

Abstract
More precise identification and treatment monitoring of prediabetic/diabetic individuals will require additional biomarkers to complement existing diagnostic tests. Candidates include hyperglycemia-induced adducts such as advanced glycation end products (AGEs) of proteins, lipids, and DNA. The potential for DNA-AGEs as diabetic biomarkers was examined in a longitudinal study using the Leprdb/db animal model of metabolic syndrome. The DNA-AGE, N2(1-carboxyethyl)-2′-deoxyguanosine (CEdG) was quantified by mass spectrometry using isotope dilution from urine and tissue of hyperglycemic and normoglycemic mice. Hyperglycemic mice (fasting plasma glucose, FPG, ≥200 mg/dL) displayed a higher median urinary CEdG value (238.4±112.8 pmol/24 h) than normoglycemic mice (16.1±11.8 pmol/24 h). Logistic regression analysis revealed urinary CEdG to be an independent predictor of hyperglycemia. Urinary CEdG was positively correlated with FPG in hyperglycemic animals and with HbA1c for all mice. Average tissue-derived CEdG was also higher in hyperglycemic mice (18.4 CEdG/106 dG) than normoglycemic mice (4.4 CEdG/106 dG). Urinary CEdG was significantly elevated in Leprdb/db mice relative to Leprwt/wt, and tissue CEdG values increased in the order Leprwt/wt <Leprwt/db < Leprdb/db. These data suggest that urinary CEdG measurement may provide a non-invasive quantitative index of glycemic status and augment existing biomarkers for the diagnosis and monitoring of diabetes.
Graphical Abstract
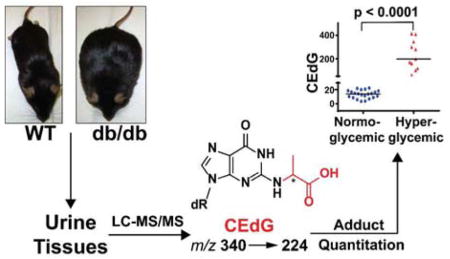
Introduction
Of the approximately 29 million people with diabetes in the United States, it is estimated that 30% are undiagnosed.1 Therefore, identifying diabetic individuals earlier and more accurately remains an unmet clinical need. Delayed clinical intervention for diabetes increases the severity of micro- and macrovascular complications as demonstrated by the Diabetes Control and Complications Trial (DCCT) and the subsequent long-term follow up study the Epidemiology of Diabetes Intervention and Complications (EDIC) trial.2,3 These studies demonstrated that frequent blood glucose measurements and intensive insulin therapy could significantly improve patient outcomes. However, such vigilant biomonitoring and therapeutic intervention is not a practical goal for the majority of patients with diabetes, and the development of improved diagnostic tools to identify patients most likely to benefit from intensive treatment is an ongoing challenge. Because of the inherent difficulties of accurate glucose monitoring4, e.g., wide fluctuations in daily plasma levels, glucose instability, and variability in measurement methods and standards, validated long-term biomarkers of glycemic control have come into routine clinical practice. The primary example is hemoglobin A1c (HbA1c), the Amadori adduct of the N-terminal valine of the hemoglobin β-chain.5 Its measurement has since become the gold standard for the monitoring of glycemic control and more recently as a diagnostic index for diabetes.6 HbA1c measurement reflects the average glucose over ~ 120 days, the mean lifetime of the erythrocyte.7 Although HbA1c levels ≥ 6.5% (48 mmol/mol) significantly correlate with diabetes, overreliance on HbA1c can lead to misdiagnosis in a significant percentage of the population. For example, the Finnish Diabetes Prevention Study showed that a diagnostic criteria of HbA1c ≥ 6.5% failed to identify ~ 60% of patients with type 2 diabetes originally identified by two consecutive oral glucose tolerance tests.8 Although there is a linear relationship between mean blood glucose and HbA1c, ~30% of patients with type 2 diabetes have HbA1c levels above or below values predicted from mean plasma glucose.9 Such individuals are classified as “high or low glycators” and consistently show these variations over time, suggesting an intrinsic biological origin of this phenomenon. The failure of HbA1c to invariably correlate with glycemic status has been attributed to a variety of factors that may influence HbA1c formation and persistence within erythrocytes. These include undiagnosed hemoglobinopathies, inter-individual variation in glucose transporter (GLUT) activity,10 erythrocyte turnover,7 and genetic differences attributed to ethnic background and other unidentified factors.11–13 Whatever the origin of the discrepancies between HbA1c measurement and standardized glucose tests, other potential biomarkers of glucose control should be developed to complement existing methods and refine our ability to more accurately diagnose and monitor diabetes.
Advanced glycation end products (AGEs) of proteins, formed from reactions of carbohydrate-derived α-oxoaldehydes such as methylglyoxal (MG) with amino acid nucleophiles, are elevated as a result of hyperglycemia and have been previously investigated as clinical biomarkers of glucose control and diabetic complications. Although there are numerous reports describing the correlation of various protein AGEs with hyperglycemia and diabetic complications,14–17 predictive power depends upon the exact combination of AGEs studied and the assay matrix (e.g. serum, collagen) examined.16, 18, 19 No general consensus has emerged regarding which specific AGEs might be of optimal value for monitoring glucose control or predicting diabetic complications. Together with the lack of uniform methodology for measurement, this has made it difficult to standardize diagnostic endpoints. For these reasons, protein-AGE determination in the clinical setting has only seen limited use.
Other potential candidates for biomarkers of glycemia are the DNA-AGEs. The stability of DNA, its uniform cellular distribution, and its substantially longer lifetime relative to proteins suggests that DNA-AGE measurement could provide a more long-term assessment of glycemic control. In contrast to the multiplicity of amino acid AGEs described in humans, there are only two DNA-AGEs detectable in blood and urine; N2-(1-carboxyethyl)- 2′-deoxyguanosine (CEdG) and a cyclic diol arising from direct addition of MG at 1, N2 of guanine (cMG-dG).20,21 In contrast to CEdG, whose stability in DNA and as a deoxynucleoside is comparable to 2′-deoxyguanosine,22, 23 cMG-dG is significantly less stable and less suitable as a potential biomarker.20,22,24 Methods to detect CEdG include 32P post-labeling, polyclonal antibody-based analysis, and liquid chromatography electrospray ionization tandem mass spectrometry (LC-ESI-MS/MS).24–26 Of these methods, LC-ESI-MS/MS coupled with stable isotope dilution provides the most accurate quantification of CEdG. Using this approach, we previously reported the first measurement of CEdG in a high-dose streptozotocin (STZ) rat model of type 1 diabetes and demonstrated increased levels relative to normoglycemic controls.22 To further elucidate the physiological relevance of DNA-AGEs in type 2 models of diabetes, in the present work we measured CEdG in Leprwt/wt (wt), Leprwt/db (wt/db), and Leprdb/db (db/db) mice, which provided a range of FPG from 70 to 978 mg/dL and HbA1c levels from 3–15% (9 to 140 mmol/mol). Mice with a Leprdb gene have an alternatively spliced variant of the leptin receptor, which is highly expressed in the hypothalamus and is resistant to the effects of the leptin hormone.27 This leads to the development of obesity, hyperglycemia, hyperlipidemia and other metabolic abnormalities within a few weeks of birth, which mimic the pathology found in type 2 diabetes.28 Heterozygous mice (wt/db) typically do not exhibit a gross diabetic phenotype; however, they exhibit several distinguishing metabolic traits relative to wt animals including a decreased rate of glucose oxidation and slower rates of catabolism.29
CEdG excreted in the urine of wt, wt/db and db/db mice was measured over a period of 36 weeks and recorded contemporaneously with FPG and HbA1c. CEdG was significantly elevated in hyperglycemic compared to normoglycemic mice. Results revealed a positive correlation of urinary CEdG with FPG and HbA1c, as well as with the protein AGEs carboxymethyl and carboxyethyl lysine (CML and CEL, respectively). These data provide a rationale for applying CEdG measurement as a clinical tool for the diagnosis and management of metabolic disease.
DNA-AGEs likely enter circulation via DNA repair and/or degradation during cell turnover. CEdG remaining in genomic DNA poses significant hazards since it contributes to genomic instability,30–32 which may substantially increase the risk for certain cancers. This is particularly true of diabetic individuals as they have been reported to be compromised in DNA repair.33–35 We therefore examined CEdG levels in a subset of organs at risk for increased cancer incidence in metabolic disease, including pancreas, kidney, colon and liver; and we report here the first measurement of DNA-AGEs in tissue from animal models of diabetes. This analysis revealed a significantly higher level of CEdG in organs isolated from hyperglycemic mice compared to normoglycemic controls.
Experimental Procedures
Materials
CEdG calibrators and internal standards were synthesized as previously described.22 LC-MS grade water with 0.1% formic acid (FA), acetonitrile (ACN), and ammonium hydroxide (NH4OH) were purchased from Sigma Aldrich. Oasis MCX 1 cc solid phase extraction (SPE) columns were purchased from Waters Corporation. LC-MS Chromasolv® methanol (MeOH) was obtained from Fluka. 0.22 μm syringe filters, 4 mm, were purchased from Thermo Scientific.
Animal Care
Leprwt/db mice of C57BL/6J stock from The Jackson Laboratories were bred for five generations with C57BL/6 mice obtained in the City of Hope Animal Resource Center. All Lepr genotypes (wt, wt/db, and db/db) were generated from brother-sister mating of Leprwt/db mice. Animals were housed in light-controlled conditions (10 h light/14 h dark cycle) at 22 °C for a maximum of nine months. All animals were provided with unlimited access to commercial chow (PicoLab Rodent Diet 20 #5053) and water. All procedures were approved under City of Hope IACUC Protocol #02016.
Genotyping
Mouse DNA was isolated from 1–2 mm tail sections (200 μL 1X PBND Buffer [50 mM KCl, 10 mM Tris-HCl pH 8.3, 2.5 mM MgCl2, 0.1 mg/mL Gelatin, 0.45% v/v NP-40, 0.45% v/v Tween-20] and 0.05 μg/μL Proteinase K) and digested overnight at 55 °C. The Lepr site was amplified from 0.5 μL DNA with 1.25 U MyTaq polymerase (Bioline) in a 50 μL volume using Lepr-forward (5′-CCAACTTCCCAACAGTCCAT-3′) and Lepr-reverse primers (5′-TGCCCTGAAAATCAAGCATA-3′). The presence of the db mutation was identified by digestion of 25 μL PCR product with 5 units of Hpy166II (New England Biolabs) in a 40 μL volume for 30 min at 37 °C. The Lepr G→T mutation was revealed as 18bp, 38bp, and 131bp bands (versus 38bp and 149bp for the wt allele).
Urine, Blood, and Tissue Sample Collection
A total of 38 mice were analyzed in this study (db/db n=11; wt/db n=16; wt n=11). Measurements commenced at 4 weeks of age immediately following completion of weaning. Mice were placed in metabolic cages (Nalgene) every 4 weeks for 24 h urine collection and provided with food and water ad libitum. The total volume of urine was recorded and stored at −20 °C. Immediately following urine collection, mice were placed in clean cages and fasted for 6 h (10:30–16:30). Blood was collected after fasting by a small incision at the tip of the tail to measure FPG (Accu-Chek Aviva Blood Glucose Meter, Roche Diagnostics). 7μL of blood was collected for HbA1c measurement using the Mouse Hemoglobin A1c assay (Crystal Chem Inc.). Equal numbers of male and female mice were used. No sex specific differences in CEdG or FPG levels were observed throughout the course of the study (Supporting Information Figure S1). For tissue collection, mice were euthanized and perfused with PBS (pH 8) to obtain liver, kidney, pancreas, and colon tissues. Samples were then immediately flash-frozen in liquid N2 and stored at −80 °C.
LC-ESI-MS/MS Analysis of CEdG in Urine
Following thawing, 100 μL of urine was added to 50 μL of 7.5 ng/mL (R, S)-15N5-CEdG and 400 μL 10% FA in H2O. Oasis MCX 1 cc SPE columns were conditioned with 1 mL MeOH and equilibrated with 1 mL 0.1% FA in H2O prior to sample loading. Columns were washed with 2 mL MeOH and 2 mL 2% FA in H2O. CEdG was eluted with 1 mL of 2% NH4OH in MeOH, dried by vacuum centrifugation, and resuspended in 100 μL 0.1% FA in H2O. Calibration standards were processed in parallel to urine samples. Liquid chromatography was performed using an Agilent 1290 Infinity Binary UHPLC with an Agilent alkyl reversed-phase ZORBAX SB-Aq column (2.1 × 50mm, 1.8μm) (40 °C) using mobile phases A (0.1% FA in H2O) and B (0.1% FA in ACN). Analytes were eluted using the following gradient: 0–4 min, 3–10% B; 4–4.5 min, 10–100% B; 4.5–5 min, 100-3% B, at a flow rate of 0.4 mL/min. R- and S-CEdG eluted at 1.9 and 2.4 min, respectively (Figure 1A). Isotope-dilution LC-ESI-MS/MS was performed in positive ion mode using an Agilent 6400 triple quadrupole mass spectrometer with multiple reaction monitoring to observe mass transitions m/z 340.1 → 224.1 (CEdG) and m/z 345.1 → 229.1 (15N5-CEdG, Figure 1B). The relative MS response of a fixed amount of (R, S)-15N5-CEdG to increasing concentrations of (R, S)-CEdG was used to generate a standard curve (R2 > 0.99). Sample CEdG concentrations were determined using isotope dilution with fitting to the standard curve using the Agilent MassHunter Workstation Quantitative Analysis software. The lower limit of detection was 0.01 ng/mL (30 pM), while the lower limit of quantification, defined as a peak height of ≥ 5× baseline noise, was 0.1 ng/mL (0.3 nM). Inter- and intraday accuracy of the assay across the range of the standard curve was established to be 96 and 94% of target concentrations, respectively. The assay was also determined to be unbiased with both inter- and intraday precision within ±6%. Intra-run coefficients of variation (CV) were ≤ 9% and ≤ 8% for R- and S-CEdG, respectively, while the corresponding values for the inter-run CV were ≤ 9% and ≤ 7%. The final volume of urine excreted over 24 h was used to calculate total pmol CEdG, expressed as pmol CEdG/24 h.
Figure 1. LC-ESI-MS/MS chromatogram of (R, S) CEdG.

A. Representative ion chromatogram of (R, S)-CEdG in urine (top panel) and 15N5-(R, S)-CEdG isotopic standard (bottom panel). Mass transitions used for identification and quantification indicated in the inset. B. Structure of the DNA-AGE CEdG (m/z 340). Dashed arrow indicates the primary fragmentation giving rise to the mass transitions indicated in A. Asterisk (*) indicates the chiral center.
Tissue DNA Isolation and Digestion
Tissues were homogenized and DNA isolated as previously described22 with the following modifications: Liver and kidney (0.05 g to 0.1 g) were homogenized in Buffer A (0.3 M sucrose, 60 mM KCl, 15 mM NaCl, 60 mM Tris-HCl, pH 8, 0.5 mM spermidine, 0.15 mM spermine, 2 mM EDTA) containing 0.5% Nonidet P-40 (NP-40). Nuclei were pelleted at 1100 × g for 12 min at 4 °C, after which supernatant was removed. The pellet was resuspended with 0.5 mL Buffer A then vortexed; 3 mL Buffer B (150 mM NaCl, 5 mM EDTA, pH 7.8), 3 mL Buffer C (20 mM Tris-HCl, pH 8, 20 mM NaCl, 20 mM EDTA, 1% SDS, and 80 μg/mL Proteinase K) were added with mixing after each step. For DNA isolation from pancreas and colon, tissue samples (0.05 to 0.1 g) were placed in a mortar with liquid nitrogen, and ground to a powder prior to processing using the procedure described above. 100 μg of isolated tissue DNA was spiked with 15N5-(R, S) CEdG (final concentration of 3.75 ng/mL), and heated to 95 °C for 5 min followed by snap cooling on ice. DNA was digested as previously described.32
HPLC analysis of dG from, genomic DNA
Genomic DNA was analyzed using an Agilent 1100 HPLC system equipped with a 10 × 250 mm, 5 μm XBridge Prep C18 column (Waters). Nucleosides were separated using mobile phases A (H2O with 0.1% FA) and B (ACN with 0.1% FA). The following gradient was used: 0–15 min, 0–9% B; 15–55 min, 9.0–9.5% B; 55–60 min, 9.5–90% B; 60–70 min, 90%B; 70–75 min, 90–0% B; 75–80 min 0% B at 2 mL/min. Chromatograms and peak area measurements were analyzed using Agilent Chemstation software.
LC-ESI-MS/MS Analysis of CML and CEL
CML and CEL were concentrated from urine and analyzed by the Analytical Pharmacology Core at City of Hope as previously described.36
Statistical Analyses of CEdG in Urine
Statistical analyses between groups were performed using one-way ANOVA or Student’s t-test. The diagnostic value of CEdG measurement to predict hyperglycemia was analyzed using R- statistical software employing a logistic regression analysis with a cutoff value set at the median value of all CEdG values measured (17 pmol/24 h). Normoglycemic animals were defined as 0 while hyperglycemic mice were defined as 1. Probability log odds were determined and the intercept for values greater than 17 were found to significantly predict hyperglycemia (FPG > 200 mg/dL. The slope of the indicator variable (values of CEdG less than 17) was found to be −4.0943, with values lower than 17 to significantly predict normoglycemia. Expressed in another way, the logistic regression result showed that the indicator variable when CEdG < 17 is statistically significant (p = 0.0016). This means the indicator variable for CEdG expression <17 with a value of 0, versus CEdG ≥17 with a value of 1, changes the log odds of being diabetic by −4.09. This data is presented in Supporting Information Table S2. We have also included results for the 95% confidence interval analysis as Supporting Information Table S3.
Correlations (FPG vs. CEdG; HbA1c vs. CEdG) were determined by plotting the average CEdG values for individual mice with contemporaneous measurements. Spearman or Pearson correlation coefficients were determined using GraphPad Prism.
Genomic DNA Statistical Analyses
CEdG values from genomic DNA for individual mice were separated by organ and glycemic status/genotype. As the variance in the raw CEdG values precluded accurate ANOVA analysis, these numbers were first converted to their natural log values. Comparisons between genotypes when organs were averaged for each individual animal were analyzed by taking the natural log of each CEdG value and then analyzing the differences using one-way ANOVA. To determine the effect of either organ or genotype on differences observed, average CEdG values for each organ or genotype were totaled and the natural log of each number calculated. One-way ANOVA was then used to determine statistical significance.
Results
CEdG is significantly elevated in urine of hyperglycemic mice
CEdG from 24 h urine collections was quantified using stable isotope dilution LC-ESI-MS/MS. The R- and S-stereoisomers of CEdG were cleanly resolved under the chromatographic conditions (Figure 1A). Mass transitions m/z 340→224 and 345→ 229 for (R, S)-CEdG and 15N5-(R, S)-CEdG, respectively, were used for identification and quantification (Figure 1B). Separately measured values for R- and S-CEdG, expressed as picomoles (pmol) of CEdG excreted over the 24 h urine collection period (pmol/24 h), were summed to provide a total CEdG measurement. Urinary CEdG levels differed significantly between hyperglycemic and normoglycemic animals (Figure 2A). Each data point corresponds to an average CEdG value for an individual mouse measured monthly over a 36 week period. Mice with FPG ≥ 200 mg/dL (11 mM) had a mean value of 238.4 ± 112.8 pmol CEdG/24 h compared to 16.1 ± 11.8 pmol CEdG/24 h for animals with FPG < 200 mg/dL. There was no overlap in CEdG levels between the two groups. The median urinary CEdG value for all animals measured over the duration of the study was 17 pmol/24 h (Figure 2B). Logistic regression analysis was performed to determine whether CEdG values above the median were predictive of hyperglycemia. A CEdG level ≥17 pmol/24 h was shown to be a significant predictor of hyperglycemia (p = 0.0016; Supporting Information Table S2) as an isolated value, with a 95% confidence level (Supporting Information Table S3).
Figure 2. CEdG is elevated as a result of hyperglycemia.

CEdG was quantified as described in Experimental Procedures and time-averaged values (repeated measures analysis) were calculated for each individual animal. A. CEdG levels were compared between normoglycemic (n = 22; FPG < 200 mg/dL) and hyperglycemic (n = 11; FPG ≥ 200 mg/dL) mice. Significance was determined using a non-parametric unpaired t-test. B. Mean CEdG levels were plotted for each animal and the median value was determined to be 17 pmol/24 h. C. Mean CEdG values grouped according to genotype, significance calculated as in A.
Because there was overlap of FPG and HbA1c values between wt, wt/db, and db/db mice (Table 1), it was of interest to determine whether CEdG measurement in urine could distinguish between genotypes. While db/db mice excreted significantly greater amounts of CEdG, compared to both wt and wt/db animals (Figure 2C, p < 0.001), the mean and median values for the latter two groups were not statistically different (Table 1). Followed over time for individual mice, CEdG in urine was found to increase significantly over time for the db/db animals (p = 0.0023), whereas there was no significant increase for the wt and wt/db animals (Supporting Information Figure S2 and Table S1).
Table 1.
Summary of Metabolic Data
| Leprdb/db | Leprwt/db | Leprwt/wt | All genotypes | |
|---|---|---|---|---|
| CEdG (pmol/24 h) | ||||
| Mean | 235.0 | 14.9 | 13.9 | 104.1 |
| SEM | 35.1 | 2.4 | 2.3 | 8.7 |
| Median | 197.1 | 13.6 | 14.3 | 17.1 |
| Range | 23.1–543.1 | 2.4–131.9 | 1.1–99.9 | 1.1–543.1 |
| N | 81 | 71 | 78 | 230 |
| HbA1c (%) | ||||
| Mean | 9.4 (80)* | 5.0 (31) | 4.7 (28) | 6.8 (51) |
| SEM | 0.7 (7.3) | 0.1 (1.3) | 0.2 (1.7) | 0.4 (4.4) |
| Median | 9.1 (76) | 5.0 (32) | 4.8 (30) | 5.3 (34) |
| Range | 3.2–15.2(11–143) | 3.7–5.8 (17–40) | 3.5–5.7 (15–39) | 3.2–15.2 (11–143) |
| N | 27 | 22 | 15 | 64 |
| FPG (mg/dL) | ||||
| Mean | 412.6 | 154.0 | 147.4 | 237.9 |
| SEM | 18.0 | 4.9 | 3.8 | 9.5 |
| Median | 408 | 143 | 141 | 164 |
| Range | 106–978 | 86–448 | 70–239 | 70–978 |
| N | 103 | 98 | 108 | 309 |
| Mass (g) | ||||
| Mean | 58.5 | 31.0 | 25.0 | 51.9 |
| SEM | 1.8 | 0.6 | 0.5 | 3.4 |
| Median | 64.7 | 31.6 | 23.7 | 31.3 |
| Range | 28.3–89.0 | 24.0–37.5 | 21.8–30.2 | 21.8–89.0 |
| N | 92 | 76 | 109 | 277 |
Data representing the mean, standard error of the mean (SEM), median, range, and number of measurements (n) for each parameter over the course of 36 weeks for all animals analyzed.
Values in parentheses = mmol/mol HbA1c.
Urinary CEdG is correlated with FPG and HbA1c
FPG and HbA1c measurements were obtained immediately after urine collection for CEdG following a 6 h fast. The db/db animals had the highest values of FPG and HbA1c, with mean values of 413 mg/dL and 9% (75 mmol/mol) respectively, vs. 147 mg/dL and 4.6% (27 mmol/mol) for wt animals (Table 1). Heterozygous wt/db and wt mice were indistinguishable based on their respective FPG or HbA1c values. A minimum expectation of any proposed biomarker for diabetes is that it shows some correlation with established biomarkers of metabolic disease. To examine the correlation between HbA1c and FPG with CEdG, plots of time-averaged measurements obtained contemporaneously for normoglycemic and hyperglycemic mice were analyzed using repeated measures analysis. Figure 3A shows significant correlation between averaged CEdG and FPG (p ≤ 0.001) while Figure 3B reveals a similar relationship between CEdG and HbA1c (p ≤ 0.001).
Figure 3. Correlation of CEdG with FPG and HbA1c.

A. Average CEdG and FPG values for individual animals were determined using repeated measures analysis. Correlations were calculated with Spearman’s coefficient (r = 0.6621). B. Average CEdG and HbA1c calculated as in A, r = 0.8016.
CEdG from tissue DNA is elevated in hyperglycemia and differentiates Lepr genotypes
Genomic DNA was isolated from pancreas, kidney, colon, and liver from wt, wt/db, and db/db mice from 28–36 weeks of age and analyzed for CEdG. CEdG levels were normalized to the amount of dG present within each sample and expressed as CEdG/106 dG. To examine total organ differences between hyperglycemic and normoglycemic mice, CEdG values were averaged over colon, kidney, pancreas and liver for individual mice and plotted according to glycemic status in Figure 4A. CEdG was significantly elevated in tissues from hyperglycemic mice (p < 0.0001). Data from Figure 4A was also stratified according to Lepr genotype, which revealed a trend of increasing CEdG from wt to wt/db and db/db mice (Figure 4B). One-way ANOVA revealed a statistically significant difference between wt and db/db genotypes (p ≤ 0.0001) as well as wt/db and db/db (p ≤ 0.05). The distribution of CEdG in genomic DNA from individual tissues is shown in Figure 4C, displaying a clear trend of CEdG levels in the order db/db > wt/db > wt. A statistically significant variation (p ≤ 0.01) in CEdG levels was observed between db/db and wt mice in pancreas, colon, and kidney while liver displayed an even greater difference (p ≤ 0.0001). In kidney a significant increase in CEdG was observed between db/db and wt/db mice (p ≤ 0.05).
Figure 4. CEdG from tissue is elevated in hyperglycemic and Lepr mutant mice.

DNA was isolated from pancreas, liver, colon, and kidney and analyzed for CEdG as described in Experimental Procedures. CEdG values were averaged across tissues from individual mice and grouped according to A. glycemic status or B. genotype. C. CEdG measurements from pancreas, liver, colon, and kidney from individual mice stratified by genotype. D. One-way ANOVA with Tukey’s modification was used to analyze CEdG differences between genotypes (irrespective of tissue, last column) and between tissue (regardless of genotype, bottom row). †CEdG/106 dG. See text for details. Significance values: *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001.
To minimize the effect of variance on inter-individual CEdG levels between organs, average CEdG values were calculated for each organ and differences between genotypes were analyzed using one-way ANOVA with Tukey’s modification (last column in Figure 4D). This analysis highlighted significant differences between all genotypes, most strikingly even between wt and wt/db mice (p ≤ 0.01). Neither urinary CEdG, FPG or HbA1c measurements could make this distinction (Table 1).
Amino Acid AGEs CML and CEL are elevated in hyperglycemic mice and correlate with CEdG
The amino acid AGEs CEL and CML were measured in urine of age-matched (28 weeks) hyperglycemic and normoglycemic mice by LC-ESI-MS/MS with isotope dilution and their relationship to both FPG and CEdG was examined (Figure 5). For mice with FPG ≥ 200 mg/dL, CML and CEL were significantly elevated relative to normoglycemic animals (Figure 5A). However, FPG did not appear to correlate with either CML or CEL (Figure 5B). When the relationship between CEdG and CML/CEL for all animals was examined, a significant positive correlation was observed, more significantly for CML than CEL (Figure 5C, p < 0.0001). For the hyperglycemic subset of mice, CML and CEL were also positively correlated with CEdG (Figure 5D).
Figure 5. CEL and CML are elevated in urine of hyperglycemic mice and correlate with CEdG.

A. Mean values of CEL and CML measured in urine from aged-matched hyperglycemic and normoglycemic wt (9), wt/db (9), and db/db (11) mice using LC-ESI-MS/MS. FPG was measured at the cessation of 24 h urine collection. Two-way ANOVA revealed a significant increase in CEL and CML in hyperglycemic mice (****p < 0.0001). B. CML (n=32) and CEL (n=32) vs. FPG. No significant correlation was observed. CML, r = 0.2458; CEL, r = 0.2267. C. Plot of CML and CEL vs. CEdG for all mice; CML, r = 0.7009 (****p < 0.0001); CEL, r = 0.4902 (**p = 0.0044). D. CML (n=32) and CEL (n=32) vs. CEdG from hyperglycemic mice; CML, r = 0.5589 (*p = 0.0471); CEL, r = 0.5805 (*p = 0.0375).
Discussion
Novel biomarkers of metabolic disease should complement existing clinical methodology by increasing diagnostic precision, particularly in patients difficult to identify as diabetic using common tests.8 The ability to accurately identify and monitor patients likely to become diabetic and to predict specific complications before patients become symptomatic remain significant challenges in diabetes care. For example, while HbA1c is an independent predictor of both mild and severe proliferative retinopathy,37 cardiovascular disease (CVD) is not clearly associated with HbA1c.38–40 Some groups have reported that deviations in measured HbA1c from values predicted from FPG can predict increased risk for retinopathy and nephropathy,41, 42 while analogous models based on fructosamine measurement were reported to provide a more accurate algorithm for the prediction of nephropathy.43 In general, efforts to find improved diabetic biomarkers have often focused on glycation and advanced glycation end products of proteins rather than nucleic acids.
Hyperglycemia increases circulating levels of glucose-derived α-oxoaldehydes such as MG which react non-enzymatically with proteins,44,45 lipids,46 and DNA24,47,48 to form AGEs, potentially modifying or inactivating their function. Unlike Amadori adducts such as HbA1c and fructosamine, which are formed reversibly in equilibrium with glucose, AGEs are typically irreversible and possess long lifetimes. Since CEdG is only known to be formed from the reaction of MG with dG it may be considered a stable surrogate biomarker of MG exposure.24,47,49 Methylglyoxal has been shown to be significantly elevated in both type 1 and type 2 diabetes.50, 51 Several endogenous sources of MG have been described including the non-enzymatic decomposition of glucose52 and its Amadori adducts,53 ketone body metabolism,54 and glycolysis.55 All of these processes are exacerbated by diabetes, and elevated circulating MG has not been clearly associated with any other diseases or environmental toxin exposure. For these reasons we anticipate that DNA-AGEs may be a specific biomarker for diabetes.
Protein AGEs in serum and collagen are also significantly elevated in obese/diabetic individuals56,57 and have been proposed as potential biomarkers of diabetic complications.14–17 In the Joslin Medalist study of patients with type 1 diabetes for ≥ 50 years, a specific combination of plasma protein AGEs was shown to be more accurate than HbA1c for the prediction of complications.58 However, it is difficult to compare data from various correlative studies on protein AGEs because different quantitative and semi-quantitative methods including GC and LC-MS, HPLC, ELISA, and spectrofluorimetry have been used. Moreover, the biological matrix used for analysis appears to be critical as well, e.g., large variations in protein AGE levels have been reported in collagen calling into question the relevance of skin measurements for diabetic complications.59
Since all nucleated cells contain DNA, and DNA has a longer lifetime than protein, measurement of the DNA-AGE CEdG may allow for more precise assessment of long term glycemic stress. We observed a significant (17-fold) increase in the median value of urinary CEdG in animals with FPG ≥ 200 mg/dL relative to normoglycemic controls. Moreover, consideration of CEdG values alone allowed for a prediction of hyperglycemia with >95% confidence. These observations are consistent with our previous work using a high dose STZ diabetic rat model, which demonstrated elevated CEdG levels in urine relative to non-diabetic animals.22 Significant increases in CEdG for both type 1 diabetes and db/db models suggest the direct influence of hyperglycemia on DNA-AGE formation, supported by the linear relationship of CEdG and FPG (Figure 3A). In contrast to the narrow range of CEdG values observed for normoglycemic mice, values for the hyperglycemic animals were widely dispersed (Figure 2A). This suggests that stratification of CEdG values may provide diagnostic information relevant to diabetes-related pathologies. For example, individuals in the highest quartile may be at the greatest risk for microvascular and macrovascular complications or cancers associated with metabolic disease. This possibility will be more properly addressed in a clinical trial. The large increase in CEdG levels observed for the db/db group over time is striking. The age of the animal alone did not appear to make a substantial contribution, since increases in CEdG over time were insignificant in wt or wt/db mice (SI Figure S2). This would suggest that increasing MG generation due to progressive diabetic pathology was the main contributing factor.
Quantification of CEdG in tissue DNA revealed differences between wt and wt/db mice not apparent using the standard FPG or HbA1c markers. Organ data revealed a clear increase in CEdG between wt and wt/db mice when aggregate tissue CEdG measurements were considered (p < 0.01, Figure 4D), even though wt/db animals had FPG, HbA1c, and urinary CEdG values that were indistinguishable from wt (Table 1). The disparity between the CEdG in urine vs tissue may reflect differences between circulating levels of CEdG resulting from DNA repair and/or cell turnover vs local tissue accumulation. The wt/db mice may be considered to exhibit borderline metabolic disease, as they have been shown to have higher rates of glucose oxidation, slower rates of catabolism, and are significantly more prone to gestational diabetes relative to wt animals.29,60 The data suggest that MG-induced AGE accumulation can occur in tissue even with a relatively mild diabetic phenotype and contribute to DNA damage despite FPG and HbA1c levels within the normal range. Since it was not practical to assay tissue DNA from all organs, the subset sampled was chosen on the basis of data indicating increased diabetes-associated cancer susceptibility.61 This tissue sampling bias may have highlighted differences not apparent in the analysis of CEdG in urine. Genetic and epigenetic dysregulation due to the presence of long-lived CEdG modification of DNA may contribute to genomic instability and/or play a role in the phenomenon of metabolic memory in diabetes.62
It is of interest that the DNA-AGEs CEdG and the protein AGEs CML and CEL were linearly correlated in our animal model (Figure 5C), but only CEdG bore a significant relationship to FPG (Figure 3A vs. 5B). This suggests that measurement of protein and DNA-AGEs may bear different relationships to clinical endpoints of metabolic disease. In summary, the stable DNA-AGE CEdG was significantly elevated in mouse models of diabetes, was able to predict diabetes as a single parameter, correlated with FPG and HbA1c, and was substantially increased in organs of wt/db and db/db mice relative to wt. These observations have catalyzed longitudinal clinical trials to examine CEdG as a biomarker for metabolic disease.
Supplementary Material
Acknowledgments
Funding Sources
This work was supported by NIH grants R01CA176611 (JT/TRO) and PA-12-49 Research Supplement to Promote Diversity in Health-Related Research to RJ (3R01CA176611-03S2). Research reported in this publication includes work performed in the Mass Spectrometry and Analytical Pharmacology Cores at City of Hope supported by the National Cancer Institute of the National Institutes of Health under award number P30CA033572. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
We would like to thank Dr. Debbie Thurmond for helpful comments and discussions. The editorial assistance of Alexandra Ciminera of the Irell and Manella Graduate School of Biological Sciences, City of Hope, is gratefully acknowledged. We thank Dr. Gabriel Gugiu and Roger Moore of the Mass Spectrometry Core Facility at City of Hope for technical assistance. The advice of Drs. Leanne Goldstein and Jeffrey Longmate, (Department of Information Sciences) regarding statistical analyses is gratefully appreciated.
ABBREVIATIONS
- ACN
Acetonitrile
- AGEs
Advanced glycation end products
- CEdG
N2-(1-carboxyethyl)-2′-deoxyguanosine
- CEL
Carboxyethyl lysine
- cMG-dG
1, N2-(1,2-dihydroxy-2-methyl)-2′-deoxyguanosine
- CML
Carboxymethyl lysine
- CV
Coefficients of variation
- CVD
Cardiovascular disease
- db/db
Leprdb/db
- DCCT
Diabetes control and complications trial
- EDIC
Epidemiology of diabetes interventions and complications
- FA
Formic acid
- FPG
Fasting plasma glucose
- GLUT
Glucose transporter
- HbA1c
Hemoglobin A1c
- Lepr
Leptin receptor
- LC-ESI-MS/MS
Liquid chromatography electrospray ionization tandem mass spectrometry
- MeOH
Methanol
- MG
Methylglyoxal
- NH4OH
Ammonium hydroxide
- wt
Leprwt/wt
- wt/db
Leprwt/db
Footnotes
Figures describing CEdG differences between genders (Figure S1) and CEdG variation over time (Figure S2) are presented. Tables describing linear mixed mode analysis for determination of time dependence on CEdG (Table S1); coefficients (Table S2) and 95% confidence interval (Table S3) determined regarding the logistic regression model are presented. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.CDC. Estimates of diabetes and its burdern in the US. Department of Health and Human Services; 2014. [Google Scholar]
- 2.The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The Diabetes Control and Complications Trial Research Group. The New England journal of medicine. 1993;329:977–986. doi: 10.1056/NEJM199309303291401. [DOI] [PubMed] [Google Scholar]
- 3.Epidemiology of Diabetes Interventions and Complications (EDIC). Design, implementation, and preliminary results of a long-term follow-up of the Diabetes Control and Complications Trial cohort. Diabetes care. 1999;22:99–111. doi: 10.2337/diacare.22.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gambino R. Glucose: a simple molecule that is not simple to quantify. Clinical chemistry. 2007;53:2040–2041. doi: 10.1373/clinchem.2007.094466. [DOI] [PubMed] [Google Scholar]
- 5.Rahbar S. An abnormal hemoglobin in red cells of diabetics. Clinica chimica acta; international journal of clinical chemistry. 1968;22:296–298. doi: 10.1016/0009-8981(68)90372-0. [DOI] [PubMed] [Google Scholar]
- 6.International Expert C. International Expert Committee report on the role of the A1C assay in the diagnosis of diabetes. Diabetes care. 2009;32:1327–1334. doi: 10.2337/dc09-9033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cohen RM, Franco RS, Khera PK, Smith EP, Lindsell CJ, Ciraolo PJ, Palascak MB, Joiner CH. Red cell life span heterogeneity in hematologically normal people is sufficient to alter HbA1c. Blood. 2008;112:4284–4291. doi: 10.1182/blood-2008-04-154112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pajunen P, Peltonen M, Eriksson JG, Ilanne-Parikka P, Aunola S, Keinanen-Kiukaanniemi S, Uusitupa M, Tuomilehto J, Lindstrom J Finnish Diabetes Prevention S. HbA(1c) in diagnosing and predicting Type 2 diabetes in impaired glucose tolerance: the Finnish Diabetes Prevention Study. Diabetic medicine: a journal of the British Diabetic Association. 2011;28:36–42. doi: 10.1111/j.1464-5491.2010.03183.x. [DOI] [PubMed] [Google Scholar]
- 9.Pupillo M, De Berardis G, Antenucci D, Minnucci A, Nicolucci A. Glycated haemoglobin or mean blood glucose as indicators of metabolic control in Type 2 diabetes? Diabetes research and clinical practice. 2008;80:e1–3. doi: 10.1016/j.diabres.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 10.Khera PK, Joiner CH, Carruthers A, Lindsell CJ, Smith EP, Franco RS, Holmes YR, Cohen RM. Evidence for interindividual heterogeneity in the glucose gradient across the human red blood cell membrane and its relationship to hemoglobin glycation. Diabetes. 2008;57:2445–2452. doi: 10.2337/db07-1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ziemer DC, Kolm P, Weintraub WS, Vaccarino V, Rhee MK, Twombly JG, Narayan KM, Koch DD, Phillips LS. Glucose-independent, black-white differences in hemoglobin A1c levels: a cross-sectional analysis of 2 studies. Annals of internal medicine. 2010;152:770–777. doi: 10.7326/0003-4819-152-12-201006150-00004. [DOI] [PubMed] [Google Scholar]
- 12.Bleyer AJ, Hire D, Russell GB, Xu J, Divers J, Shihabi Z, Bowden DW, Freedman BI. Ethnic variation in the correlation between random serum glucose concentration and glycated haemoglobin. Diabetic medicine: a journal of the British Diabetic Association. 2009;26:128–133. doi: 10.1111/j.1464-5491.2008.02646.x. [DOI] [PubMed] [Google Scholar]
- 13.Snieder H, Sawtell PA, Ross L, Walker J, Spector TD, Leslie RD. HbA(1c) levels are genetically determined even in type 1 diabetes: evidence from healthy and diabetic twins. Diabetes. 2001;50:2858–2863. doi: 10.2337/diabetes.50.12.2858. [DOI] [PubMed] [Google Scholar]
- 14.Brownlee M, Cerami A, Vlassara H. Advanced glycosylation end products in tissue and the biochemical basis of diabetic complications. The New England journal of medicine. 1988;318:1315–1321. doi: 10.1056/NEJM198805193182007. [DOI] [PubMed] [Google Scholar]
- 15.Sell DR, Lapolla A, Odetti P, Fogarty J, Monnier VM. Pentosidine formation in skin correlates with severity of complications in individuals with long-standing IDDM. Diabetes. 1992;41:1286–1292. doi: 10.2337/diab.41.10.1286. [DOI] [PubMed] [Google Scholar]
- 16.Beisswenger PJ, Makita Z, Curphey TJ, Moore LL, Jean S, Brinck-Johnsen T, Bucala R, Vlassara H. Formation of immunochemical advanced glycosylation end products precedes and correlates with early manifestations of renal and retinal disease in diabetes. Diabetes. 1995;44:824–829. doi: 10.2337/diab.44.7.824. [DOI] [PubMed] [Google Scholar]
- 17.Miura J, Yamagishi S, Uchigata Y, Takeuchi M, Yamamoto H, Makita Z, Iwamoto Y. Serum levels of non-carboxymethyllysine advanced glycation endproducts are correlated to severity of microvascular complications in patients with Type 1 diabetes. Journal of diabetes and its complications. 2003;17:16–21. doi: 10.1016/s1056-8727(02)00183-6. [DOI] [PubMed] [Google Scholar]
- 18.Misselwitz J, Franke S, Kauf E, John U, Stein G. Advanced glycation end products in children with chronic renal failure and type 1 diabetes. Pediatric nephrology. 2002;17:316–321. doi: 10.1007/s00467-001-0815-9. [DOI] [PubMed] [Google Scholar]
- 19.Schleicher ED, Wagner E, Nerlich AG. Increased accumulation of the glycoxidation product N(epsilon)-(carboxymethyl)lysine in human tissues in diabetes and aging. The Journal of clinical investigation. 1997;99:457–468. doi: 10.1172/JCI119180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thornalley PJ, Waris S, Fleming T, Santarius T, Larkin SJ, Winklhofer-Roob BM, Stratton MR, Rabbani N. Imidazopurinones are markers of physiological genomic damage linked to DNA instability and glyoxalase 1-associated tumour multidrug resistance. Nucleic acids research. 2010;38:5432–5442. doi: 10.1093/nar/gkq306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li H, Nakamura S, Miyazaki S, Morita T, Suzuki M, Pischetsrieder M, Niwa T. N2-carboxyethyl-2′-deoxyguanosine, a DNA glycation marker, in kidneys and aortas of diabetic and uremic patients. Kidney international. 2006;69:388–392. doi: 10.1038/sj.ki.5000064. [DOI] [PubMed] [Google Scholar]
- 22.Synold T, Xi B, Wuenschell GE, Tamae D, Figarola JL, Rahbar S, Termini J. Advanced glycation end products of DNA: quantification of N2-(1-Carboxyethyl)-2′-deoxyguanosine in biological samples by liquid chromatography electrospray ionization tandem mass spectrometry. Chemical research in toxicology. 2008;21:2148–2155. doi: 10.1021/tx800224y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cao H, Jiang Y, Wang Y. Stereospecific synthesis and characterization of oligodeoxyribonucleotides containing an N2-(1-carboxyethyl)-2′-deoxyguanosine. Journal of the American Chemical Society. 2007;129:12123–12130. doi: 10.1021/ja072130e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frischmann M, Bidmon C, Angerer J, Pischetsrieder M. Identification of DNA adducts of methylglyoxal. Chemical research in toxicology. 2005;18:1586–1592. doi: 10.1021/tx0501278. [DOI] [PubMed] [Google Scholar]
- 25.Schneider M, Thoss G, Hubner-Parajsz C, Kientsch-Engel R, Stahl P, Pischetsrieder M. Determination of glycated nucleobases in human urine by a new monoclonal antibody specific for N2-carboxyethyl-2′-deoxyguanosine. Chemical research in toxicology. 2004;17:1385–1390. doi: 10.1021/tx049929d. [DOI] [PubMed] [Google Scholar]
- 26.Vaca CE, Fang JL, Conradi M, Hou SM. Development of a 32P-postlabelling method for the analysis of 2′-deoxyguanosine-3′-monophosphate and DNA adducts of methylglyoxal. Carcinogenesis. 1994;15:1887–1894. doi: 10.1093/carcin/15.9.1887. [DOI] [PubMed] [Google Scholar]
- 27.Lee GH, Proenca R, Montez JM, Carroll KM, Darvishzadeh JG, Lee JI, Friedman JM. Abnormal splicing of the leptin receptor in diabetic mice. Nature. 1996;379:632–635. doi: 10.1038/379632a0. [DOI] [PubMed] [Google Scholar]
- 28.King AJ. The use of animal models in diabetes research. British journal of pharmacology. 2012;166:877–894. doi: 10.1111/j.1476-5381.2012.01911.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coleman DL. Obesity genes: beneficial effects in heterozygous mice. Science. 1979;203:663–665. doi: 10.1126/science.760211. [DOI] [PubMed] [Google Scholar]
- 30.Bucala R, Lee AT, Rourke L, Cerami A. Transposition of an Alu-containing element induced by DNA-advanced glycosylation endproducts. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:2666–2670. doi: 10.1073/pnas.90.7.2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wuenschell GE, Tamae D, Cercillieux A, Yamanaka R, Yu C, Termini J. Mutagenic potential of DNA glycation: miscoding by (R)- and (S)-N2-(1-carboxyethyl)-2′-deoxyguanosine. Biochemistry. 2010;49:1814–1821. doi: 10.1021/bi901924b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tamae D, Lim P, Wuenschell GE, Termini J. Mutagenesis and repair induced by the DNA advanced glycation end product N2-1-(carboxyethyl)-2′-deoxyguanosine in human cells. Biochemistry. 2011;50:2321–2329. doi: 10.1021/bi101933p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blasiak J, Arabski M, Krupa R, Wozniak K, Zadrozny M, Kasznicki J, Zurawska M, Drzewoski J. DNA damage and repair in type 2 diabetes mellitus. Mutation research. 2004;554:297–304. doi: 10.1016/j.mrfmmm.2004.05.011. [DOI] [PubMed] [Google Scholar]
- 34.Tyson J, Caple F, Spiers A, Burtle B, Daly AK, Williams EA, Hesketh JE, Mathers JC. Inter-individual variation in nucleotide excision repair in young adults: effects of age, adiposity, micronutrient supplementation and genotype. The British journal of nutrition. 2009;101:1316–1323. doi: 10.1017/S0007114508076265. [DOI] [PubMed] [Google Scholar]
- 35.Tempera I, Cipriani R, Campagna G, Mancini P, Gatti A, Guidobaldi L, Pantellini F, Mandosi E, Sensi M, Quesada P, Mario UD, D’Erme M, Morano S. Poly(ADP-ribose)polymerase activity is reduced in circulating mononuclear cells from type 2 diabetic patients. Journal of cellular physiology. 2005;205:387–392. doi: 10.1002/jcp.20414. [DOI] [PubMed] [Google Scholar]
- 36.Figarola JL, Scott S, Loera S, Xi B, Synold T, Weiss L, Rahbar S. Prevention of early renal disease, dyslipidaemia and lipid peroxidation in STZ-diabetic rats by LR-9 and LR-74, novel AGE inhibitors. Diabetes/metabolism research and reviews. 2005;21:533–544. doi: 10.1002/dmrr.550. [DOI] [PubMed] [Google Scholar]
- 37.Klein R, Klein BE, Moss SE, Davis MD, DeMets DL. Glycosylated hemoglobin predicts the incidence and progression of diabetic retinopathy. Jama. 1988;260:2864–2871. [PubMed] [Google Scholar]
- 38.Haffner SM, Lehto S, Ronnemaa T, Pyorala K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. The New England journal of medicine. 1998;339:229–234. doi: 10.1056/NEJM199807233390404. [DOI] [PubMed] [Google Scholar]
- 39.Morgan CL, Currie CJ, Peters JR. Relationship between diabetes and mortality: a population study using record linkage. Diabetes care. 2000;23:1103–1107. doi: 10.2337/diacare.23.8.1103. [DOI] [PubMed] [Google Scholar]
- 40.Stratton IM, Adler AI, Neil HA, Matthews DR, Manley SE, Cull CA, Hadden D, Turner RC, Holman RR. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. Bmj. 2000;321:405–412. doi: 10.1136/bmj.321.7258.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McCarter RJ, Hempe JM, Gomez R, Chalew SA. Biological variation in HbA1c predicts risk of retinopathy and nephropathy in type 1 diabetes. Diabetes care. 2004;27:1259–1264. doi: 10.2337/diacare.27.6.1259. [DOI] [PubMed] [Google Scholar]
- 42.Rohlfing CL, Wiedmeyer HM, Little RR, England JD, Tennill A, Goldstein DE. Defining the relationship between plasma glucose and HbA(1c): analysis of glucose profiles and HbA(1c) in the Diabetes Control and Complications Trial. Diabetes care. 2002;25:275–278. doi: 10.2337/diacare.25.2.275. [DOI] [PubMed] [Google Scholar]
- 43.Cohen RM, Holmes YR, Chenier TC, Joiner CH. Discordance between HbA1c and fructosamine: evidence for a glycosylation gap and its relation to diabetic nephropathy. Diabetes care. 2003;26:163–167. doi: 10.2337/diacare.26.1.163. [DOI] [PubMed] [Google Scholar]
- 44.Lo TW, Westwood ME, McLellan AC, Selwood T, Thornalley PJ. Binding and modification of proteins by methylglyoxal under physiological conditions. A kinetic and mechanistic study with N alpha-acetylarginine, N alpha-acetylcysteine, and N alpha-acetyllysine, and bovine serum albumin. The Journal of biological chemistry. 1994;269:32299–32305. [PubMed] [Google Scholar]
- 45.Thornalley PJ, Battah S, Ahmed N, Karachalias N, Agalou S, Babaei-Jadidi R, Dawnay A. Quantitative screening of advanced glycation endproducts in cellular and extracellular proteins by tandem mass spectrometry. The Biochemical journal. 2003;375:581–592. doi: 10.1042/BJ20030763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nakagawa K, Oak JH, Higuchi O, Tsuzuki T, Oikawa S, Otani H, Mune M, Cai H, Miyazawa T. Ion-trap tandem mass spectrometric analysis of Amadori-glycated phosphatidylethanolamine in human plasma with or without diabetes. Journal of lipid research. 2005;46:2514–2524. doi: 10.1194/jlr.D500025-JLR200. [DOI] [PubMed] [Google Scholar]
- 47.Papoulis A, al-Abed Y, Bucala R. Identification of N2-(1-carboxyethyl)guanine (CEG) as a guanine advanced glycosylation end product. Biochemistry. 1995;34:648–655. doi: 10.1021/bi00002a032. [DOI] [PubMed] [Google Scholar]
- 48.Shapiro R, Cohen BI, Shiuey SJ, Maurer H. On the reaction of guanine with glyoxal, pyruvaldehyde, and kethoxal, and the structure of the acylguanines. A new synthesis of N2-alkylguanines. Biochemistry. 1969;8:238–245. doi: 10.1021/bi00829a034. [DOI] [PubMed] [Google Scholar]
- 49.Ochs S, Severin T. Reaction of 2′-Deoxyguanosine with Glyceraldehyde. Liebigs Ann Chem. 1994:851–853. [Google Scholar]
- 50.Han Y, Randell E, Vasdev S, Gill V, Gadag V, Newhook LA, Grant M, Hagerty D. Plasma methylglyoxal and glyoxal are elevated and related to early membrane alteration in young, complication-free patients with Type 1 diabetes. Molecular and cellular biochemistry. 2007;305:123–131. doi: 10.1007/s11010-007-9535-1. [DOI] [PubMed] [Google Scholar]
- 51.Beisswenger PJ, Howell SK, Touchette AD, Lal S, Szwergold BS. Metformin reduces systemic methylglyoxal levels in type 2 diabetes. Diabetes. 1999;48:198–202. doi: 10.2337/diabetes.48.1.198. [DOI] [PubMed] [Google Scholar]
- 52.Hayashi T, Mase S, Namiki M. Formation of 3-Carbon Sugar Fragment at an Early Stage of the Browning Reaction of Sugar with Amines or Amino-Acids. Agr Biol Chem Tokyo. 1986;50:1959–1964. [Google Scholar]
- 53.Hayashi CM, Nagai R, Miyazaki K, Hayase F, Araki T, Ono T, Horiuchi S. Conversion of Amadori products of the Maillard reaction to N-epsilon-(carboxymethyl) lysine by short-term heating: Possible detection of artifacts by immunohistochemistry. Laboratory Investigation. 2002;82:795–807. doi: 10.1097/01.lab.0000018826.59648.07. [DOI] [PubMed] [Google Scholar]
- 54.Casazza JP, Felver ME, Veech RL. The metabolism of acetone in rat. The Journal of biological chemistry. 1984;259:231–236. [PubMed] [Google Scholar]
- 55.Richard JP. Acid-Base Catalysis of the Elimination and Isomerization-Reactions of Triose Phosphates. Journal of the American Chemical Society. 1984;106:4926–4936. [Google Scholar]
- 56.Bourajjaj M, Stehouwer CD, van Hinsbergh VW, Schalkwijk CG. Role of methylglyoxal adducts in the development of vascular complications in diabetes mellitus. Biochemical Society transactions. 2003;31:1400–1402. doi: 10.1042/bst0311400. [DOI] [PubMed] [Google Scholar]
- 57.Fosmark DS, Torjesen PA, Kilhovd BK, Berg TJ, Sandvik L, Hanssen KF, Agardh CD, Agardh E. Increased serum levels of the specific advanced glycation end product methylglyoxal-derived hydroimidazolone are associated with retinopathy in patients with type 2 diabetes mellitus. Metabolism: clinical and experimental. 2006;55:232–236. doi: 10.1016/j.metabol.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 58.Sun JK, Keenan HA, Cavallerano JD, Asztalos BF, Schaefer EJ, Sell DR, Strauch CM, Monnier VM, Doria A, Aiello LP, King GL. Protection from retinopathy and other complications in patients with type 1 diabetes of extreme duration: the joslin 50-year medalist study. Diabetes care. 2011;34:968–974. doi: 10.2337/dc10-1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jakus V, Rietbrock N. Advanced glycation end-products and the progress of diabetic vascular complications. Physiological research/Academia Scientiarum Bohemoslovaca. 2004;53:131–142. [PubMed] [Google Scholar]
- 60.Yamashita H, Shao J, Ishizuka T, Klepcyk PJ, Muhlenkamp P, Qiao L, Hoggard N, Friedman JE. Leptin administration prevents spontaneous gestational diabetes in heterozygous Lepr(db/+) mice: effects on placental leptin and fetal growth. Endocrinology. 2001;142:2888–2897. doi: 10.1210/endo.142.7.8227. [DOI] [PubMed] [Google Scholar]
- 61.Emerging Risk Factors C. Seshasai SR, Kaptoge S, Thompson A, Di Angelantonio E, Gao P, Sarwar N, Whincup PH, Mukamal KJ, Gillum RF, Holme I, Njolstad I, Fletcher A, Nilsson P, Lewington S, Collins R, Gudnason V, Thompson SG, Sattar N, Selvin E, Hu FB, Danesh J. Diabetes mellitus, fasting glucose, and risk of cause-specific death. The New England journal of medicine. 2011;364:829–841. doi: 10.1056/NEJMoa1008862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Reddy MA, Zhang E, Natarajan R. Epigenetic mechanisms in diabetic complications and metabolic memory. Diabetologia. 2015;58:443–455. doi: 10.1007/s00125-014-3462-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.