

Abstract
Previous studies have shown that activation of ventral midbrain NMDA receptors is required for the initiate of sensitization to amphetamine. In view of the recent evidence that neurotensin modulates ventral midbrain glutamate neurotransmission, we tested the hypothesis that neurotensin is acting upstream to glutamate to initiate sensitization to the behavioral and neurochemical effects of amphetamine. During a first testing phase, adult male rats implanted with bilateral VM cannulae were injected every second day for three days with D-[Tyr11]neurotensin (1.5 nmol/side), the preferred NMDA GluN2A/B antagonist, RS-CPP (40 or 120 pmol/side), the selective GluN2B antagonist, Ro04-5595 (200 or 1200 pmol/side), RS-CPP (40 or 120 pmol/side) + D-[Tyr11]neurotensin (1.5 nmol/side) or Ro04-5595 (200 or 1200 pmol/side) + D-[Tyr11]neurotensin (1.5 nmol/side) and locomotor activity was measured immediately after the injection. Five days after the last central injection, the locomotor response or the expression of phosphorylated extracellular signal-regulated kinase (pERK1/2) in neurons of different limbic nuclei was measured following a systemic injection of amphetamine sulfate (0.75 mg/kg, ip). Results show that amphetamine induced significantly stronger locomotor activity and pERK1/2 expression in the nucleus accumbens shell and infralimbic cortex in neurotensin pre-exposed animals than in controls (vehicle pre-exposed). These sensitization effects initiated by neurotensin were prevented by RS-CPP, but not Ro04-5595. These results support the hypothesis that neurotensin is stimulating glutamate neurotransmission to initiate neural changes that sub-serve amphetamine sensitization and that glutamate is acting on NMDA receptors that are mostly likely composed of GluN2A, but not GluN2B, subunits.
Keywords: Accumbens, Amphetamine, ERK, Glutamate, Neurotensin, Prefrontal Cortex
Introduction
Animals and humans that were pre-exposed to the psychostimulant drug, amphetamine, are more sensitive to its pharmacological and behavioral effects than naïve subjects, a phenomenon that is known as sensitization or reverse tolerance (Boileau et al, 2006; Vezina, 2007). Psychostimulant sensitization is an enduring phenomenon hypothesized to play a key role in vulnerability to addiction and relapse to compulsive drug intake (Robinson and Berridge, 1993; Steketee and Kalivas, 2011). While the exact mechanism(s) that sub-serve(s) the initiation of amphetamine sensitization is/are still incomplete, previous studies have revealed that it is the action of amphetamine on neurotransmitter release in the ventral midbrain that initiates the neural changes that lead to the sensitized state. Repeated microinjections of amphetamine into the ventral midbrain (VM), but not into the ventral striatum nor into the prefrontal cortex, initiate sensitization to the stimulant effect of amphetamine on locomotor activity and on dopamine release in the nucleus accumbens (Hooks et al, 1992; Kalivas and Weber, 1988; Vezina, 1993). Based on the current knowledge, amphetamine is initiating sensitization by increasing VM dopamine release which activates dopamine D1-like receptor leading to enhanced glutamate release from VM afferent terminals that originate from the prefrontal cortex (Bjijou et al, 1996; Stewart and Vezina, 1989; Cador et al, 1999). Glutamate is hypothesized to act on N-Methyl-D-Aspartate receptors (NMDAR) to enhance basic fibroblast growth factor production leading to neural plastic changes that sub-serve enhanced sensitivity to the psychostimulant drug (Cador et al, 2002; Flores et al, 1998).
More recent findings also suggest that neurotensin, a tridecapeptide found in a dense network of nerve terminals that innervates VM neurons, is most likely to be part of this neural cascade. Repeated central injections of neurotensin (NT), and its active analog, D-[Tyr11]NT, lead to an enhancement of amphetamine-induced-locomotor activity (Rompré, 1997), an effect that is also prevented by excitotoxic lesions of the prefrontal cortex, a cortical region that send glutamatergic efferent to VM neurons (Blackburn et al, 2004; Sesack and Pickel, 1992). Furthermore, Panayi et al (2005) have shown that the initiation of amphetamine sensitization by repeated systemic amphetamine injections is prevented by blockade of VM NT receptors. Electrophysiological results showing that NT enhances glutamatergic excitatory post-synaptic current in the VM neurons (Kempadoo et al., 2013; Bose et al., 2015; Rouibi et al., 2016), suggest that it may act upstream to glutamate to initiate amphetamine sensitization. The present study was aimed at testing this hypothesis. We investigated whether repeated VM NT microinjections sensitize to systemic amphetamine-induced locomotor activity and determined whether this sensitization effect was dependent upon activation of VM NMDAR. Two NMDAR antagonists were used, RS-CPP an antagonist at NMDAR composed of GluN2 subunits, and Ro04-5595, a selective antagonist at the GluN2B subunit of the NMDAR. These antagonists were chosen on the basis of previous studies showing that RS-CPP is effective at blocking the initiation of amphetamine sensitization when it is injected into the VM and that GluN2 (2A and 2B) subunits play a role in synaptic plasticity in VM neurons (Bjijou et al, 1996; Schilström et al, 2006). We also determined whether repeated VM NT microinjections sensitize to amphetamine-induced activation of extracellular signal-regulated kinase (ERK1/2) pathway in different regions of the limbic system and whether this effect was also dependent upon VM NMDAR activation. It is well established that amphetamine activates ERK1/2 in limbic regions known to play a key role in drug addiction, and that this signaling pathway is involved in long-term synaptic plasticity (Sweatt, 2004) initiated by drugs of abuse (Pan et al, 2011; Valjent et al, 2006; Zhai et al, 2008). Such an enhancement of amphetamine-induced ERK1/2 would be indicative of a major role of VM NT in the development of abnormal behaviors related to an alteration of limbic neurotransmission such as drug addiction.
Experimental procedures
Subjects
Adult male Long-Evans (Charles River Canada) rats weighing between 300–350g at the time of surgery were used. They were housed 1 (after surgery) or 2 per cage in a temperature (22°C) and humidity (40%) controlled room with a 12h light/dark cycle (lights on 06:00). They were allowed to habituate for 5 to 7 days to the housing environment before surgery and had free access to food and water. All procedures were in accordance with guidelines of the Canadian Council on Animal Care and all efforts were made to minimize suffering and number of animals used.
Surgery
Following the habituation period to the housing environment, rats were deeply anaesthetized with isoflurane (2.5–3.5%, O2 0.6L/min) and mounted on a stereotaxic apparatus. The surface of the skull was exposed between lambda and bregma and burr holes were made into the cranium in each hemisphere at the point of insertion of the guide cannulae (HRS Scientific, Montreal, Canada, model C315G). Four miniatures screws were threaded into the bone and guide cannulae were implanted using the following flat skull stereotaxic coordinates: 5.3–5.6 mm posterior to bregma, 1.6–1.8 mm lateral to the midline (10–12° medio-lateral angle) and 6.4 mm below the surface of the skull (Paxinos and Watson, 1998). The cannulae were closed with an obturator of the same length and were anchored to the skull with dental acrylic. A 0.05 mL of duplocillin LA containing 15,000 I.U. of penicillin was administered (i.m.) to prevent infections. The analgesic anaphen (5 mg/kg, sc) was administered at the end of the surgery and 24 h later.
Behavioral tests
The experimental paradigm consisted of a habituation phase, a training phase and a sensitization test (see Supplementary Figure S1). On the first day of the habituation phase, the animals were placed into the test cage for 45 min. On the second day, they were all injected with saline into the VM and placed in the test cage for 120 min. Bilateral microinjections were made by inserting into each guide cannula an injection cannula (model C315I) that extended 2 mm beyond the tip of the guide. Each cannula was connected with polyethylene tubing to a 2-μl microsyringe and a volume of 0.5 μl of sterile 0.9% saline was injected into each hemisphere simultaneously with a micro-infusion pump over a period of 60 sec; cannulae were left in place for an additional 60 sec to allow diffusion into the tissue. This first injection was aimed at habituating the animals to the procedure. On the third day, the animals were placed into the test cage for 120 min without any treatment. The training phase began the following day (day 4). During this phase, independent groups of animals were injected three times, every second day (day 4, 6 and 8), with vehicle (0.5 μl/side), D-[Tyr11]neurotensin [NT, 1.5 nmol/0.5 μl/side], RS-CPP (40 or 120 pmol/0.5 μl/side), Ro04-5595 (200 or 1200 pmol/0.5 μl/side), RS-CPP (40 or 120 pmol/0.5 μl/side) + NT (1.5 nmol/0.5 μl/side) or Ro04-5595 (200 or 1200 pmol/0.5 μl/side) + NT (1.5 nmol/0.5 μl/side) using the injection procedure described above. They were placed into the test cage immediately after the injection and locomotor activity was measured for 120 min. Five days after the last day of the training phase (day 13, sensitization test), all the animals were injected with a single dose of amphetamine (0.75 mg/kg, ip) and locomotor activity was measured for 120 min (behavioral sensitization test) or the animals were sacrificed after 15 min (neurochemical sensitization test, see below). Behavioral tests were conducted during the light cycle between 08:30 and 16:30 in a room separate from the housing colony with the light turned off; a 70–75 dB white noise was used to mask any external noise. We used the NT analog, D-[Tyr11]NT, because it is resistant to enzymatic degradation (Checler et al., 1983) and mimics several behavioral, neurochemical and physiological effects of NT. When injected repeatedly into the cerebral ventricle, D-[Tyr11]NT produces a stronger amphetamine sensitization response and potentiation of brain stimulation reward than NT at an equimolar concentration (Rompré, 1997; Rompré and Bauco, 2001). Ventral midbrain infusion of NT and [D-Tyr11]NT stimulates locomotor activity (Rompre and Bauco, 2003) and enhances mesoaccumbens DA release (Steinberg et al. 1995; Sotty et al. 2000a). But, when injected unilaterally, [D-Tyr11]NT is more effective than NT at inducing a circling behavior (Steinberg et al., 1995) but less effective than NT at enhancing mesoprefontal DA release (Sotty et al. 2000a). A recent study also shows that the NT analog modulates glutamatergic neurotransmission in different populations of VM neurons by acting on different NT receptors (Rouibi et al., 2015).
Apparatus
Locomotor activity was measured using an Opto-Varmex Auto Track System (Columbus Instruments, Columbus, OH, USA). Test cage consisted of four Plexiglass walls of 43 cm (wide) × 43 cm (length) and 33 cm (height) and a wire-mesh floor; they were equipped with two arrays of 15 infrared photocells located 1.5 and 14.5 cm above the wire-mesh floor to detect horizontal and vertical movements respectively. Computer software quantified ambulatory activity by calculating the distance traveled beyond a virtual box of 9.6 × 9.6 cm (3 × 3 photocells) drawn around the animal; the computer determined the location of the animal within the box 10 times per second. Movements detected within the virtual box were considered as non-ambulatory and were quantified as time (in sec) during which photocell beam interruptions were detected. Vertical activity was quantified as the total number of photocell beam interruptions produced by rearing (see Elmer et al, 1996 for validation data on these measures of activity).
Histology
At the end of the experiment, behaviorally tested animals were deeply anesthetized with urethane (2 g/kg, i.p.) and transcardially perfused with 0.9% saline followed by 10% formalin. Brains were removed, stored in 10% formalin and subsequently sliced in serial 40-μm sections that were stained with formal-thionin solution. Locations of the injection sites were determined under light microscopic examination. Only animals that had both injection sites within the ventral tegmental area, near or in the rostral and caudal linear nuclei, the paranigral, parabrachial and the interfascicular nuclei between 5.0 and 5.8 mm behind bregma (Paxinos and Watson, 1986) were included in the analyses.
Immunohistochemistry
On the day of the sensitization test (day 13), some animals in the following groups: vehicle (0.5 μl/side), NT (1.5 nmol/0.5 μl/side), RS-CPP (120 pmol/0.5 μl/side), Ro04-5595 (1200 pmol/0.5 μl/side), RS-CPP (120 pmol/0.5 μl/side) + NT (1.5 nmol/0.5 μl/side), and Ro04-5595 (1200 pmol/0.5 μl/side) + NT (1.5 nmol/0.5 μl/side) were sacrificed 15 min after the amphetamine injection; additional animals that were injected centrally with vehicle or NT (1.5 nmol/0.5 μl/side) alone during the training phase were injected systemically with the vehicle. They were anesthetized with isoflurane and perfused with phosphate-buffer (0.1 M, pH 7.4) paraformaldehyde (4%); the brains were removed and frozen at −80°C. Brains were subsequently sliced in 30 μm coronal sections with a cryostat. Slices were rinsed three times in H2O2 and in PBS 0.1M pH7.4; free floating slices were then blocked 30 min in a blocking solution (5% Goat serum, 1% Triton in PBS 0.1M) before being incubated overnight at 4°C with the primary antibody (anti-phospho Thr202-Tyr204 ERK1/2 1:500; Cell Signaling Technology). After three PBS rinses, the slices were incubated with the secondary antibody (anti-rabit IgG-Biotine 1:1000, Vector Lab) for 90 min and rinsed three times with PBS. Slices were treated with the avidin-biotin complex (ABC kit, Vector Lab.) for 90 min and immunoreactivity was revealed by 3,3-diaminobenzimide tetrahydrochloride (DAB) solution (Sigma–Aldrich) and H2O2 in TRIS 0.05M pH7.4. After washes, slices were mounted on slides and cover-slipped with DPX mounting medium (Sigma–Aldrich). Positive immunolabeled cells were manually counted by an observer blind to treatment using a bright field microscope (Zeiss axioskop). In each region, the amount of pERK1/2 positive neurons (evaluated on the basis of a cytoplasmic and nuclear staining) was counted on at least four different 30 μm slices for each brain region, for each animal, to give an averaged number of pERK1/2 positive cells for a given area (0.2 square millimeter or, the total surface of the structure analyzed). Representative images were taken with a SPOT RT Slider camera 2.3.1 and the SPOT imaging software 4.4 (SPOT imaging solution™, Sterling Height, MI).
Drugs
The NMDAR antagonists, RS-CPP [(RS)-3-(2-Carboxypiperazin-4-yl)-propyl-1-phosphonic acid)] and Ro04-5595 hydrochloride 1-[2-(4-Chlorophenyl)ethyl]-1,2,3,4-tetrahydro-6-methoxy-2-methyl-7-isoquinolinol hydrochloride) were purchased from Tocris Bioscience (Ellisville, MI, USA) and [D-Tyr11]neurotensin-(1-13) from Bachem (Sunnydale, CA, USA). They were dissolved in sterile 0.9% saline and stored frozen at −20°C in 40–50 μl aliquots. Peptide and drug solutions were thawed just before testing and used only once. Amphetamine sulphate was dissolved in saline and injected intraperitoneally in a volume of 1 ml/kg. Drug and peptide doses were chosen on the basis of previous studies (Bauco and Rompré, 2004; Bergeron and Rompré, 2013; Cador et al, 1999; Kalivas and Duffy, 1990).
Statistical analysis
Measures of locomotor activity (distance traveled, time of non-ambulatory activity and vertical counts) and number of pERK positive neurons, were totaled for all subjects and group means were calculated. Data were analyzed with a two-way ANOVA (treatment x day) with repeated measures on day (training phase) or a one-way ANOVA (sensitization test and immunohistochemistry results) and differences among means were determined with Duncan’s multiple range post-hoc tests. Student T-test was used to compare means of a given treatment obtained at different days. The accepted value for significance was P < 0.05 (Statistica V5.0, StatSoft; IBM SPSS Version 23).
RESULTS
Histology
Location of the injection sites for each group of animals that were included in the behavioral and the neurochemical analyses are shown in Figure 1 and 2 respectively. Sites were located within the ventral tegmental area between a rostro-caudal region extending between 5.0 and 5.8 mm posterior to bregma, a region that is densely innervated by NT-containing terminals and NT receptors (Geisler and Zahm, 2006; Szigethy and Beaudet, 1989).
Figure 1.

Location of injection sites for each animal included in the behavioral study. Filled circles indicate the location of the injections for those animals that were injected with the vehicle, CPP or Ro04-5595 alone (left panels). Filled stars indicate the location of the injections for those animals that were injected with the neurotensin (NT), CPP + NT or Ro04-5595 + NT alone (right panels). Illustrations are modified drawings from Paxinos and Watson (1986; numbers in the middle represent the anterior-posterior distance from bregma.
Figure 2.

Location of injection sites for each animal included in the immunohistochemical study. See legend of figure 1 for details.
Training Phase
Microinjections of NT into the VM produced a significant increase in ambulatory and non ambulatory activity, an effect that was stronger after the third injection on day 5 (Figure 3, top and middle panels). The ANOVA yielded a significant treatment by day interaction (Ambulatory activity, F5,87 = 9.0 p < 0.001; non-ambulatory activity, F5,87 = 4,4 p < 0.001) and post-hoc test confirmed that the magnitude of the activity measured after the third NT injection (Day 5) was significantly higher than that measured after the first injection on day 1. Blockade of VM NMDAR with CPP did not attenuate the stimulant effect of NT (Figure 3, top left panel), but slightly reduced, at the low dose, the enhancement that was observed from day 1 to day 5. When administered alone, RS-CPP had no effect on ambulatory activity, but slightly enhanced non-ambulatory activity; this effect did not change with repeated injections (Figure 3, middle panels). Repeated VM injections of NT had very similar effects on non-ambulatory activity (Figure 3, middle panels). After the first injection, vertical activity was slightly increased by NT but this effect reached statistical significance only after the third injection (Figure 3, bottom panels). The ANOVA yielded a significant treatment by day interaction (F5,87 = 3.34, p < 0.05) and post-hoc test confirmed that vertical activity measured on day 5 after NT was significantly higher than on day 1. CPP did not block the stimulant effect of NT on vertical activity and when given alone it produced no change in vertical movements.
Figure 3.

Ambulatory (top panels), non-ambulatory (middle panels) and vertical (bottom panels) activity measured over the two-hour test period during the training phase after the first (left panels) and the third (right panels) VM microinjections of vehicle (VEH, n = 22), 1.5 nmol of D-Tyr[11]Neurotensin (NT, n = 23), 40 pmol of CPP + NT (CPP40 + NT, n = 6), 120 pmol of CPP + NT (CPP120 + NT, n = 18), 40 pmol (CPP40, n = 8) or 120 of CPP (CPP120, n = 16) alone. Each bar represents the group mean ± s.e.m. Symbols indicate a statistically significant difference with respective VEH (*p < 0.05; **p < 0.01; ***p < 0.001) with respective NT group (+p < 0.05; +++p < 0.001) or between the first and the third injections for the corresponding treatment group (#p < 0.05; ##p < 0.01; ###p < 0.001). See method section for details.
The selective GluN2B antagonist, Ro04-5595, when co-administered with NT did not block its stimulant effect on ambulatory and non-ambulatory activity nor its enhancement following repeated injections (Figure 4, top and middle panels); on the contrary, it enhanced at the high dose. The ANOVA yielded a significant treatment by day interaction (ambulatory activity, F5,76 = 11.1, p < 0.001; non-ambulatory activity, F5,76 = 4.1, p < 0.01) and post-hoc test confirmed that the level of activity measured following co-injection of the high dose of Ro04-5595 with NT was significantly higher than NT alone after the first injection. After the third injection, it enhanced the stimulant effect of NT at both doses. When administered alone, Ro04-5595 did not alter ambulatory and non-ambulatory activity neither on day 1 nor on day 5. NT-induced enhancement of vertical activity was also enhanced by the high dose of Ro04-5595, an effect that increased with repeated injections (F5,76 = 5.7, p < 0.001; Figure 3, lower panels). Each dose of Ro04-5595 failed to alter vertical activity when administered alone.
Figure 4.

Ambulatory (top panels), non-ambulatory (middle panels) and vertical (bottom panels) activity measured over the two-hour test period during the training phase after the first (left panels) and the third (right panels) VM microinjections of vehicle (VEH, n = 22), 1.5 nmol of D-Tyr[11]Neurotensin (NT, n = 23), 200 pmol of Ro04-5595 + NT (Ro200 + NT, n = 5), 1200 pmol of Ro04-5595 + NT (Ro1200 + NT, n = 14), 200 pmol (Ro200, n = 5) or 120 of Ro04-5595 (Ro1200, n = 13) alone. Each bar represents the group mean ± s.e.m. Symbols indicate a statistically significant difference with respective VEH (*p < 0.05; ***p < 0.001) with respective NT group (+p < 0.05; +++p < 0.001) or between the first ant the third injections for the corresponding treatment group (##p < 0.01; ###p < 0.001). See method section for details.
Sensitization test
Locomotor activity
The amphetamine test performed five days after the last VM microinjections, revealed that animals pre-exposed to NT displayed higher ambulatory (F5,53 = 5.8, p < 0.001), non-ambulatory (F5,53 = 4.0, p < 0.01) and vertical activity (F5,53 = 8.1, p < 0.001) than the animals that were pre-exposed to the vehicle (Figure 5). This sensitization effect was not observed in the animals pre-exposed to a combination of each dose of CPP with NT as their levels of activity following amphetamine injection were not different than the activity of the animals pre-exposed to vehicle (P > 0.05). There is also a significant difference in the response to amphetamine between the group pre-exposed to NT alone and the two groups pre-exposed to CPP + NT (except low dose and non-ambulatory activity) showing that CPP blocked the development of amphetamine sensitization by NT. This blockade cannot be attributed to a reduction effect of CPP on the sensitivity to amphetamine as pre-exposure to each dose of CPP alone had no consequence on amphetamine-induced locomotor activity.
Figure 5.

Ambulatory (top panel), non-ambulatory (middle panel) and vertical (bottom panel) activity measured over the two-hour test period during the sensitization phase after a systemic injection of amphetamine from the groups pre-exposed to the vehicle (VEH, n=14), D-Tyr[11]Neurotensin (NT, n=16), CPP (40 or 120 pmol) with NT (CPP40+NT, n=5; CPP120+NT, n=8) and CPP alone (CPP40, n=8; CPP120, n=7). Each bar represents the group mean ± s.e.m. Symbols indicate a statistically significant difference with VEH (**p < 0.01) or with NT (+p < 0.05; ++p < 0.01).
Contrary to CPP, the selective GluN2B antagonist, Ro04-5595, did not block the development of sensitization to amphetamine-induced locomotor activity (Figure 6). The ANOVA yielded a significant effect of pre-treatment (ambulatory activity: F5,51 = 11.1, p < 0.001; non-ambulatory activity: F5,51 = 7.8, p <0.001; vertical activity: F5,51 = 7.7, p < 0.001) and post-hoc test showed that animals pre-treated with the low or with the high dose of Ro04-5595 in combination with NT showed an enhanced locomotor response to amphetamine similar to those pre-treated with NT alone. In addition, repeated VM injections of each dose of the GluN2B antagonist alone did not alter the response to amphetamine.
Figure 6.

Ambulatory (top panel), non-ambulatory (middle panel) and vertical (bottom panel) activity measured over the-two hour test period during the sensitization phase after a systemic injection of amphetamine from the group pre-exposed to the vehicle (VEH, n=14), D-Tyr[11]Neurotensin (NT, n=16), Ro04-5595 (200 or 1200 pmol) with NT (Ro200+NT, n=7; Ro1200+NT, n=7) and Ro04-5595 alone (Ro200, n=6; Ro1200, n=7). Each bar represents the group mean ± s.e.m. Stars indicate a statistically significant difference with VEH (*p < 0.05; **p < 0.01; ***p < 0.001).
pERK1/2 immunohistochemistry
Results of the immunohistological analyses performed on different groups of animals treated with amphetamine 5 days after the last VM microinjection (day 13) are presented in Figure 7 and 8. In the in the shell of the nucleus accumbens and in infralimbic part of the prefrontal cortex (Fig. 7 B,C) the number of pERK1/2 positive cells was significantly higher in the animals pre-exposed to NT than in those pre-exposed to the vehicle (infralimbic, F5,34 = 4.78, p < 0.01; shell, F5,34 = 11.77, p < 0.001) (Fig. 7C). The sensitization effect of NT on amphetamine-induced expression of pERK1/2 in these regions was blocked by CPP, but not by Ro04-5595, immunohistochemical results that parallel the behavioral results. Pre-exposure to CPP and to Ro04-5595 alone did not alter amphetamine-induced pERK1/2 in the infralimbic cortex as the number of positive cells was not different than vehicle. Similar results were obtained in the shell with the except that pre-exposure to Ro04-5595 increased amphetamine-induced pERK1/2 in this region. In addition, the NT and vehicle pre-exposed groups injected with saline do not differ (P > 0.05).
Figure 7.
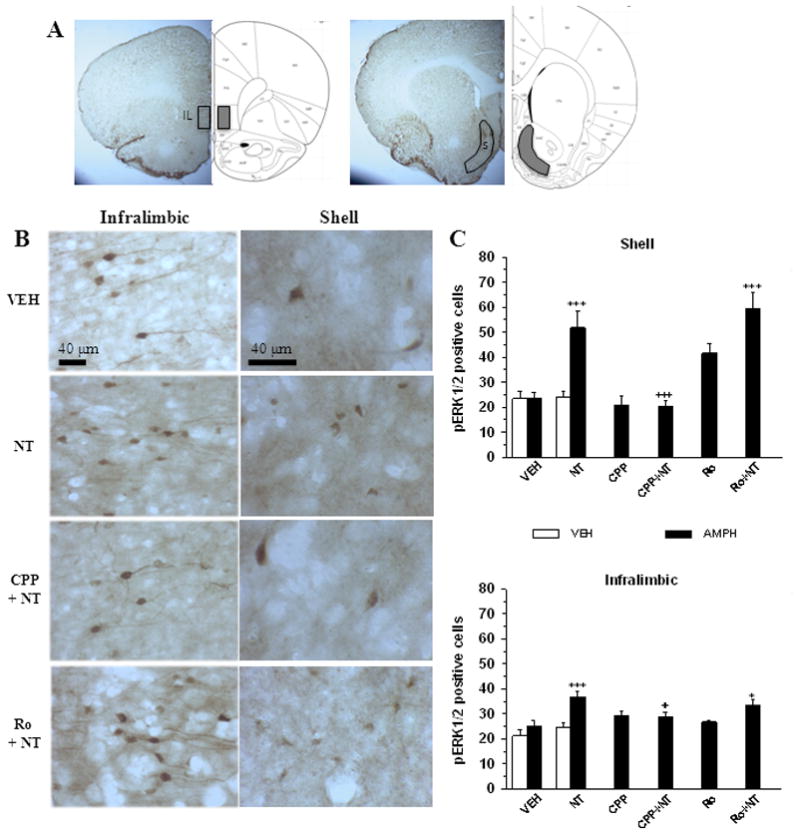
Amphetamine-induced pERK1/2 in the shell of the nucleus accumbens and the infralimbic cortex in animals pre-exposed to VM neurotensin. A. Representative coronal brain slices depicting the regions of infralimbic cortex (IL) and nucleus accumbens shell (S) where pERK1/2 immunoreactivity were sampled. B. Representative photomicrophaphs illustrating pERK1/2 immunoreactivity in the infralimbic cortex and shell of the nucleus accumbens in coronal brain slices from animals injected with systemic amphetamine (0.75 mg/kg, i.p.). Legend on the left indicates the treatment that each animal received during the training phase: Vehicle (VEH); D-Tyr[11]Neurotensin (NT); CPP + NT (CPP + NT); Ro04-5595 + NT (Ro + NT). C. Group means (± s.e.m) total number (shell) or total number per 0.2 mm2 (infralimbic) of pERK1/2 positive cells quantified in the shell (top panel) and the infralimbic cortex (bottom panel) after systemic vehicle (VEH, white) or amphetamine (AMPH, black) in different groups of animals that were injected during the training phase with VEH (n = 7 in each group), 1.5 nmol/side of D-Tyr[11]Neurotensin (NT; n = 7 in each group), 120 pmol/side of CPP with or without NT (CPP + NT and CPP, n = 6 in each group), or 1200 pmol/side of Ro04-5595 with or without NT (Ro + NT, n = 7; Ro, n = 6). Symbols indicate a statistically significant difference with VEH + AMPH group (*p < 0.05; ***p < 0.001) or with NT + AMPH (+p < 0.05; +++p < 0.001). See text for details.
Figure 8.

Group means (± s.e.m) total or total number per 0.2 mm2 (prelimbic) of pERK1/2 positive cells measured in different limbic nuclei after systemic vehicle (VEH) or amphetamine (AMPH) administration in different group of animals that were injected intra-VM during the training phase with VEH, 1.5 nmol/side of D-Tyr[11]Neurotensin (NT), 120 pmol/side of CPP with or without NT (CPP + NT and CPP), or 1200 pmol/side of Ro04-5595 with or without NT (Ro + NT). Stars indicate a statistically significant difference with the VEH (**p < 0.01; ***p < 0.001) for the VEH and NT pr-injected groups. See legend of figure 7 and text for details.
Immuhistochemical analyses were performed in several other limbic brain regions following an injection of saline and amphetamine on day 13 (Figure 8). In the nucleus accumbens core, the medial VM and the peduncopontine tegmental nucleus amphetamine enhanced pERK1/2 in both the VEH and NT pre-injected group (saline vs amphetamine, P < 0.05). Such an increase in pERK1/2 was not seen in the prelimbic cortex, the lateral VM and the laterodorsal tegmental nucleus, following amphetamine injection. For each of these regions, the number of pERK positive neurons measured after amphetamine injection was near in each group and consistent the ANOVA yielded to effect of pretreatment (P > 0.05).
Discussion
NMDAR-dependent sensitization to the locomotor activity
The present study was aimed at testing the hypothesis that NT is acting upstream to glutamate in the VM to initiate sensitization to the behavioral effect of systemic amphetamine. A first important finding that supports the hypothesis is that repeated VM microinjections of NT lead to an enhancement of the locomotor stimulant effect of amphetamine. Animals that were pre-treated with VM NT displayed a stronger locomotor response to systemic amphetamine than those pretreated with VM vehicle. The second important finding that supports the hypothesis is that sensitization effect induced by NT was blocked by the selective NMDAR antagonist, CPP.
The induction of amphetamine sensitization by VM NT is consistent with previous results showing senstization is induced by repeated intracerebroventricular injections of NT, or its active analog, D-[Tyr11]NT, and prevented by VM microinjection of the NTS1/NTS2 receptor antagonist, SR142948 (Panayi et al., 2005; (Rompré, 1997). It is inconsistent, however, with the lack of effect of repeated VM microinjections of NT on the stimulant effect of systemic amphetamine on locomotion reported by Elliott and Nemeroff (1986). In this latter study, however, the amphetamine test was performed 24h after the last VM NT microinjection, a time that is most likely insufficient to allow neural changes that sub-serve the expression of sensitization to take place (Vanderscuren et al, 1999). The mechanism by which NT induces amphetamine sensitization remains speculative. A first hypothesis is a desensitization of the dopamine D2 autoreceptors. Both in vitro and in vivo experiments have shown that activation of VM NT receptors results in a reduction of D2 autoreceptor sensitivity (Shi and Bunney, 1992; Thibault et al, 2011), a phenomenon that is also observed following repeated amphetamine treatment (White and Kalivas, 1998; Vanderschuren and Kalivas, 2000). Since it is a short lasting effect, it has been proposed to account for the sensitization effect measured during the short- (days), but not the long-term withdrawal (weeks). In the present study, we performed a sensitization test five days after withdrawal from NT, a period that is within the time course of D2 autoreceptor subsensitivity. To determine whether the sensitization effect last longer, we carried out an additional experiment with two new groups that injected on three occasions, every second day, with the vehicle or NT 3; the amphetamine test was performed three weeks after withdrawal (after the last VM injection). Results show that the sensitization effect of NT is persistent (supplementary Figure S2) and is unlikely due solely to D2 autoreceptor subsensitivity. It thus appears likely that neurotensin initiates the same VM neural cascade that is initiated by amphetamine itself leading to the long-term neural changes that subserve sensitization. It has been shown previously that activation of VM NMDAR is a critical step in this cascade. Indeed, amphetamine sensitization is blocked by VM co-injection of the NMDAR antagonist, CPP with amphetamine (Cador et al, 1999; Vezina and Queen, 2000). Consistently, our results show that NMDAR activation is required for the initiation of amphetamine sensitization by NT. Such a role for NMDAR is consistent with the modulation that NT exerts on VM glutamate neurotransmission. Recent studies, for instance, reported an enhancement of glutamatergic excitatory post-synaptic currents (EPSCs) recorded from VM neurons by endogenous and exogenous NT (Kempadoo et al, 2013; Bose et al 2015). Neurotensin, however, induced a biphasic effect, an enhancement at low concentration and a reduction in EPSCs at high concentration (Kempadoo et al, 2013). An attenuation of glutamatergic EPSCs by NT has also been reported by Kortleven and Trudeau (2012) in mice. Interestingly, Roubi et al (2015) have reported that NT analog, D-[Tyr11]NT, produces opposite effects on EPSCs recorded from different population of VM neurons. In those neurons that display a hyperpolarization-activated cationic (Ih) current, D-[Tyr11]NT attenuated the EPSCs while in those that do not have a Ih current it enhanced it. Because, VM neurons that lack an Ih current are non-dopamine neurons, it is possible that D-[Tyr11]NT acted on these to initiate sensitization. Some support for this hypothesis comes from Luo et al’s results (2010) showing that a glutamatergic-dependent sensitization to cocaine can still be induced after deletion of NMDAR from VM dopamine neurons. It is well established that cocaine, like amphetamine, sensitization is dependent upon VM NMDAR activation (see Vandeschuren and Kalivas, 2000).
Roubi et al (2015) also found that the enhancement glutamatergic EPSCs in non-dopamine neurons by D-[Tyr11]NT is blocked by the NTS1/NTS2 antagonist, SR142948, but not the preferred NTS1 antagonist, SR48692. This finding together with that of Panayi et al (2005) showing that the initiation of amphetamine sensitization is blocked by VM co-injection of SR142948 strongly suggest that D-[Tyr11]NT acted on VM NTS2 receptors leading to enhancement of glutamatergic EPSCs in non-dopamine neurons.
A NMDAR is composed of four sub-units, two GluN1 sub-units forming a hetero-or trihetero-tetramer with two additional sub-units that could be GluN2A-D and/or GluN3A or 3B (McBain and Mayer, 1994). Electrophysiological studies suggest VM dopamine neurons expressed NMDARs that contain GluN2B and/or GluN2D (Jones and Gibb, 2005; Suarez et al, 2010). The hypothesis that VM dopamine neurons are not involved in the initiation of amphetamine sensitization by NT is supported by the results we obtained with Ro04-5595. Unlike CPP, this selective GluN2B antagonist was ineffective at blocking the sensitization effect of NT. It is unlikely that the ineffectiveness of Ro04-5595 was due to the range of doses that we used. During the training phase, for instance (see below), Ro04-5595 enhanced the acute stimulant effect of NT on locomotor activity, an effect that increased with repeated injections at both doses.
NMDAR-dependent sensitization to pERK1/2 activation
Another important finding is that repeated VM NT microinjections sensitize to amphetamine-induced pERK1/2 in the shell of the nucleus accumbens and the infralimbic part of the prefrontal cortex. Moreover, this neurochemical sensitization effect of amphetamine was blocked by CPP, but not by Ro04-5595, which paralleled behavioral results. Previous studies have shown that acute amphetamine enhances pERK1/2 in the nucleus accumbens and the prefrontal cortex, but in general this enhancement is observed at much higher doses that the one used in the present study (0.75 mg/kg; see Zhai et al 2008). This further reinforces the hypothesis that VM NT produced an enhancement of the sensitivity of this limbic circuitry to the psychostimulant drug. The MAP kinase pathway plays a key role in drugs-induced neuro-adaptations that underlie enhanced drug reward and conditioned drug effects and the shell region of the nucleus accumbens mediates amphetamine reward (Sellings and Clarke, 2003). Direct microinjections of amphetamine into this nucleus induce a conditioned place-preference that is dependent upon ERK1/2 activation (Gerdjikov et al, 2004). An increase of VM NT neurotransmission is thus likely to lead to long-term enhancement of amphetamine reward. While the precise signaling cascade mediating ERK1/2 activation in the prefrontal cortex following acute amphetamine needs further studies, it’s phosphorylation in the nucleus accumbens has been attributed to a dopamine/glutamate receptors interplay (Pascoli et al, 2014; Valjent et al, 2005). It is interesting to note that the other region in which we found a sensitize pERK1/2 activation is the infralimbic cortex, a cortical area that sends glutamatergic efferents to the shell (see Voorn et al, 2004).
NMDAR-independent stimulation of locomotor activity by VM NT
During the training phase, the NT analog that we used, D-[Tyr11]NT, produced a significant enhancement of locomotor activity an effect that is potentiated with repeated injections. Such a progressive increase was shown previous with NT itself and its c-terminal fragment NT-8-13 (Kalivas and Taylor, 1988). This shows that the NT analog can fully mimics the effect of the native neuropeptide. Interestingly, NMDAR antagonists were ineffective at attenuating the acute stimulant effect of NT on locomotor activity, hence showing that NMDAR are not involved. This represents a complete dissociation between the mechanisms that sub-serve the acute effect of NT and the neural plastic changes that it induces. Such a dissociation further reinforces the hypothesis that the acute effect of treatment, observed during the training phase, is not predictive of its long-term consequences. Microinjection of amphetamine into the nucleus accumbens for instance enhances locomotor activity but failed to induce a long-term sensitized effect. In the opposite, VM microinjections of amphetamine failed to alter locomotor activity but lead to a sensitization effect (See Kalivas and Vanderschuren, 2000; Vezina 2007). The most likely mechanism by which acute NT stimulates locomotor activity is an activation of NT receptors on mesolimbic dopamine neurons. A large number of NT receptors are expressed on dopamine neurons and their activation stimulates dopamine cell firing and release in the nucleus accumbens (Boudin et al, 1996; Leonetti et al, 2002; St-Gelais et al, 2004). Such an increase in dopamine neurotransmission likely accounts for the increase in locomotor activity (see Bozart and Wise, 1987).
Neurotensin is a potent modulator of limbic neurotransmission. Depending upon its site of action it may enhance or attenuate behavioural effects produce by psychostimulant drugs. Here we show that its action within the VM leads to a change in sensitivity of the limbic circuitry to a psychostimulant drug. Moreover, the selective increase of pERK1/2 levels in two limbic nuclei that play a key role in motivation, learning and cognition provides additional evidence that an increase in VM neurotensinergic neurotransmission is most likely to contribute to the development of drug addiction.
Supplementary Material
Diagram illustrating the experimental paradigm. On day 2, all the animals received bilateral ventral midbrain (VM) microinjections of sterile saline (0.5μl/side). On day 4 (1st), 6 (2nd) and 8 (3rd injection) of the training phase different groups received bilateral VM microinjections according to the design described in the method section. On day 13 (sensitization phase), all the animals were injected with amphetamine sulphate (0.75 mg/kg, ip). On each injection day the animals were put in the test cage for two hours.
Ambulatory (top panel), non-ambulatory (middle panel) and vertical (bottom panel) activity measured 21 days after the training phase following a systemic injection of amphetamine (0.75 mg/kg, ip) in two different groups. The legend on the x-axis indicates the pretreatment (VM microinjection) that each group received during the training phase: Vehicle (VEH, n = 6) or D-Tyr[11]Neurotensin (NT, n = 7). Each bar represents the mean (± s.e.m) total activity measured over the two-hour test period. Stars indicate a statistically significant difference with VEH (*p < 0.05; ***p < 0.001).
Acknowledgments
Funding
This work was supported by the Canadian Institutes of Health Research Funding (grant #102572).
Authors express their special thanks to Claude Bouchard for is excellent assistance.
Footnotes
Author disclosure
DV, DL and PPR designed the experiments; DV and DL carried out the neurochemical studies and analyzed the data; PPR analyzed the behavioral data. All authors contributed to the final version of the paper.
Conflict of interest
None
References
- Bauco P, Rompré PP. Central neurotensin receptor activation produces differential behavioral responses in Fischer and Lewis rats. Psychopharmacology (Berl) 2003;168:253–261. doi: 10.1007/s00213-003-1436-8. [DOI] [PubMed] [Google Scholar]
- Bergeron S, Rompré PP. Blockade of ventral midbrain NMDA receptors enhances brain stimulation reward: a preferential role for GluN2A subunits. Eur Neuropsychopharmacol. 2013;23:1623–1635. doi: 10.1016/j.euroneuro.2012.12.005. [DOI] [PubMed] [Google Scholar]
- Bjijou Y, Stinus L, Le Moal M, Cador M. Evidence for selective involvement of dopamine D1 receptors of the ventral tegmental area in the behavioural sensitization induced by intra-ventral tegmental area injections of D-amphetamine. J Pharmacol Exp Ther. 1996;277:1177–1187. [PubMed] [Google Scholar]
- Blackburn A, Dewar KM, Bauco P, Rompré PP. Excitotoxic lesions of the prefrontal cortex attenuate the potentiation of amphetamine-induced locomotion by repeated neurotensin receptor activation. Brain Res. 2004;998:184–193. doi: 10.1016/j.brainres.2003.11.022. [DOI] [PubMed] [Google Scholar]
- Boileau I, Dagher A, Leyton M, Gunn RN, Baker GB, Diksic M. Modeling sensitization to stimulants in humans: an [11C]raclopride/positron emission tomography study in healthy men. Arch Gen Psychiatry. 2006;63:1386–1395. doi: 10.1001/archpsyc.63.12.1386. [DOI] [PubMed] [Google Scholar]
- Boudin H, Pélaprat D, Rostène W, Beaudet A. Cellular distribution of neurotensin receptors in rat brain: immunohistochemical study using an antipeptide antibody against the cloned high affinity receptor. J Comp Neurol. 1996;373:76–89. doi: 10.1002/(SICI)1096-9861(19960909)373:1<76::AID-CNE7>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Cador M, Bjijou Y, Cailhol S, Stinus L. D-amphetamine-induced behavioural sensitization: implication of a glutamatergic medial prefrontal cortex-ventral tegmental area innervation. Neuroscience. 1999;94:705–721. doi: 10.1016/s0306-4522(99)00361-9. [DOI] [PubMed] [Google Scholar]
- Checler F, Vincent JP, Kitabgi P. Neurotensin analogs [D-TYR11] and [D-PHE11]neurotensin resist degradation by brain peptidases in vitro and in vivo. J Pharmacol Exp Ther. 1983;227:743–748. [PubMed] [Google Scholar]
- Elliott PJ, Nemeroff CB. Repeated neurotensin administration in the ventral tegmental area: effects on baseline and D-amphetamine-induced locomotor activity. Neurosci Lett. 1986;68:239–244. doi: 10.1016/0304-3940(86)90149-7. [DOI] [PubMed] [Google Scholar]
- Elmer GI, Brockington A, Gorelick DA, Carrol FI, Rice KC, Matecka D. Cocaine cross-sensitization to dopamine uptake inhibitors: unique effects of GNR12909. Pharmacol Biochem Behav. 1996;53:911–918. doi: 10.1016/0091-3057(95)02143-4. [DOI] [PubMed] [Google Scholar]
- Flores C, Stewart J. Basic fibroblast growth factor as a mediator of the effects of glutamate in the development of long-lasting sensitization to stimulant drugs: studies in the rat. Psychopharmacology (Berl) 2000;151:152–165. doi: 10.1007/s002130000417. [DOI] [PubMed] [Google Scholar]
- Geisler S, Zahm DS. On the retention of neurotensin in the ventral tegmental area (VTA) despite destruction of the main neurotensinergic afferents of the VTA--implications for the organization of forebrain projections to the VTA. Brain Res. 2006;1087:87–104. doi: 10.1016/j.brainres.2006.02.108. [DOI] [PubMed] [Google Scholar]
- Gerdjikov TV, Ross GM, Beninger RJ. Place preference induced by nucleus accumbens amphetamine is impaired by antagonists of ERK or p38 MAP kinases in rats. Behav Neurosci. 2004;118:740–750. doi: 10.1037/0735-7044.118.4.740. [DOI] [PubMed] [Google Scholar]
- Hooks MS, Jones GH, Liem BJ, Justice JB., Jr Sensitization and individual differences to IP amphetamine, cocaine, or caffeine following repeated intracranial amphetamine infusions. Pharmacol Biochem Behav. 1992;43:815–823. doi: 10.1016/0091-3057(92)90413-a. [DOI] [PubMed] [Google Scholar]
- Jones S, Gibb AJ. Functional NR2B- and NR2D-containing NMDA receptor channels in rat substantia nigra dopaminergic neurones. J Physiol. 2005;569:209–221. doi: 10.1113/jphysiol.2005.095554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW, Duffy P. Effect of acute and daily neurotensin and enkephalin treatments on extracellular dopamine in the nucleus accumbens. J Neurosci. 1990;10:2940–2949. doi: 10.1523/JNEUROSCI.10-09-02940.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW, Weber B. Amphetamine injection into the ventral mesencephalon sensitizes rats to peripheral amphetamine and cocaine. J Pharmacol Exp Ther. 1988;245:1095–1102. [PubMed] [Google Scholar]
- Kempadoo KA, Tourino C, Cho SL, Magnani F, Leinninger GM, Stuber GD. Hypothalamic neurotensin projections promote reward by enhancing glutamate transmission in the VTA. J Neurosci. 2013;33:7618–7626. doi: 10.1523/JNEUROSCI.2588-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortleven C, Bruneau LC, Trudeau LE. Neurotensin inhibits glutamate-mediated synaptic inputs onto ventral tegmental area dopamine neurons through the release of the endocannabinoid 2-AG. Neuropharmacology. 2012;63:983–991. doi: 10.1016/j.neuropharm.2012.07.037. [DOI] [PubMed] [Google Scholar]
- Leonetti M, Brun P, Sotty F, Steinberg R, Soubrie P, Bert L. The neurotensin antagonist SR142948A blocks the efflux of dopamine evoked in nucleus accumbens by neurotensin ejection into the ventral tegmental area. Naunyn-Schmiedeberg’s Arch Pharmacol. 2002;365:427–433. doi: 10.1007/s00210-002-0574-6. [DOI] [PubMed] [Google Scholar]
- Luo Y, Good CH, Diaz-Ruiz O, Zhang Y, Hoffman AF, Shan L. NMDA receptors on non-dopaminergic neurons in the VTA support cocaine sensitization. PLoS One. 2010;5:e12141. doi: 10.1371/journal.pone.0012141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBain CJ, Mayer ML. N-methyl-D-aspartic acid receptor structure and function. Physiol Rev. 1994;74:723–760. doi: 10.1152/physrev.1994.74.3.723. [DOI] [PubMed] [Google Scholar]
- Pan B, Zhong P, Sun D, Liu QS. Extracellular signal-regulated kinase signaling in the ventral tegmental area mediates cocaine-induced synaptic plasticity and rewarding effects. J Neurosci. 2011;31:11244–11255. doi: 10.1523/JNEUROSCI.1040-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panayi F, Colussi-Mas J, Lambas-Senas L, Renaud B, Scarna H, Berod A. Endogenous neurotensin in the ventral tegmental area contributes to amphetamine behavioral sensitization. Neuropsychopharmacol. 2005;30:871–879. doi: 10.1038/sj.npp.1300638. [DOI] [PubMed] [Google Scholar]
- Pascoli V, Cahill E, Bellivier F, Caboche J, Vanhoutte P. Extracellular signal-regulated protein kinases 1 and 2 activation by addictive drugs: A signal toward pathological adaptation. Biol Psychiatry. 2014;76:917–926. doi: 10.1016/j.biopsych.2014.04.005. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. Academic Press; New York: 1986. [Google Scholar]
- Robinson TE, Berridge KC. The neural basis of drug craving: an incentive-sensitization theory of addiction. Brain Res Rev. 1993;18:247–291. doi: 10.1016/0165-0173(93)90013-p. [DOI] [PubMed] [Google Scholar]
- Rompre PP. Repeated activation of neurotensin receptors sensitizes to the stimulant effect of amphetamine. Eur J Pharmacol. 1997;328:131–134. doi: 10.1016/s0014-2999(97)00159-3. [DOI] [PubMed] [Google Scholar]
- Roubi K, Bose P, Rompré PP, Warren RA. Ventral midbrain NTS1 receptors mediate conditioned reward induced by the neurotensin analog, D-Tyr[11]neurotensin. Front Neurosci. 2015;9:470. doi: 10.3389/fnins.2015.00470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiltröm B, Yaka R, Argilli E, Survana N, Schumann J, Chen BT. Cocaine enhances NMDA receptor-mediated currents in ventral tegmental area cells via dopamine D5 receptor-dependent redistribution of NMDA receptors. J Neurosci. 2006;26:8549–8558. doi: 10.1523/JNEUROSCI.5179-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellings LH, Clarke PB. Segregation of amphetamine reward and locomotor stimulation between nucleus accumbens medial shell and core. J Neurosci. 2003;23:6295–6303. doi: 10.1523/JNEUROSCI.23-15-06295.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sesack SR, Pickel VM. Prefrontal cortical efferents in the rat synapse on unlabeled neuronal targets of catecholamine terminals in the nucleus accumbens septi and on dopamine neurons in the ventral tegmental area. J Comp Neurol. 1992;320:145–160. doi: 10.1002/cne.903200202. [DOI] [PubMed] [Google Scholar]
- Shi WS, Bunney BS. Neurotensin attenuates dopamine D2 agonist quinpirole-induced inhibition of midbrain dopamine neurons. Neuropharmacolgy. 1990;29:1095–1097. doi: 10.1016/0028-3908(90)90119-c. [DOI] [PubMed] [Google Scholar]
- St-Gelais F, Legault M, Bourque MJ, Rompre PP, Trudeau LE. Role of calcium in neurotensin-evoked enhancement in firing in mesencephalic dopamine neurons. J Neurosci. 2004;24:2566–2574. doi: 10.1523/JNEUROSCI.5376-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steketee JD, Kalivas PW. Drug wanting: behavioral sensitization and relapse to drug-seeking behavior. Pharmacol Rev. 2011;63:348–365. doi: 10.1124/pr.109.001933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart J, Vezina P. Microinjections of Sch-23390 into the ventral tegmental area and substantia nigra pars reticulata attenuate the development of sensitization to the locomotor activating effects of systemic amphetamine. Brain Res. 1989;495:401–406. doi: 10.1016/0006-8993(89)90236-9. [DOI] [PubMed] [Google Scholar]
- Suarez F, Zhao Q, Monaghan DT, Jane DE, Jones S, Gibb AJ. Functional heterogeneity of NMDA receptors in rat substantia nigra pars compacta and reticulata neurones. Eur J Neurosci. 2010;32:359–367. doi: 10.1111/j.1460-9568.2010.07298.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweatt JD. Mitogen-activated protein kinases in synaptic plasticity and memory. Curr Opin Neurobiol. 2004;14:311–317. doi: 10.1016/j.conb.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Szigethy E, Beaudet A. Correspondence between high affinity 125I-neurotensin binding sites and dopaminergic neurons in the rat substantia nigra and ventral tegmental area: a combined radioautographic and immunohistochemical light microscopic study. J Comp Neurol. 1989;276:128–137. doi: 10.1002/cne.902790111. [DOI] [PubMed] [Google Scholar]
- Thibault D, Albert PR, Pineyro G, Trudeau LE. Neurotensin triggers dopamine D2 receptor desensitization through a protein kinase C and beta-arrestin1-dependent mechanism. J Biol Chem. 2011;286:9174–9184. doi: 10.1074/jbc.M110.166454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valjent E, Corvol JC, Trzaskos JM, Girault JA, Herve D. Role of ERK pathway in psychostimulant-induced locomotor sensitization. BMC Neurosci. 2006;7:20. doi: 10.1186/1471-2202-7-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valjent E, Pascoli V, Svenningsson P, Paul S, Enslen H, Corvol JC. Regulation of a protein phosphatase cascade allows convergent dopamine and glutamate signals to activate ERK in the striatum. Proc Natl Acad Sci USA. 2005;102:491–496. doi: 10.1073/pnas.0408305102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderschuren LJ, Kalivas PW. Alterations in dopaminergic and glutamatergic transmission in the induction and expression of behavioral sensitization: a critical review of preclinical studies. Psychopharmacology (Berl) 2000;151:99–120. doi: 10.1007/s002130000493. [DOI] [PubMed] [Google Scholar]
- Vezina P. Amphetamine injected into the ventral tegmental area sensitizes the nucleus accumbens dopaminergic response to systemic amphetamine: an in vivo microdialysis study in the rat. Brain Res. 1993;605:332–337. doi: 10.1016/0006-8993(93)91761-g. [DOI] [PubMed] [Google Scholar]
- Vezina P. Sensitization, drug addiction and psychopathology in animals and humans. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31:1553–1555. doi: 10.1016/j.pnpbp.2007.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezina P, Stewart J. Amphetamine administered to the ventral tegmental area but not to the nucleus accumbens sensitizes rats to systemic morphine: lack of conditioned effects. Brain Res. 1990;516:99–106. doi: 10.1016/0006-8993(90)90902-n. [DOI] [PubMed] [Google Scholar]
- Vezina P, Queen AL. Induction of locomotor sensitization by amphetamine requires the activation of NMDA receptors in the rat ventral tegmental area. Psychopharmacology (Berl) 2000;151:184–191. doi: 10.1007/s002130000463. [DOI] [PubMed] [Google Scholar]
- Voorn P, Vanderschuren LJ, Groenewegen HJ, Robbins TW, Pennartz MA. Putting a spin on the dorsal-ventral divide of the striatum. Trends Neurosci. 2004;27:468–474. doi: 10.1016/j.tins.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Wise RA, Bozarth MA. A psychomotor stimulant theory of addiction. Psychol Rev. 1987;94:469–492. [PubMed] [Google Scholar]
- White FJ, Kalivas PW. Neuroadaptations involved in amphetamine and cocaine addiction. Drug Alcohol Depend. 1998;51:151–153. doi: 10.1016/s0376-8716(98)00072-6. [DOI] [PubMed] [Google Scholar]
- Zhai H, Li Y, Wang X, Lu L. Drug-induced alterations in the extracellular signal-regulated kinase (ERK) signalling pathway: Implications for reinforcement and reinstatement. Cell Mol Neurobiol. 2008;25:157–172. doi: 10.1007/s10571-007-9240-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Diagram illustrating the experimental paradigm. On day 2, all the animals received bilateral ventral midbrain (VM) microinjections of sterile saline (0.5μl/side). On day 4 (1st), 6 (2nd) and 8 (3rd injection) of the training phase different groups received bilateral VM microinjections according to the design described in the method section. On day 13 (sensitization phase), all the animals were injected with amphetamine sulphate (0.75 mg/kg, ip). On each injection day the animals were put in the test cage for two hours.
Ambulatory (top panel), non-ambulatory (middle panel) and vertical (bottom panel) activity measured 21 days after the training phase following a systemic injection of amphetamine (0.75 mg/kg, ip) in two different groups. The legend on the x-axis indicates the pretreatment (VM microinjection) that each group received during the training phase: Vehicle (VEH, n = 6) or D-Tyr[11]Neurotensin (NT, n = 7). Each bar represents the mean (± s.e.m) total activity measured over the two-hour test period. Stars indicate a statistically significant difference with VEH (*p < 0.05; ***p < 0.001).