2 map [85] (WxB) and the interspecific DvsS BC2 map obtained by backcrossing a hybrid between peach and P. davidiana as donor with peach as recurrent [81]. Both maps indicate > 80% coverage for each individual chromosome, globally 93% and 95% of Peach v2.0 length, respectively. The physical distances covered by these two latter maps were recalculated based on the Peach v2.0 assembly. The ANOVA comparison was then applied giving significant differences among the seven maps (F = 7.17: p = 2.249x10 −5). The lowest recombination frequency, displayed by the recurrent parent (1.941 cM/Mb), was not significantly different (see Post hoc Tuckey’s pairwise comparison test, Table 6) from that of the two interspecific hybrids (TxE and DvsS, 2.148 and 1.954 cM/Mb, respectively). The PxF F1 map, having the highest recombination frequency (3.057 cM/Mb), was significantly different from the two interspecific maps (TxE and DvsS) (p = 0.0266, p = 0.0013 respectively) as well as from PxF recurrent map (p = 0.0002) but not significantly different from the other intraspecific maps (CxA and WxB, 2.564 and 2.553 cM/Mb, respectively). The extreme reduction of recombination rate, observed in the PxF recurrent parent (IF7310828, derived from ‘JH Hale’ x ‘Bonanza’) is comparable to that of the interspecific crosses. ‘JH Hale’ is an old cultivar, one of the founders of the so-called modern Western germplasm, carrying the male sterility allele (Ps) while ‘Bonanza’ is a dwarf accession. The IF7310828 recurrent parent is classified as an accession with intermediate tree vigor (semidwarf) [86] belonging to the Western subgroup [20]. No differences in the recombination frequency were observed among the other peach F1 individuals (PxF, CxA, WxB). The other parents of the crosses analyzed in this study have different origin: ‘Contender’, ‘Ambra’ and ‘Bounty’ all derive from Western breeding programs while Ferganensis belongs to the Eastern subgroup [20] and NJ Weeping is believed to have Japanese origin [85]. Our results are in agreement with those reported in Arabidopsis thaliana [87] where recombination rates, calculated in 17 F2 populations derived from 18 accessions, do not correlate with genetic distances between intraspecific parental accessions. An exception in this study is the PxF recurrent parent, which displays a reduction in recombination rate similar to that of the interspecific crosses.
Table 6.
Tuckey's pairwise comparison test among the different maps
| DvsS | WxB | CxA | PxF recurrent | PxF F1 | PxF | |
|---|---|---|---|---|---|---|
| TxE | 0.9335 | 0.6958 | 0.9596 | 0.2531 | 0.0266 | 0.2337 |
| PxF | 0.0201 | 0.9843 | 0.7918 | 0.0006 | 0.9592 | |
| PxF F1 | 0.0013 | 0.5816 | 0.2327 | 0.0002 | ||
| PxF recurrent parent | 0.8660 | 0.0055 | 0.0301 | |||
| CxA | 0.4103 | 0.9959 | ||||
| WxB | 0.1362 |
Significant p-values (α = 0.05) are reported in bold
Conclusions
In this paper an improved and refined version of the peach genome assembly based on high quality linkage maps and resequencing data is presented. This new assembly release has been improved in terms of completeness and accuracy, including the increase of mapped and oriented sequences, repositioning of misassembled portions, enhancement of contiguity and correction of base errors. High density (referring to the number of markers used) and high resolution (referring to the number of the seedlings of the mapping progeny) maps are important tools to assist WGS efforts. In fact, even if unanchored WGS assemblies are able to catch the full gene complement, they defect in depicting the whole genome view thus being of little use for comparative genomics. The late high-throughput genotyping technologies such as SNP arrays or genotyping-by-sequencing platforms are essential for developing saturated and high-resolution maps in short times with minimal cost, even in species with a narrow genetic base like peach and other self-pollinating species. Third generation and NGS technologies can be efficiently used in newly or already available genome sequences to obtain a high quality assembly.
Acknowledgments
The authors thank the Centre for Applied Biomedical Research (CRBA) of Bologna for valuable contributions to the Sequenom analyses and Elisa Banchi from FEM for performing the SNP genotyping on iScan platform. The authors also thank Fondazione Cassa di Risparmio in Bologna for supporting CRBA.
Funding
The work conducted in Italy was funded by the Ministero delle Politiche Agricole Alimentari e Forestali -Italy (MiPAAF, http://www.politicheagricole.it) through the project “DRUPOMICS: “Sequenziamento del genoma del pesco ed utilizzo della sequenza in programmi di miglioramento della qualità del frutto del pesco e della resistenza alle malattie" (Grant # DM14999/7303/08) and the European Union-funded project “FruitBreedomics: “Integrated approach for increasing breeding efficiency in fruit tree crops" (Grant #FP7-265582; http://fruitbreedomics.com/; http://ec.europa.eu/research/fp7/index_en.cfm). The work conducted by the US Department of Energy Joint Genome Institute, was supported by the Office of Science of the US Department of Energy under Contract no. DE-AC02-05CH11231.
Availability of data and materials
The dataset supporting the conclusions of this article are partly included within the article and its Additional files 1, 2, 3, 4 and 5.
Further datasets supporting the conclusions of this article are available at the following repositories. The peach genome sequence can be accessed at following databases URLs:
Phytozome (https://phytozome.jgi.doe.gov/pz/portal.html#!info?alias=Org_Ppersica);
Genome Database for Rosaceae (https://www.rosaceae.org/species/prunus_persica/genome_v2.0.a1);
Applied Genomic Institute (IGA) : (http://services.appliedgenomics.org/fgb2/iga/prunus_persica_v2/gbrowse/prunus_persica_v2/).
This Whole Genome Shotgun project had been deposited at DDBJ/EMBL/GenBank under the accession AKXU00000000. The version described in this paper is accessible at the accession AKXU02000000
Illumina short reads are deposited into NCBI Short Read Archive under accession numbers SRX2273835, SRX2381075, SRX2392647, SRX2392648, SRX2392650
SSR primer sequences were deposited into NCBI probe database under accession numbers from Pr032816704 to Pr032816814.
Authors’ contributions
IV, ST and JS conceived and designed the experiments; JJ, LD, SM, GP EV, RP, VA LG, LR, SS and JG performed the experiments; IV, JJ, LD, SM, EV, SS, JG, ST, MTD and JS analyzed the data; IV, LR, DB, MT, SS, JG, ST and JS contributed reagents/materials/analysis tools; IV, JJ and ST wrote the manuscript. LD, SM, GP, EV, RP, DB, MT, ST, MTD and JS critically revised the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
SSR markers developed in this study from Peach v1.0. For each primer, the locus name, primer sequences, position in Peach v1.0 and v2.0, repeated motif, polymorphism in TxE (monomorphic, BIN or fully mapped) and location in genic or intergenic regions are reported. Table S2. SNP markers developed in this study from the resequencing of the CxA F1 parent. For each primer, the locus name, position in Peach v1.0 and v2.0, primer sequences, BIN position and location in genic or intergenic regions are reported. Table S3. Illumina short reads libraries. All libraries were sequenced as 2x250s using Illumina sequencing. Reads were filtered on quality, screening for reads that were predominantly simple sequence, and residual phix contamination. Table S4. TxE linkage map. For each locus, the genetic position in linkage groups and physical position in Peach v2.0 are reported. Only integrated markers are reported. Table S5. Sequenom SNP development and their efficiency with the SNP calling parameters used in this study. A posteriori efficiency was calculated using more stringent parameters as in Verde et al. 2013. Table S6. CxA linkage map. For each locus, the genetic position in linkage groups and physical position in Peach v2.0 are reported. Table S7. PxF linkage map. For each locus, the genetic position in linkage groups and physical position in Peach v2.0 are reported. Table S8. Linkage map of PxF F1 parent. For each locus, the genetic position in linkage groups and physical position in Peach v2.0 are reported. Table S9. Linkage map of PxF recurrent parent (IF7310828). For each locus, the genetic position in linkage groups and physical position in Peach v2.0 are reported. Table S10. Composition of the 8 Peach v2.0 pseudomolecules. Start and end of each scaffold, total scaffold length, scaffold denomination in v2.0 and in v1.0, v1.0 position of each v2.0 scaffold, length of the scaffold ends uncovered with markers and mapping and orientation information achieved with the linkage maps used in this study, are reported. Table S11. Linkage map of LG6 of MDxSD. For each locus, the genetic position (cM) in linkage groups and physical position in Peach v2.0 are reported. Table S12. Peach v2.0 assembly statistics (N/L) prior to the pseudomolecules build. Table S13. Peach v2.0 assembly statistics post chromosome-scale build. (XLSX 391 kb)
Anchoring of the peach scaffolds to the three genetic maps. Colored bars represent the 8 linkage groups: pink for PxF, purple for TxE and blue for CxA. WGS scaffolds were positioned in each pseudomolecule (Pp01 to Pp08) with the corresponding genetic markers and are depicted in three different colors (dark blue, pink and pale blue); genetic markers are in the same colors of the corresponding WGS scaffolds. The zero (0) denotes the six scaffolds placed with random orientation along the pseudomolecules. The asterisk (*) indicates the two scaffolds with random order. The crosshatch (#) indicates the two scaffolds with the wrong order in Peach v2.0 that need to be inverted in a future release. (PDF 495 kb)
MareyMap plot of PxF linkage maps (including the F1 and recurrent parent maps). Vertical bars indicate the putative position of the centromere. The solid line represents the recombination rate plotted along the 8 pseudomolecules calculated using the cubic spline method. (PDF 405 kb)
MareyMap plot of TxE linkage map. Vertical bars indicate the putative position of the centromere. The solid line represents the recombination rate plotted along the 8 pseudomolecules calculated using the cubic spline method. (PDF 236 kb)
MareyMap plot of CxA linkage map. Vertical bars indicate the putative position of the centromere. The solid line represents the recombination rate plotted along the 8 pseudomolecules calculated using the cubic spline method. (PDF 233 kb)
References
- 1.Myers EW, Sutton GG, Delcher AL, Dew IM, Fasulo DP, Flanigan MJ, et al. A Whole-Genome Assembly of Drosophila. Science. 2000;287:2196–204. doi: 10.1126/science.287.5461.2196. [DOI] [PubMed] [Google Scholar]
- 2.Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, et al. The Sequence of the Human Genome. Science. 2001;291:1304–51. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 3.Tuskan GA, Difazio S, Jansson S, Bohlmann J, Grigoriev I, Hellsten U, et al. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray) Science. 2006;313:1596–1604. doi: 10.1126/science.1128691. [DOI] [PubMed] [Google Scholar]
- 4.Jaillon O, Aury JM, Noel B, Policriti A, Clepet C, Casagrande A, et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature. 2007;449:463–7. doi: 10.1038/nature06148. [DOI] [PubMed] [Google Scholar]
- 5.International Rice Genome Sequencing Project The map-based sequence of the rice genome. Nature. 2005;436:793–800. doi: 10.1038/nature03895. [DOI] [PubMed] [Google Scholar]
- 6.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 7.The Arabidopsis Genome Initiative Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000;408:796–815. doi: 10.1038/35048692. [DOI] [PubMed] [Google Scholar]
- 8.Schnable PS, Ware D, Fulton RS, Stein JC, Wei F, Pasternak S, et al. The B73 Maize Genome: Complexity, Diversity, and Dynamics. Science. 2009;326:1112–5. doi: 10.1126/science.1178534. [DOI] [PubMed] [Google Scholar]
- 9.The International Wheat Genome Sequencing Consortium (IWGSC) A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science. 2014;345:1251788. doi: 10.1126/science.1251788. [DOI] [PubMed] [Google Scholar]
- 10.Velasco R, Zharkikh A, Affourtit J, Dhingra A, Cestaro A, Kalyanaraman A, et al. The genome of the domesticated apple (Malus x domestica Borkh.) Nat Genet. 2010;42:833–9. doi: 10.1038/ng.654. [DOI] [PubMed] [Google Scholar]
- 11.Velasco R, Zharkikh A, Troggio M, Cartwright DA, Cestaro A, Pruss D, et al. A High Quality Draft Consensus Sequence of the Genome of a Heterozygous Grapevine Variety. PLoS ONE. 2007;2:e1326. doi: 10.1371/journal.pone.0001326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Dijk EL, Auger H, Jaszczyszyn Y, Thermes C. Ten years of next-generation sequencing technology. Trends Genet. 2014;30:418–26. doi: 10.1016/j.tig.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 13.Miller JR, Koren S, Sutton G. Assembly algorithms for next-generation sequencing data. Genomics. 2010;95:315–27. doi: 10.1016/j.ygeno.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Henson J, Tischler G, Ning Z. Next-generation sequencing and large genome assemblies. Pharmacogenomics. 2012;13:901–15. doi: 10.2217/pgs.12.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lewin HA, Larkin DM, Pontius J, O’Brien SJ. Every genome sequence needs a good map. Genome Res. 2009;19:1925–8. doi: 10.1101/gr.094557.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mascher M, Stein N. Genetic anchoring of whole-genome shotgun assemblies. Front Genet. 2014;5:208. doi: 10.3389/fgene.2014.00208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fierst JL. Using linkage maps to correct and scaffold de novo genome assemblies: methods, challenges, and computational tools. Front Genet. 2015;6:220. doi: 10.3389/fgene.2015.00220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gore MA, Chia J-M, Elshire RJ, Sun Q, Ersoz ES, Hurwitz BL, et al. A First-Generation Haplotype Map of Maize. Science. 2009;326:1115–7. doi: 10.1126/science.1177837. [DOI] [PubMed] [Google Scholar]
- 19.Cao J, Schneeberger K, Ossowski S, Günther T, Bender S, Fitz J, et al. Whole-genome sequencing of multiple Arabidopsis thaliana populations. Nat Genet. 2011;43:956–63. doi: 10.1038/ng.911. [DOI] [PubMed] [Google Scholar]
- 20.Verde I, Abbott AG, Scalabrin S, Jung S, Shu S, Marroni F, et al. The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat Genet. 2013;45:487–94. doi: 10.1038/ng.2586. [DOI] [PubMed] [Google Scholar]
- 21.Bianco L, Cestaro A, Linsmith G, Muranty H, Denancé C, Théron A, et al. Development and validation of the Axiom ® Apple480K SNP genotyping array. Plant J. 2016;86:62–74. doi: 10.1111/tpj.13145. [DOI] [PubMed] [Google Scholar]
- 22.Bassil NV, Davis TM, Zhang H, Ficklin S, Mittmann M, Webster T, et al. Development and preliminary evaluation of a 90 K Axiom® SNP array for the allo-octoploid cultivated strawberry Fragaria × ananassa. BMC Genomics. 2015;16:155. doi: 10.1186/s12864-015-1310-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lepoittevin C, Bodénès C, Chancerel E, Villate L, Lang T, Lesur I, et al. Single-nucleotide polymorphism discovery and validation in high-density SNP array for genetic analysis in European white oaks. Mol Ecol Resour. 2015;15:1446–59. doi: 10.1111/1755-0998.12407. [DOI] [PubMed] [Google Scholar]
- 24.Geraldes A, DiFazio SP, Slavov GT, Ranjan P, Muchero W, Hannemann J, et al. A 34 K SNP genotyping array for Populus trichocarpa: Design, application to the study of natural populations and transferability to other Populus species. Mol Ecol Resour. 2013;13:306–23. doi: 10.1111/1755-0998.12056. [DOI] [PubMed] [Google Scholar]
- 25.Gunderson KL. Whole-genome genotyping on bead arrays. Methods Mol. Biol. 2009;529:197–213. doi: 10.1007/978-1-59745-538-1_13. [DOI] [PubMed] [Google Scholar]
- 26.Gunderson KL, Steemers FJ, Lee G, Mendoza LG, Chee MS. A genome-wide scalable SNP genotyping assay using microarray technology. Nat Genet. 2005;37:549–54. doi: 10.1038/ng1547. [DOI] [PubMed] [Google Scholar]
- 27.Bielenberg DG, Rauh B, Fan S, Gasic K, Abbott AG, Reighard GL, et al. Genotyping by Sequencing for SNP-Based Linkage Map Construction and QTL Analysis of Chilling Requirement and Bloom Date in Peach [Prunus persica (L.) Batsch] Plos One. 2015;10:e0139406. doi: 10.1371/journal.pone.0139406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen C, Mitchell SE, Elshire RJ, Buckler ES, El-Kassaby YA. Mining conifers’ mega-genome using rapid and efficient multiplexed high-throughput genotyping-by-sequencing (GBS) SNP discovery platform. Tree Genet Genomes. 2013;9:1537–44. doi: 10.1007/s11295-013-0657-1. [DOI] [Google Scholar]
- 29.Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, et al. A Robust, Simple Genotyping-by-Sequencing (GBS) Approach for High Diversity Species. PLoS ONE. 2011;6:e19379. doi: 10.1371/journal.pone.0019379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Torkamaneh D, Belzile F. Scanning and Filling: Ultra-Dense SNP Genotyping Combining Genotyping-By-Sequencing, SNP Array and Whole-Genome Resequencing Data. PLOS ONE. 2015;10:e0131533. doi: 10.1371/journal.pone.0131533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Verde I, Bassil N, Scalabrin S, Gilmore B, Lawley CT, Gasic K, et al. Development and evaluation of a 9 K SNP array for peach by internationally coordinated SNP detection and validation in breeding germplasm. PLoS One. 2012;7:e35668. doi: 10.1371/journal.pone.0035668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peace C, Bassil N, Main D, Ficklin S, Rosyara UR, Stegmeir T, et al. Development and evaluation of a genome-wide 6 K SNP array for diploid sweet cherry and tetraploid sour cherry. PloS One. 2012;7:e48305. doi: 10.1371/journal.pone.0048305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bianco L, Cestaro A, Sargent DJ, Banchi E, Derdak S, Di Guardo M, et al. Development and Validation of a 20K Single Nucleotide Polymorphism (SNP) Whole Genome Genotyping Array for Apple (Malus × domestica Borkh) PLoS ONE. 2014;9:e110377. doi: 10.1371/journal.pone.0110377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roberts RJ, Carneiro MO, Schatz MC. The advantages of SMRT sequencing. Genome Biol. 2013;14:405. doi: 10.1186/gb-2013-14-6-405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Voskoboynik A, Neff NF, Sahoo D, Newman AM, Pushkarev D, Koh W, et al. The genome sequence of the colonial chordate, Botryllus schlosseri. eLife. 2013;2:e00569. doi: 10.7554/eLife.00569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee H, Gurtowski J, Yoo S, Marcus S, McCombie WR, Schatz M. Error correction and assembly complexity of single molecule sequencing reads. BioExiv 006395. 2014;http://dx.doi.org/10.1101/006395.
- 37.Chin C-S, Alexander DH, Marks P, Klammer AA, Drake J, Heiner C, et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods. 2013;10:563–9. doi: 10.1038/nmeth.2474. [DOI] [PubMed] [Google Scholar]
- 38.Quail M, Smith ME, Coupland P, Otto TD, Harris SR, Connor TR, et al. A tale of three next generation sequencing platforms: comparison of Ion torrent, Pacific Biosciences and illumina MiSeq sequencers. BMC Genomics. 2012;13:341. doi: 10.1186/1471-2164-13-341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ming R, VanBuren R, Wai CM, Tang H, Schatz MC, Bowers JE, et al. The pineapple genome and the evolution of CAM photosynthesis. Nat Genet. 2015;47:1435–42. doi: 10.1038/ng.3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Redwan RM, Saidin A, Kumar SV. The draft genome of MD-2 pineapple using hybrid error correction of long reads. DNA Res. 2016;23:427–39. doi: 10.1093/dnares/dsw026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li X, Kui L, Zhang J, Xie Y, Wang L, Yan Y, et al. Improved hybrid de novo genome assembly of domesticated apple (Malus x domestica) GigaScience. 2016;5:35. doi: 10.1186/s13742-016-0139-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zapata L, Ding J, Willing E-M, Hartwig B, Bezdan D, Jiao W-B, et al. Chromosome-level assembly of Arabidopsis thaliana L er reveals the extent of translocation and inversion polymorphisms. Proc Natl Acad Sci. 2016;113:E4052–60. doi: 10.1073/pnas.1607532113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chain PSG, Grafham DV, Fulton RS, FitzGerald MG, Hostetler J, Muzny D, et al. Genome Project Standards in a New Era of Sequencing. Science. 2009;326:236–7. doi: 10.1126/science.1180614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aranzana M, Pineda A, Cosson P, Dirlewanger E, Ascasibar J, Cipriani G, et al. A set of simple-sequence repeat (SSR) markers covering the Prunus genome. Theor Appl Genet. 2003;106:819–825. doi: 10.1007/s00122-002-1094-y. [DOI] [PubMed] [Google Scholar]
- 45.Dirlewanger E, Graziano E, Joobeur T, Garriga-Calderé F, Cosson P, Howad W, et al. Comparative mapping and marker-assisted selection in Rosaceae fruit crops. Proc Natl Acad Sci U S A. 2004;101:9891–6. doi: 10.1073/pnas.0307937101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Joobeur T, Viruel MA, De Vicente MC, Jauregui B, Ballester J, Dettori MT, et al. Construction of a saturated linkage map for Prunus using an almond x peach F2 progeny. Theor Appl Genet. 1998;97:1034–1041. doi: 10.1007/s001220050988. [DOI] [Google Scholar]
- 47.Howad W, Yamamoto T, Dirlewanger E, Testolin R, Cosson P, Cipriani G, et al. Mapping with a few plants: using selective mapping for microsatellite saturation of the Prunus reference map. Genetics. 2005;171:1305–9. doi: 10.1534/genetics.105.043661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Illa E, Eduardo I, Audergon JM, Barale F, Dirlewanger E, Li X, et al. Saturating the Prunus (stone fruits) genome with candidate genes for fruit quality. Mol Breed. 2011;28:667–82. doi: 10.1007/s11032-010-9518-x. [DOI] [Google Scholar]
- 49.Foulongne M, Pascal T, Arús P, Kervella J. The potential of Prunus davidiana for introgression into peach [Prunus persica (L.) Batsch] assessed by comparative mapping. Theor Appl Genet. 2003;107:227–38. doi: 10.1007/s00122-003-1238-8. [DOI] [PubMed] [Google Scholar]
- 50.Verde I, Lauria M, Dettori MT, Vendramin E, Balconi C, Micali S, et al. Microsatellite and AFLP markers in the Prunus persica [L.(Batsch)] x P. ferganensis BC1 linkage map: saturation and coverage improvement. Theor Appl Genet. 2005;111:1013–21. doi: 10.1007/s00122-005-0006-3. [DOI] [PubMed] [Google Scholar]
- 51.Eduardo I, Pacheco I, Chietera G, Bassi D, Pozzi C, Vecchietti A, et al. QTL analysis of fruit quality traits in two peach intraspecific populations and importance of maturity date pleiotropic effect. Tree Genet Genomes. 2011;7:323–35. doi: 10.1007/s11295-010-0334-6. [DOI] [Google Scholar]
- 52.Dettori MT, Quarta R, Verde I. A peach linkage map integrating RFLPs, SSRs, RAPDs, and morphological markers. Genome. 2001;44:783–90. doi: 10.1139/g01-065. [DOI] [PubMed] [Google Scholar]
- 53.Quarta R, Dettori MT, Verde I, Gentile A. Genetic analysis of agronomic traits and genetic linkage mapping in a BC1 peach population using RFLPs and RAPDs. Acta Hort ISHS. 1998;465:51–9. doi: 10.17660/ActaHortic.1998.465.3. [DOI] [Google Scholar]
- 54.Mercado JA, El Mansouri I, Jiménez-Bermúdez S, Pliego-Alfaro F, Quesada MA. A convenient protocol for extraction and purification of DNA from Fragaria. Vitro Cell. Dev Biol Plant. 1999;35:152–3. doi: 10.1007/s11627-999-0025-x. [DOI] [Google Scholar]
- 55.Dettori MT, Micali S, Giovinazzi J, Scalabrin S, Verde I, Cipriani G. Mining microsatellites in the peach genome: development of new long-core SSR markers for genetic analyses in five Prunus species. SpringerPlus. 2015;4:337. doi: 10.1186/s40064-015-1098-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goodstein DM, Shu S, Howson R, Neupane R, Hayes RD, Fazo J, et al. Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 2012;40:D1178–D1186. doi: 10.1093/nar/gkr944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.JGI - Phytozome v11.0 [Internet]. JGI. [Accessed 2016 Oct 18]. Available from: https://phytozome.jgi.doe.gov/pz/portal.html
- 58.Primer3 [Internet]. [Accessed 2016 Oct 18]. Available from: http://bioinfo.ut.ee/primer3-0.4.0/primer3/
- 59.Lander ES, Green P, Abrahamson J, Barlow A, Daly MJ, Lincoln SE, et al. MAPMAKER: an interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics. 1987;1:174–81. doi: 10.1016/0888-7543(87)90010-3. [DOI] [PubMed] [Google Scholar]
- 60.IGA - Istituto di Genomica Applicata [Internet]. IGA. [Accessed 18 Oct 2016]. Available from: http://www.appliedgenomics.org/.
- 61.Jung S, Ficklin SP, Lee T, Cheng CH, Blenda A, Zheng P, et al. The Genome Database for Rosaceae (GDR): year 10 update. Nucleic Acids Res 2014;42 (D1):D1237-D1244. doi: 10.1093/nar/gkt1012. Genome Databease For The Rosaceae [Internet]. [Accessed 18 Oct 2016]. Available from: https://www.rosaceae.org/. [DOI] [PMC free article] [PubMed]
- 62.Oeth P, Beaulieu M, Park C, Kosman D, del Mistro G, van den Boom D, et al. iPLEX assay: Increased plexing efficiency and flexibility for MassArray system through single base primer extension with mass-modified terminators. Seq. Appl. Note. Doc. No. 8876-006, R01 CO 050154. http://www.usc.es/cegen/wp-content/uploads/2016/03/Sequenom-iplex-assay.pdf. Accessed 06 March 2017.
- 63.van Ooijen JW, Voorrips RE. JoinMap Version 3.0: software for the calculation of genetic linkage maps. Plant Res. Int. Wagening. The Netherlands. 2001.
- 64.Kosambi DD. The estimation of map distances from recombination values. Ann Eugen. 1943;12:172–5. doi: 10.1111/j.1469-1809.1943.tb02321.x. [DOI] [Google Scholar]
- 65.van Ooijen JW. Multipoint maximum likelihood mapping in a full-sib family of an outbreeding species. Genet Res. 2011;93:343–9. doi: 10.1017/S0016672311000279. [DOI] [PubMed] [Google Scholar]
- 66.Schuler GD. Sequence mapping by electronic PCR. Genome Res. 1997;7:541–50. doi: 10.1101/gr.7.5.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJM, Birol I. ABySS: A parallel assembler for short read sequence data. Genome Res. 2009;19:1117–23. doi: 10.1101/gr.089532.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kent WJ. BLAT—the BLAST-like alignment tool. Genome Res. 2002;12:656–64. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.MareyMap package on R version 3.2.4 revised [Internet]. Compr. R Arch. Netw. [Accessed 2016 Oct 18]. Available from: https://cran.r-project.org/
- 72.Neumann P, Navrátilová A, Koblížková A, Kejnovský E, Hřibová E, Hobza R, et al. Plant centromeric retrotransposons: a structural and cytogenetic perspective. Mob DNA. 2011;2:4. doi: 10.1186/1759-8753-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hammer Ø, Harper DA, Ryan PD. PAST: Paleontological Statistics Software Package for Education and Data Analysis. Palaeontol Electron. 2001;4:1–9. [Google Scholar]
- 74.Testone G, Condello E, Verde I, Nicolodi C, Caboni E, Dettori MT, et al. The peach (Prunus persica L. Batsch) genome harbours 10 KNOX genes, which are differentially expressed in stem development, and the class 1 KNOPE1 regulates elongation and lignification during primary growth. J Exp Bot. 2012;63:5417–35. doi: 10.1093/jxb/ers194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vendramin E, Dettori MT, Giovinazzi J, Micali S, Quarta R, Verde I. A set of EST-SSRs isolated from peach fruit transcriptome and their transportability across Prunus species: PRIMER NOTE. Mol Ecol Notes. 2007;7:307–10. doi: 10.1111/j.1471-8286.2006.01590.x. [DOI] [Google Scholar]
- 76.Cabrera A, Kozik A, Howad W, Arús P, Iezzoni AF, Knaap E. Development and bin mapping of a Rosaceae Conserved Ortholog Set (COS) of markers. BMC Genomics. 2009;10:562. doi: 10.1186/1471-2164-10-562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Illa E, Sargent DJ, Girona EL, Bushakra J, Cestaro A, Crowhurst R, et al. Comparative analysis of rosaceous genomes and the reconstruction of a putative ancestral genome for the family. BMC Evol Biol. 2011;11:9. doi: 10.1186/1471-2148-11-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pirona R, Eduardo I, Pacheco I, Da Silva LC, Miculan M, Verde I, et al. Fine mapping and identification of a candidate gene for a major locus controlling maturity date in peach. BMC Plant Biol. 2013;13:166. doi: 10.1186/1471-2229-13-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Vendramin E, Pea G, Dondini L, Pacheco I, Dettori MT, Gazza L, et al. A Unique Mutation in a MYB Gene Cosegregates with the Nectarine Phenotype in Peach. PLoS One. 2014;9:e90574. doi: 10.1371/journal.pone.0090574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sánchez G, Martínez J, Romeu J, García J, Monforte AJ, Badenes ML, et al. The peach volatilome modularity is reflected at the genetic and environmental response levels in a QTL mapping population. BMC Plant Biol. 2014;14:137. doi: 10.1186/1471-2229-14-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Desnoues E, Baldazzi V, Génard M, Mauroux J-B, Lambert P, Confolent C, et al. Dynamic QTLs for sugars and enzyme activities provide an overview of genetic control of sugar metabolism during peach fruit development. J Exp Bot. 2016;67:3419–31. doi: 10.1093/jxb/erw169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Romeu JF, Monforte AJ, Sánchez G, Granell A, García-Brunton J, Badenes ML, et al. Quantitative trait loci affecting reproductive phenology in peach. BMC Plant Biol. 2014;14:52. doi: 10.1186/1471-2229-14-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Garrido-Ramos MA. Satellite DNA, in Plants: More than Just Rubbish. Cytogenet Genome Res. 2015;146:153–70. doi: 10.1159/000437008. [DOI] [PubMed] [Google Scholar]
- 84.Melters DP, Bradnam KR, Young HA, Telis N, May MR, Ruby J, et al. Comparative analysis of tandem repeats from hundreds of species reveals unique insights into centromere evolution. Genome Biol. 2013;14:R10. doi: 10.1186/gb-2013-14-1-r10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.da Silva Linge C, Bassi D, Bianco L, Pacheco I, Pirona R, Rossini L. Genetic dissection of fruit weight and size in an F2 peach (Prunus persica (L.) Batsch) progeny. Mol Breed. 2015;35:71. doi: 10.1007/s11032-015-0271-z. [DOI] [Google Scholar]
- 86.Quarta R, Scortichini M. Morphological characters and yielding efficiency of semi-dwarf peach selections. Acta Hort ISHS. 1985;173:63–8. doi: 10.17660/ActaHortic.1985.173.8. [DOI] [Google Scholar]
- 87.Salomé PA, Bomblies K, Fitz J, Laitinen RA, Warthmann N, Yant L, et al. The recombination landscape in Arabidopsis thaliana F2 populations. Heredity. 2012;108:447–55. doi: 10.1038/hdy.2011.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Schmutz J, Cannon SB, Schlueter J, Ma J, Mitros T, Nelson W, et al. Genome sequence of the palaeopolyploid soybean. Nature. 2010;463:178–83. doi: 10.1038/nature08670. [DOI] [PubMed] [Google Scholar]
- 89.Ming R, Hou S, Feng Y, Yu Q, Dionne-Laporte A, Saw JH, et al. The draft genome of the transgenic tropical fruit tree papaya (Carica papaya Linnaeus) Nature. 2008;452:991–6. doi: 10.1038/nature06856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.International Brachypodium Initiative Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature. 2010;463:763–8. doi: 10.1038/nature08747. [DOI] [PubMed] [Google Scholar]
- 91.Paterson AH, Bowers JE, Bruggmann R, Dubchak I, Grimwood J, Gundlach H, et al. The Sorghum bicolor genome and the diversification of grasses. Nature. 2009;457:551–6. doi: 10.1038/nature07723. [DOI] [PubMed] [Google Scholar]
- 92.Banks JA, Nishiyama T, Hasebe M, Bowman JL, Gribskov M, dePamphilis C, et al. The Selaginella genome identifies genetic changes associated with the evolution of vascular plants. Science. 2011;332:960–3. doi: 10.1126/science.1203810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rensing SA, Lang D, Zimmer AD, Terry A, Salamov A, Shapiro H, et al. The Physcomitrella Genome Reveals Evolutionary Insights into the Conquest of Land by Plants. Science. 2008;319:64–9. doi: 10.1126/science.1150646. [DOI] [PubMed] [Google Scholar]
- 94.Sato S, Tabata S, Hirakawa H, Asamizu E, Shirasawa K, Isobe S, et al. The tomato genome sequence provides insights into fleshy fruit evolution. Nature. 2012;485:635–41. doi: 10.1038/nature11119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.D’Hont A, Denoeud F, Aury J-M, Baurens F-C, Carreel F, Garsmeur O, et al. The banana (Musa acuminata) genome and the evolution of monocotyledonous plants. Nature. 2012;488:213–7. doi: 10.1038/nature11241. [DOI] [PubMed] [Google Scholar]
- 96.Wu GA, Prochnik S, Jenkins J, Salse J, Hellsten U, Murat F, et al. Sequencing of diverse mandarin, pummelo and orange genomes reveals complex history of admixture during citrus domestication. Nat Biotechnol. 2014;32:656–62. doi: 10.1038/nbt.2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Guo S, Zhang J, Sun H, Salse J, Lucas WJ, Zhang H, et al. The draft genome of watermelon (Citrullus lanatus) and resequencing of 20 diverse accessions. Nat Genet. 2012;45:51–8. doi: 10.1038/ng.2470. [DOI] [PubMed] [Google Scholar]
- 98.Amborella Genome Project. Albert VA, Barbazuk WB, dePamphilis CW, Der JP, Leebens-Mack J, et al. The Amborella Genome and the Evolution of Flowering Plants. Science. 2013;342:1241089. doi: 10.1126/science.1241089. [DOI] [PubMed] [Google Scholar]
- 99.Young ND, Debellé F, Oldroyd GED, Geurts R, Cannon SB, Udvardi MK, et al. The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature. 2011;480:520–4. doi: 10.1038/480162a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Garcia-Mas J, Benjak A, Sanseverino W, Bourgeois M, Mir G, Gonzalez VM, et al. The genome of melon (Cucumis melo L.) Proc Natl Acad Sci. 2012;109:11872–7. doi: 10.1073/pnas.1205415109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Denoeud F, Carretero-Paulet L, Dereeper A, Droc G, Guyot R, Pietrella M, et al. The coffee genome provides insight into the convergent evolution of caffeine biosynthesis. Science. 2014;345:1181–4. doi: 10.1126/science.1255274. [DOI] [PubMed] [Google Scholar]
- 102.Wang K, Wang Z, Li F, Ye W, Wang J, Song G, et al. The draft genome of a diploid cotton Gossypium raimondii. Nat Genet. 2012;44:1098–103. doi: 10.1038/ng.2371. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The dataset supporting the conclusions of this article are partly included within the article and its Additional files 1, 2, 3, 4 and 5.
Further datasets supporting the conclusions of this article are available at the following repositories. The peach genome sequence can be accessed at following databases URLs:
Phytozome (https://phytozome.jgi.doe.gov/pz/portal.html#!info?alias=Org_Ppersica);
Genome Database for Rosaceae (https://www.rosaceae.org/species/prunus_persica/genome_v2.0.a1);
Applied Genomic Institute (IGA) : (http://services.appliedgenomics.org/fgb2/iga/prunus_persica_v2/gbrowse/prunus_persica_v2/).
This Whole Genome Shotgun project had been deposited at DDBJ/EMBL/GenBank under the accession AKXU00000000. The version described in this paper is accessible at the accession AKXU02000000
Illumina short reads are deposited into NCBI Short Read Archive under accession numbers SRX2273835, SRX2381075, SRX2392647, SRX2392648, SRX2392650
SSR primer sequences were deposited into NCBI probe database under accession numbers from Pr032816704 to Pr032816814.