


Abstract
As proteomics initiatives mature, the need will arise for the multiple visualization of proteins and supramolecular complexes within their true context, in situ. Single-stranded DNA and RNA aptamers can be used for low resolution imaging of cellular receptors and cytoplasmic proteins by light microscopy (LM). These techniques, however, cannot be applied to the imaging of nuclear antigens as these single-stranded aptamers bind endogenous RNA and DNA with high affinity. To overcome this problem, we have developed a novel method for the in situ detection of proteins using double-stranded DNA oligonucleotides. To demonstrate this system we have utilized the prokaryotic DNA-binding proteins LacI and TetR as peptide tags to image fusion proteins in situ using dsDNA oligonucleotides encoding either the Lac or Tet operator. Using fluorescent and fluorogold dsDNA oligonucleotides, we localized within the nucleus a TetR–PML fusion protein within promyelocytic leukaemia protein (PML) bodies by LM and a LacI–SC35 fusion protein within nuclear speckles by correlative light and electron microscopy (LM/EM). Isolation of LacI–SC35 was also accomplished by using biotinylated dsDNA and streptavidin sepharose. The use of dsDNA oligonucleotides should complement existing aptamer in situ detection techniques by allowing the multiple detection and localization of nuclear proteins in situ and at high resolution.
INTRODUCTION
Proteins in cells are generally detected and localized with antibodies directed against the proteins themselves or to fused peptide tags (e.g. Flag-tag or HA-tag) (1). Antibodies can be functionalized with fluorescent molecules or gold particles for the detection of the proteins by light (LM) and electron microscopy (EM). To visualize the localization and dynamics of specific proteins in living cells, natural and engineered fluorescent proteins (FPs) [e.g. green fluorescent protein (GFP)] have been used as tags (2,3). Both antibodies and FPs have been attractive reagents for cell biologists because of their versatility and ease of use, though several limitations exist with these systems. One limitation is the small repertoire of bright and spectrally distinct proteins or fluorophores that are available. In addition, a specific combination of excitation filter, emission filter and dichroic mirror is required to visualize the fluorescence emission from a particular fluorophore or combination of fluorophores during epifluorescence microscopy, with the result that detection of three different proteins is often the practical limit. Furthermore, the in situ detection of three or more proteins simultaneously, using antibodies requires that each antibody be produced in a different and immunologically distinct animal so that secondary antibodies conjugated to each fluorophore do not cross-hybridize, resulting in false-positive detection. FPs cannot be used for EM but antibodies tagged with electron dense materials do provide the ability to localize proteins at high spatial resolution. The combination of these techniques via correlative LM and EM has provided great insight into cellular function by bridging the resolution gap between observations made by LM and the ultrastructure of complexes containing particular protein molecules (4,5). Unfortunately, the practical limit of simultaneous protein localization by EM is only two; achieved by using two different sizes of gold particles, e.g. 5 and 10 nm gold. In particular, no system currently exists for the multiple and simultaneous detection of proteins by EM. Thus, as proteomics efforts mature from the cataloguing of proteins within complexes to the actual localization of these proteins in situ, new reagents will be needed that can provide for multiple and simultaneous (multiplex) detection of proteins by both LM and EM.
Many of the problems faced by cell biologists when imaging proteins in situ are shared by biochemists developing diagnostic methods for the detection of multiple protein components in complex biological solutions. Over the past decade, a technology based on oligonucleotides has been developed that demonstrates the potential for multiplex detection of proteins in both solution and in situ. This technology relies on single-stranded (ss) DNA or RNA oligonucleotides called aptamers, which can bind protein targets with high affinity (6). However, the use of the term aptamer, which is derived from the latin root apt (meaning ‘to fit’), has not been specifically limited to single-stranded RNA and DNA but has been used to describe any synthetic polymer that binds a target ligand, including peptides (7). Aptamers that recognize a specific protein are selected by multiple rounds of binding, isolation and amplification using a procedure termed systematic evolution of ligands by exponential enrichment (SELEX)(8,9). The chemistry for the production and functionalization of oligonucleotides is well developed. Consequently, once aptamers are selected, they can be functionalized using a wide variety of fluorophores, as well as cobalt or iron paramagnetic particles, gold, radio-isotopes and biotin. Labelled aptamers have been used much in the same way as antibodies, for example in ELISAs, sandwich assays, and Western blotting (10–12). Aptamers have been selected that bind a number of eukaryotic transcription factors (13) as well as non-DNA binding proteins such as thrombin (14,15). Aptamers are generally much smaller than even Fab fragments of antibodies (<10 kDa on average compared with 50 kDa, respectively), indicating that they would exhibit high penetrance within fixed biological material and should provide superior spatial resolution when used as probes for EM (16). In addition, nucleic acid aptamers also provide the potential for multiplex detection of proteins in situ. Unfortunately, ssDNA and RNA aptamers have a propensity to bind cellular RNA and DNA with high affinity, precluding their use as detection reagents for nuclear proteins (16). Therefore, nucleic acid-based imaging reagents that can be used for high resolution imaging of nuclear proteins need to be developed.
In this paper we describe a method for the in situ detection and isolation of proteins that can be used to successfully image nuclear antigens by LM and EM. This method is based on the ability of a peptide tag to bind a double-stranded DNA oligonucleotide (dsDNA oligo) with high affinity. As a proof-of-principle, we have chosen to use two well-known prokaryotic DNA binding proteins, the Lac repressor (LacI) and the Tet repressor (TetR) as our peptide tags and the symmetric dsDNA Lac operator (O-Sym) or the dsDNA Tet operator (Tet-O) as our detecting oligos. We use these dsDNA oligos to detect a LacI-tagged splicing factor, SC35 and a TetR-tagged promyelocytic leukaemia protein (PML). In addition, we use the O-Sym dsDNA oligo to isolate LacI-tagged SC35 from cell extracts via biotinylated dsDNA oligos and streptavidin sepharose. Lastly, using fluorescent dsDNA coupled to gold particles we demonstrate the utility of dsDNA oligos for correlative LM/EM. Our results demonstrate that LacI-tagged SC35 and TetR-tagged PML localize adequately to nuclear speckles or PML nuclear bodies, and that we can detect these proteins in situ using small (41 bp) dsDNA oligos at nanomolar concentrations. Furthermore, we can detect both these proteins in the same cell without cross-hybridization of the detecting oligos, demonstrating the ability of dsDNA oligos for the multiplex detection of proteins in situ. Thus, our results provide the first evidence that dsDNA oligos can be used to localize proteins in situ within sub-cellular structures at high resolution.
MATERIALS AND METHODS
Vector construction
The mammalian expression vector pGD-Flag-Lac338 and its derivative pGD-Flag-Lac-SC35 were constructed as follows. Plasmid pcDNA3.1/His-C (Invitrogen) was cut with HindIII and Asp718 and ligated to an Asp718/HindIII cut PCR product (Flag-Lac338) to produce pGD-Flag-Lac338. The Flag-Lac338 PCR product encodes the amino acid sequence of the Flag epitope (MDYKDDDDK) fused to the first 338 aminoacids of the LacI and was amplified from the vector p3′SS-GFP-Lac-NLS [generous gift of A. Belmont (17)] using the following primers, LACFLAG-1 (tgacgtaagcttaggatggactataaagacgatgacgataaaccagtaacgttatacga) and LAC3R-338 (ctataaggtaccgccccctccacttccaccgcccccagaggcggtttgcgtattgggcgcca). To generate pGD-Flag-Lac-SC35, both pGD-Flag-Lac338 and the vector pBSK-SC35 were first cut with Asp718, blunt ended with Klenow in the same buffer, followed by phenol/chlorophorm extraction and precipitation, after which the DNA was resuspended and digested with BamHI. The resulting blunt end/BamHI pGD-Flag-Lac338 vector and the fragment containing the SC35 cDNA from pBSK-SC35 were ligated together. The pBSK-SC35 was generated by subcloning of the human SC35 HindIII fragment from pEGFP-SC35 (gift of Dr M. Hendzel) into HindIII cut pBlueScript(+) (Stratagene).
The mammalian expression vector pGD-HA-TET and its derivative pGD-HA-TET-PML were constructed as follows. Plasmid pcDNA3.1/His-C (Invitrogen) was cut with HindIII and Asp718 and ligated to an Asp718/HindIII cut PCR product (HA-TET) to produce pGD-HA-TET. The HA-TET PCR product encodes the amino acid sequence of the haemagglutinin (HA) epitope (MGYPYDVPDYAG) fused to the TetR and was amplified from the vector pTET-OFF (Clontech) using the following primers, TET-HA-1F (gggttttaagcttaccatgggatatccctatgatgtgccagactacgcgggaatgtctagattagataaaagt) and TET-HA-1R (tagattggatccaccgcctcctttaagttgtttttctaatccgca). To generate pGD-HA-TET-PML, both pGD-HA-TET and the vector pBSK-PML IV were first cut with BamHI and EcoRI, after which the linearized DNA fragment containing the human PML IV gene was gel purified and ligated to the linearized pGD-HA-TET. pBSK-PML IV was generated by subcloning the BamHI/EcoRI DNA fragment of the human PML IV gene from pDsRED-PML (gift of M. Hendzel) into BamHI/EcoRI digested pBlueScript(+) (Stratagene).
Oligonucleotide synthesis and modification
For construction of functionalized dsDNA oligos recognized by LacI, two 41 bp single-stranded oligonucleotides were constructed encoding the symmetrical Lac operator 19 bp core sequence O-Sym (18): O-Sym-1 (*gcgtgtgccagaattgtgagcgctcacaatttcttgaatct) and O-Sym-2 (*agattcaagaaattgtgagcgctcacaattctggctcacgc); where * represents a 5′ modification with either biotin, Cy3 or a disulfide group linked via a (CH2)6 spacer (Sigma). O-Sym-1 and 2 were resuspended to 200 μM and equal volumes of each oligo were added to a 1/5 volume of 10× annealing buffer (50 mM Tris–Hcl pH 7.5, 1 M NaCl and 0.2 mM EDTA). The oligo mixture was then boiled for 4 min at 90°C followed by slow equilibration to room temperature to allow the annealing of the two O-Sym oligos to produce a ∼91 μM solution of the dsDNA O-Sym oligonucleotide.
Similarly, functionalized dsDNA oligos recognized by TetR, were constructed from the following 41 bp oligonucleotides: Tet-O-1 (*tcgagtttactccctatcagtgatagagaacgtatgtcgcc) and Tet-O-2 (*ggcgacatacgttctctatcactgatagggagtaaactcgt); where * represents a 5′ modification with either biotin or Cy5 (Sigma). These oligonucleotides were annealed as described above to produce a ∼91 μM solution of dsDNA Tet-O oligonucleotide.
Fluorogold dsDNA oligo synthesis
The dsDNA oligo consisting of the disulfide-modified O-Sym-1 and Cy3-modified O-Sym-2 was treated with 0.04 M DTT (0.17 M Na2HPO4, pH 8.0) for 16 h at room temperature to cleave the disulfide bond. The thiol by-products and DTT were removed using a Sephadex column (NAP-5, Amersham Biosciences) equilibrated with 20 mM Na2HPO4, 150 NaCl and 1 mM EDTA, pH 6.5 (conjugation buffer). The dsDNA oligo was further purified by 70% ethanol precipitation and repeated washes. The pellet was resuspended in the conjugation buffer to a concentration of 100 μM. Then, 10 μl (1 nmol) of this solution was added to 10 nmol of monomaledo-undecagold reagent (Nanoprobes, Yaphank, NY) in 1 ml of the conjugation buffer. The mixture was incubated at room temperature with stirring for 1 h, then incubated at 4°C for 16 h. The functionalized dsDNA oligos were isolated from excess nanocrystals by ethanol precipitation with excess salmon sperm DNA, followed by repeated washes. The product was resuspended in 10 mM Tris–HCl, 0.1 mM EDTA and 150 mM KCl, pH 7.5, to achieve a final oligo concentration of ∼10 μM.
Cell culture and transfection
SK-N-SH cells were cultured according to the American Type Culture Collection (ATCC) guidelines for each cell line. Cells were split the day before transfection and 2 × 105 cells were seeded at 105 cells/ml onto 18 mm square coverslips in 8 or 6 well plates. The following day, cells were transfected with 1–2 μg of pGD-Flag-Lac338 and pGD-TET-PML DNA alone or combined per well using Lipofectamine 2000 (Invitrogen) as suggested by the manufacturer.
Protein isolation using dsDNA oligos and Western analysis
SK-N-SH neuroblastoma cells transfected with either LacI or LacI–SC35 were lysed by sonication [3 × 30 s at 20% power using an Ultrasonic Processor (Hert Systems)] in protein isolation buffer-A (PIB-A; 20 mM HEPES pH 7.5, 250 mM KCl, 10% glycerol, 1 mM phenylmethylsulfonyl fluoride (PMSF), 1× Complete protease cocktail (Roche), 1 mM NaF, 40 mM β-glycerolphosphate). The resulting lysate (PI lysate) was then centrifuged at 12 000 g for 20 min to remove cellular debris and pre-cleared by incubation of the supernatant with streptavidin sepharose beads (Invitrogen) for 1 h at 4°C followed by centrifugation at 12 000 g for 5 s to remove the sepharose beads. The precleared PI lysate was either snap frozen on dry ice or used immediately for protein isolation. For protein isolation using dsDNA oligos, streptavidin sepharose was first pre-incubated with biotinylated O-Sym oligo (100 μl of streptavidin sepharose in 1 ml of PBS containing 500 nM dsDNA oligo) and then washed 3 times with PIB-A. Then PI lysate containing 250–500 μg of total protein was incubated with 30–50 μl of streptavidin sepharose beads pre-incubated with biotinylated O-Sym oligo over night at 4°C. Mock protein isolation (Mock-PI) was carried out using streptavidin sepharose without oligos. The protein isolated using dsDNA oligos was then washed 3 times with PIB-A containing 0.5% Triton X-100 and 2 times with phosphate-buffered saline (PBS) before being boiled for SDS–PAGE and western transfer. Western blots were visualized using the SuperSignal West Pico Chemiluminescent kit (Pierce) as per the manufacturer's instructions.
Alternatively, qualitative analysis of the level of purification of LacI–SC35 by protein isolation using dsDNA oligos, versus immunoprecipitation using an antibody, was carried out by SDS–PAGE, followed by Coomassie blue staining of the acrylamide gel to visualize the isolated proteins. Briefly, 500–1000 μg of total protein lysate prepared from LacI–SC35 transfected SK-N-SH, as described above, was first precleared twice by incubation with 2 × 100 μl of streptavidin sepharose 1 h at 4°C. The precleared lysate was then incubated overnight at 4°C with continuous mixing with either 100 μl of streptavidin sepharose alone (Mock) or pre-incubated with O-Sym oligo or 50 μl of Protein G sepharose pre-incubated with 20 μg of anti-Flag epitope antibody M2 (Sigma) (immunoprecipitation or IP). In addition, unrelated dsDNA oligo (i.e. TetO) was added to a concentration of ∼1 μM to the precleared lysate during protein isolation to reduce the non-specific co-purification of other DNA binding proteins. The proteins isolated by dsDNA oligonucleotides and anti-Flag IP were then washed 3 times with A-PIB containing 0.5% Triton X-100 and 2 times with PBS before being boiled for SDS–PAGE followed by staining of the gel by Coomassie blue.
Oligonucleotide hybridization, immunofluorescence and microscopic imaging of LacI- and TetR-tagged proteins
Twenty-four to 36 h post transfection cells were fixed in 1–2% paraformaldehyde in PBS for 5 min. followed by permeabilization in 0.5% Triton X-100 for 5 min at RT. Following several washes in PBS, cells were then blocked for 20 min at RT with O-Sym Binding/Blocking (OSB) buffer (10 mM Tris–HCl pH 7.5, 0.1 mM EDTA, 150 mM KCl, 600 μg/ml of sheared herring sperm DNA and 200 μg/ml of BSA). Directly following the blocking step, cells were hybridized for 1–2 h at 37°C with the dsDNA O-Sym oligo (labelled with either Cy3, biotin or both) alone or combined with dsDNA Tet-O oligo (labelled with either Cy5, biotin or both) in OSB buffer at a concentration of 50–100 nM. After hybridization with the O-Sym or Tet-O oligo, coverslips were either washed with 3× PBS and mounted in anti-fade reagent for immediate immunofluorescence detection or were further processed for immunofluorescent localization of PML, SC35 or PRP4 kinase (PRP4K) as described previously (19). Primary antibodies directed against the Flag epitope (1:250, mouse mAb M2, Sigma), HA epitope (1:1000, mouse mAb HA.11, Zymed), PML (1:200, rabbit anti-PML antibody, Chemicon) or PRP4K [1:100, sheep antibody H143 (18)] were subsequently detected with secondary antibodies conjugated to Cy5 (1:100, donkey anti-sheep and anti-mouse Abs) or to Cy3 (1:500, donkey anti-rabbit Ab) (Jackson Immuno Laboratories). Alternatively, following hybridization with a biotinylated O-Sym oligo, the localization of the LacI- or TetR-fusion protein was visualized using Cy3-Streptavidin (1:200, Sigma). All fluorescence microscopy was performed using a Leica DMR upright microscope fitted with a Hamamatsu ORCA camera. Openlab V3.5.1 (Improvision) software was used to collect fluorescence images. Images were processed using either ImageJ V1.28 (NIH) or Photoshop 6.0/7.0 (Adobe).
Correlative light and electron spectroscopic imaging (LM/ESI) of fluorogold dsDNA oligonucleotides
SK-N-SH cells transfected with LacI–SC35 were labelled using dsDNA oligos as above using fluorogold O-Sym oligos. After labelling, cells were post-fixed (8% paraformaldehyde and 2% glutaraldehyde for 5 min at RT) and subjected to silver enhancement of the fluorogold dsDNA oligos for 30 min at RT using a either a silver enhancement kit for LM or EM (Electron Microscopy Sciences). The EM enhancement was performed 3 times, using fresh enhancement solution each time. The cells were then dehydrated in a series of graded ethanol washes (30, 50, 70 and 95%) before Quetol 651 resin (EM Science) infiltration, curing and sectioning, as described previously (5,20). Images of cells of interest were collected by both LM, as described above, and ESI, using a Tecnai 20 TEM (FEI) at 200 kV, equipped with an imaging spectrometer (Gatan). Elemental maps were generated by dividing the element-enhanced post-edge image by the pre-edge image following alignment by cross-correlation. Net ratio elemental maps were derived from pre- and post-edge images recorded at 120 and 155 eV (LII,III edge) for phosphorus, and at 385 and 415 eV (K edge) for nitrogen. The recording times required to obtain the pre-edge and post-edge images are in the range of 10–30 s. The images were processed using Digital Micrograph (Gatan) and Photoshop 6.0/7.0 (Adobe).
RESULTS
An overview of protein in situ detection and isolation using dsDNA oligos is shown in Figure 1. The LacI and the TetR were chosen as dsDNA-binding protein tags to demonstrate the ability of dsDNA oligos to image proteins in situ within cells. A LacI-fusion vector was generated (pGD-Flag-Lac338) containing the sequence of the first 338 aminoacids of LacI downstream of a single Flag-tag. This truncated form of the LacI protein is incapable of forming tetramers and is thought to form weak dimers in vivo (16). A second DNA construct, a TetR-fusion vector was constructed (pGD-HA-TET) containing the full-length Tet repressor downstream of a single HA epitope-tag. The cDNA sequence of any protein can then be cloned into these vectors for the expression of LacI- or TetR-tagged fusions of the target protein in mammalian cells. Consequently, the fusion protein can be localized using a fluorescent labelled dsDNA oligo specific for the LacI (i.e. O-Sym) or TetR protein (i.e. TET-O) (see Methods and Materials).
Figure 1.

In situ detection and isolation of proteins using dsDNA oligonucleotides. (A) Detection of proteins in situ using dsDNA oligos. Cells grown on slides are fixed and hybridized with fluorescently labelled dsDNA oligos (red or green) that can detect the presence of proteins fused to either LacI (1) or TetR (2). The fluorescent dsDNA oligos corresponding to the operator sequences for LacI or TetR (green or red, respectively) bind each bacterial fusion protein specifically without cross hybridizing resulting in the sub-nuclear localization of the tagged proteins by light microscopy. (B) Isolation and detection of proteins in vitro using dsDNA oligos. Streptavidin-sepharose beads (S) are sequentially incubated with biotinylated dsDNA oligos representing the operator sequences for LacI followed by incubation with a cell lysate containing LacI fused to a protein of interest (1). After binding the beads are washed and the resulting purified LacI-fusion protein can be observed by SDS–PAGE. M, marker, IN, input lysate, PI, isolated protein.
In situ detection of proteins within subnuclear compartments using dsDNA oligos
To demonstrate the ability of dsDNA oligos (Figure 1A) to localize proteins in situ, we chose to observe the localization of the splicing factor protein, SC35. SC35 localizes to well characterized sub-domains within the mammalian nucleus, termed nuclear speckles at the light level and interchromatin granule clusters at the ultrastructural level. These structures contain both RNAs and proteins involved in pre-mRNA splicing and metabolism (21). We cloned the cDNA sequence encoding the human SC35 gene into the LacI expression vector to create pGD-Flag-Lac-SC35. A human neuroblastoma cell line, SK-N-SH, was transfected with pGD-Flag-Lac-SC35. After 24 h post transfection, cells were fixed and analysed by immunofluorescence using antibodies directed against the Flag epitope or with the O-Sym dsDNA oligo (Figure 2A and B). This localization was compared with that of PRP4 kinase (PRP4K), an endogenous marker for nuclear speckle domains (19), by antibody detection (PRP4K, Figure 2A and B). Both the anti-Flag antibody (Flag, Figure 2A) and the O-Sym oligo (O-Sym, Figure 2B) could specifically detect cells transfected with the LacI–SC35 fusion vector. We found that the O-Sym oligo could be used at concentrations between 50 and 100 nM for the detection of LacI-fusion proteins within transfected cells. The distribution of the LacI–SC35 fusion protein was indistinguishable from that of PRP4K, indicating that the fusion protein had been targeted correctly to nuclear speckle domains.
Figure 2.

In situ localization of SC35 and PML in human SK-N-SH cells using dsDNA oligonucleotides. (A) Localization of the LacI-tagged SC35 in paraformaldehyde fixed cells transfected with pGD-Flag-Lac-SC35 using the anti-Flag antibody M2 (Flag, red) or (B) with a Cy3-labelled dsDNA oligo (O-Sym, red) specific for LacI. PRP4 kinase was also localized as an endogenous marker for splicing speckles by antibody detection (PRP4K, green). Positive co-localization between PRP4K and LacI-tagged SC35 is demonstrated by a yellow signal in the merged images. The localization of the LacI–SC35 fusion protein was observed only in transfected cells (see B, O-Sym) and showed complete co-localization with PRP4 kinase (PRP4K, green) in nuclear speckles. (C) Localization of the TetR-tagged PML in paraformaldehyde fixed cells transfected with pGD-HA-TET-PML using the anti-HA (HA, red) or (D) with a Cy5-labelled dsDNA oligo (TET-O, red) specific for TetR. Endogenous PML was also localized as a marker for PML nuclear bodies antibody detection (PML, green). Positive co-localization between PML and TetR-tagged PML is demonstrated by a yellow signal in the merged images. The localization of the TetR–PML fusion protein was observed only in transfected cells (see C, TET-O) and showed complete co-localization with PML (PML, green) in PML nuclear bodies. (E) Multiple detection and localization of LacI–SC35 and TetR–PML in cells transfected with pGD-HA-TET-PML and pGD-Flag-Lac338-Sc35. Localization of LacI–SC35 and TetR–PML was accomplished using Cy3-labelled O-Sym or Cy5-labelled TET-O dsDNA oligos, respectively. TetR–PML and LacI–SC35 do not co-localize [separate red and green signals (respectively) in merged image], thus demonstrating the utility of dsDNA oligos for the multiplex detection of proteins in situ. DNA was counterstained with DAPI (blue in merged images). Scale bars = 5 μm.
To demonstrate the utility of dsDNA oligonucleotides for multiplex detection of proteins in situ, we generated a second fusion protein vector containing PML fused with the Tet repressor (pGD-HA-TET-PML). The PML protein is a structural component of PML nuclear bodies implicated in DNA repair, apoptosis, gene regulation, and tumour suppression (22–24). PML nuclear bodies are functionally, spatially and biochemically distinct from nuclear speckles and thus a fusion protein targeted to this sub-nuclear compartment should not co-localize with a protein directed to nuclear speckles containing SC35. SK-N-SH cells were transfected with pGD-HA-TET-PML alone or in combination with pGD-Flag-Lac-SC35. After 24 h post transfection, cells were fixed and analysed by immunofluorescence using antibodies directed against the HA epitope or with the Tet-O dsDNA oligo (Figure 2C and D). This localization was compared with that of endogenous PML, using an anti-PML rabbit polyclonal antibody (PML, Figure 2C and D). Both anti-HA antibodies (HA, Figure 2A) and the Tet-O oligo (TET-O, Figure 2D) could specifically detect cells transfected with the TetR–PML fusion vector. In addition, dsDNA oligo detection of TetR–PML and LacI–SC35 was accomplished over a wide range of transient expression levels, which paralleled or exceeded antibody detection of the same fusion proteins. Although we did not quantitate absolute expression on a cell per cell basis, we estimate the fusion-protein levels to be physiologically relevant in the images shown in Figure 2 based on the nuclear morphology of these cells in reference to PML bodies and splicing speckles, which appear normal. The Tet-O oligo could be used at concentrations between 50 and 100 nM, thus equalling the sensitivity of O-Sym oligo detection of LacI-fusion proteins within transfected cells. The TetR–PML fusion protein localized correctly to PML bodies as demonstrated by co-localization with endogenous PML (Figure 2C and D). Unambiguous detection and localization of TetR–PML and LacI–SC35 was accomplished within the same cell by hybridization with both dsDNA oligos (Tet-O and O-Sym, respectively; Figure 2E). As expected, PML nuclear bodies containing TetR–PML and nuclear speckles containing LacI–SC35 did not co-localize, demonstrating the ability of dsDNA oligos to detect multiple proteins within the same cell without cross-hybridization.
Isolation of the LacI-tagged SC35 protein using dsDNA oligos
To demonstrate the ability of dsDNA oligos for protein isolation (Figure 1B), we made whole cell lysates from SK-N-SH cells transiently expressing either LacI-tagged SC35 or LacI alone. The lysates were then used in protein isolation using biotinylated O-Sym dsDNA oligos immobilized on streptavidin sepharose. The LacI protein also contains an N-terminal Flag-tag, which was used for western analysis of the LacI–SC35 isolated using dsDNA oligos (Figure 3). The level of the LacI–SC35 fusion protein in these lysates was roughly equal to the endogenous level of SC35 protein (data not shown). Both LacI–SC35 and LacI alone were isolated by the O-Sym oligo (Lanes 3, 5 of Figure 3A, respectively) but streptavidin sepharose alone (Lane 4, Figure 3A) failed to isolate LacI–SC35. The identity of the LacI–SC35 protein was confirmed by western analysis of the same blot using the anti-phospho-SR protein monoclonal antibody mAb 104 (Figure 3B), which detects a number of SR proteins including SC35 (25). We also carried out a qualitative comparison between IP using an anti-Flag epitope antibody and dsDNA oligo isolation of the LacI–SC35 fusion protein (Figure 3C). Although a comparison of the purity of the LacI–SC35 protein isolated by IP or dsDNA oligos was precluded by the relatively small recovery of protein in our experiments, the purity of the IP was compromised by the co-purification of the immunoglobulin used for IP (Figure 3C, the band marked by the asterix is the mouse IgG heavy chain).
Figure 3.
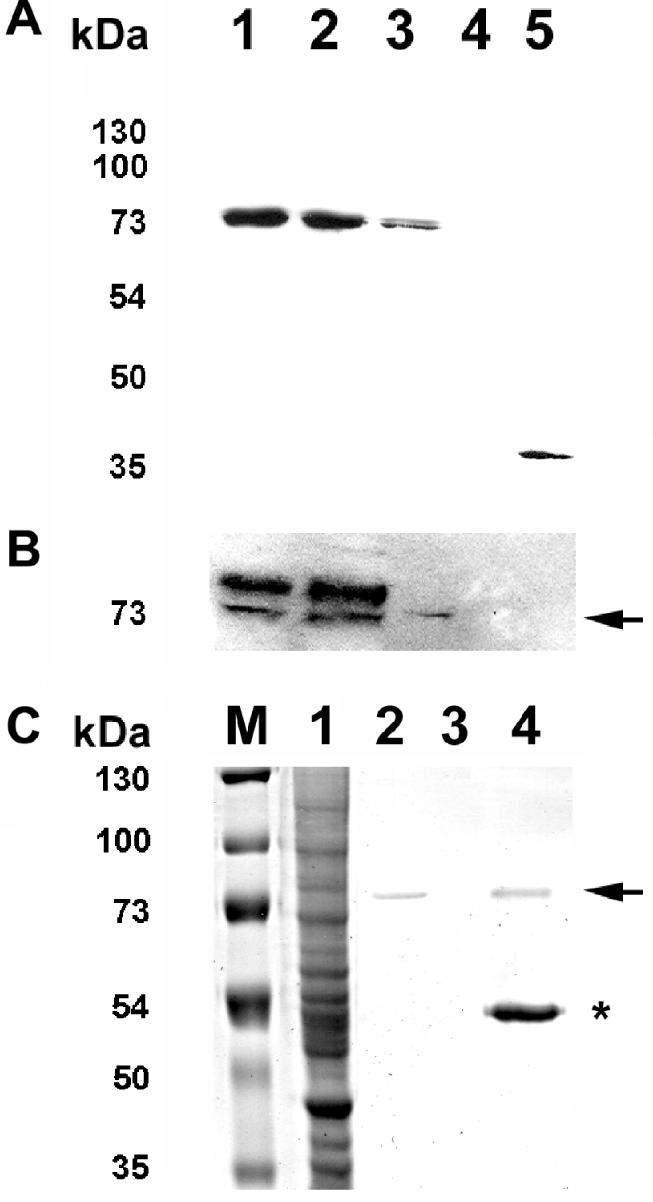
Isolation of LacI-tagged SC35 from human SK-N-SH cells using dsDNA oligonucleotides. Total cellular lysates and isolated protein (PI) lysates were prepared from SK-N-SH cells transiently expressing LacI or LacI-tagged SC35. The protein isolation of LacI and LacI-tagged SC35 from PI-lysates was carried out using streptavidin beads pre-incubated with biotinylated dsDNA oligo specific for LacI (O-Sym). Western analysis of total cellular lysate (1), PI lysate (2), protein isolated by dsDNA oligos (oligo-PI) (3), and mock oligo-PI (4) from LacI–SC35 expressing cells, or PI lysate were generated from LacI expressing cells. Proteins were detected using either anti-Flag (A) or anti-SR protein (mAb 104) (B) antibodies. In addition, a Coomassie stained SDS–PAGE gel is shown as a qualitative comparison of dsDNA oligo PI versus immunopreciptiation (IP) using an anti-Flag antibody (C). In panel C: lane M, protein molecular weight marker; lane 1, 40 μg of total lysate; lane 2, oligo-PI; lane 3, Mock oligo-PI; and lane 4, anti-Flag IP. The asterisk marks the position of co-purifying immunoglobulin (i.e. heavy chain is shown) in the Flag-IP. The black arrows indicate the position of LacI–SC35 in panels B,C. Approximately 250–500 μg or 500–1000 μg of total protein lysate was used for western or Coomassie SDS–PAGE analysis, respectively.
Correlative light and electron spectroscopic imaging using fluorogold dsDNA oligos
In order to demonstrate the utility of our dsDNA oligo detection system for correlative fluorescence and EM, a fluorogold dsDNA oligo was generated. This oligo contains a Cy3 moiety on the 5′ end of one strand of the O-Sym oligo for detection in the fluorescence microscope, and an undecagold nanoparticle on the 5′ end of the complementary strand for detection by EM. The nanogold label by itself is too small to be detected by conventional transmission EM, but can easily be enlarged for detection by silver enhancement. To illustrate the use of the doubly labelled fluorogold oligo for detection by correlative LM and EM, SK-N-SH neuroblastoma cells were transfected with pGD-Flag-Lac-SC35 and detected with the fluorogold oligo as shown in Figure 4A. The transfected cell (left panel) exhibited the distinct speckled pattern of the SC35 domain (21), illustrating the effectiveness of the fluorescent component of the oligo. We confirmed the presence of gold in the oligo by performing an extended silver enhancement on the sample and bright field imaging by the light microscope [Figure 4A (right panel)]. Whereas transfected cells are heavily labelled with silver, untransfected cells have only background levels of silver deposition.
Figure 4.

Correlative light and electron spectroscopic imaging (LM/ESI) of SK-N-SH cells expressing LacI-tagged SC35 using fluorogold dsDNA oligos. Detection of nuclear speckles (arrow) via the Cy3 fluorophore (A, left panel), and confirmation of gold conjugation to the dsDNA oligo through silver enhancement (A, right panel). Untransfected cells (arrowhead) contain background levels of silver deposition. Scale bar = 200 nm. (B) Overlay of fluorescence and low magnification ESI micrographs collected at 155 eV. With two rounds of the silver enhancement using a LM enhancement kit (B, left panel), the silver particles are visible as bright spots. The right panel is an overlay image of a nucleus containing EM-enhanced gold. Scale bar = 2 μm. An area corresponding to an IGC, as defined by a box in the left panel is analysed at high resolution, and maps of phosphorus (C, red panel) and nitrogen (C, green panel) show the ultrastructure of the IGC region, which is low in phosphorus content and contains protein-based fibrous structures. The fluorogold oligo localization is indicated with silver-enhanced gold particles false-coloured in white. The composite map (D, left panel) illustrates the position of the IGC relative to chromatin (Ch, yellow) and the nucleolus (Nu, yellow-green). The silver particles in the interior of the IGC are labelled with arrowheads, whereas those proximal to the neighbouring chromatin are indicated by arrows. The composite map (D, right panel) of the EM-enhanced nucleus (B, right panel) contains smaller but uniform silver particles, as indicated by the arrows and arrowheads. Scale bar = 5 μm.
Correlative fluorescence and ESI (5) was then carried out on LacI–SC35 transfected cells (see Materials and Methods). Physical sections were first imaged with the fluorescence microscope to detect the Cy3 label bound to the oligo. The section containing the same cell was then imaged in the electron microscope. We chose to use ESI, rather than conventional transmission EM because of its advantages in identifying protein- versus nucleic acid-based components in and around nuclear speckle domains (also known as interchromatin granule clusters or IGCs). An overlay of fluorescence microscope images and low magnification energy filtered images were obtained (Figure 4B). Only cells that expressed the LacI–SC35 fusion protein had detectable silver-enhanced fluorogold oligo and this signal was restricted to regions of Cy3 signal in the correlative fluorescence image (Figure 4B). A phosphorus map [Figure 4C (left panel)] and a nitrogen map [Figure 4C (right)] can be used to identify condensed chromatin, the nucleolus and the IGC domain. The fluorogold oligo was heavily enhanced with silver, resulting in large accumulation of silver, shown as white blobs (Figure 4C). Careful examination of the low magnification image also revealed the large silver clusters [Figure 4B (left panel)]. The composite of the phosphorus and nitrogen images [Figure 4D (left panel)] shows chromatin (yellow regions) and a morphologically dense region, corresponding to the nucleolus (yellow-green). The green structures, including the fibrous material in the IGC signifies protein-based structures. The area delineated by a dashed line corresponds to the region occupied by the IGC. The gold particles within the interior of this domain are highlighted by arrowheads, whereas the particles having close proximity to neighbouring chromatin are highlighted with arrows.
In a second nucleus presented, the gold particles were enhanced to a lesser degree [Figure 4B (right panel)], and imaged by phosphorus and nitrogen mapping at higher magnification [Figure 4D (composite shown in right panel)]. The silver particles are much smaller but more uniform, thus more suitable for ultrastructural characterization of protein components. Fluorogold dsDNA oligos revealing the presence of SC35 in the core of the IGC are indicated with arrowheads, whereas oligos revealing the presence of SC35 at the IGC:chromatin boundary are indicated with arrows.
DISCUSSION
In the present study, we have demonstrated the ability of synthetic dsDNA oligos, to detect proteins in situ within mammalian cells. As proof-of-principle, we used a dsDNA oligo based on the symmetrical operator of the LacI repressor (O-Sym) to successfully identify the presence of the LacI–SC35 fusion protein in nuclear speckles (Figure 2). Qualitative comparison indicate that in situ detection with dsDNA oligos is at least as sensitive if not superior to antibody detection of the same fusion protein using the anti-Flag antibody. Our dsDNA oligo in situ detection protocol is rapid and flexible, being ∼90 min shorter than conventional immuno-detection using primary and secondary antibodies, and easily combined with the standard antibody detection protocol without extensive sample processing. These results are in sharp contrast to a recent failure to specifically localize GFP in both fungi and mammalian cells using a ssDNA aptamer selected in vitro to bind GFP (16). Two other studies have shown the ability of fluorescently tagged, ssDNA aptamers to image endogenous proteins by LM either as cell surface markers [pigpen endothelial protein (26)] or cytoplasmic antigens [cytoplasmic ERK2 (27)]. However, to date there are no studies describing the high resolution imaging of subnuclear antigens, most likely due to the propensity of ssDNA aptamers to non specifically bind RNA within the nucleus (16). Therefore, an important conclusion from our study is that aptamers that are at least in part double stranded could provide a simple means of circumventing the propensity of ssRNA or DNA aptamers to bind endogenous RNA during the in situ detection of nuclear antigens. In addition, we have also successfully detected cytoplasmic fusion proteins using our dsDNA oligo in situ detection protocol, suggesting that the technique need not be limited to the detection of nuclear antigens (Klip, Dellaire and Bazett-Jones, unpublished observations). Thus, we present both the first peptide-oligo tag system for in situ detection and the first demonstration that double-stranded nucleic acids can be used to image proteins in situ at high resolution within sub-nuclear compartments of mammalian cells. Futhermore, our system is the first oligonucleotides-based system to demonstrate the ability for multiplex detection of proteins in situ (Figure 2).
Both the O-Sym and Tet-O dsDNA oligos were routinely used at concentrations in the 50–100 nM range to successfully detect LacI or TetR-tagged proteins (respectively) in situ. The reported affinity of the Lac repressor for the O-Sym operator in solution is much higher; binding constants have been reported in the 1–10 nM range for the dimeric form of LacI (28). The requirement for fixation before in situ detection may limit conformational changes in the LacI or TetR protein required to bind their operators with optimum efficiency. Supporting this hypothesis, longer fixation times or higher concentrations of fixative resulted in the progressive reduction in O-Sym detection sensitivity (data not shown). Nonetheless, the observed optimum binding concentration of both the O-Sym and TetR operator is in the same range as the published binding constants of many protein specific nucleic acid aptamers [i.e. in the 10 nM–10 μM range (29)]. Thus, we believe that beyond the detection of DNA binding protein tags, endogenous nuclear proteins could be detected in situ using ssDNA aptamers that are at least partially double stranded in nature and have affinities at or <100 nM for their ligand.
Transcription factors and prokaryotic DNA binding proteins such as LacI have been traditionally purified using dsDNAs encoding the binding sites of these proteins bound to sepharose (30). Similary, using our dsDNA oligo-peptide tag system we were able to isolate transiently expressed LacI-tagged SC35 from total cell lysate using biotinylated dsDNA oligos encoding the Lac Operator bound to streptavidin sepharose (Figure 3). In addition, ssDNA aptamers directed against recombinant proteins have been used in aptamer-affinity chromatography (31). For the purification of proteins for proteomics analysis such as mass spectrometry, it is preferred if the protein complexes of interest are isolated under native conditions. For example, the tandem affinity purification (TAP) tag system, which utilizes tandem IgG binding domains from protein A and the calmodulin binding peptide, allows the gentle elution of proteins under native conditions using ethylenebis (oxyethylenenitrilo tetraacetic acid) (EGTA) (32). In our dsDNA-peptide tag system, which is based on TetR or LacI fusion proteins, gentle elution from the affinity column may be accomplished by the addition of heparin (33) to the column or by using small metabolites such as tetracycline (34) or isopropyl β-d-thiogalactoside (IPTG) (30), respectively. In particular, affinity purification of proteins by nucleic acid based chromatography may provide a preferred means of isolation of recombinant proteins for therapeutic use, as antibody-based chromatography runs the risk of the introduction of antibody fragments that may induce an undesirable antigenic response in the patient (e.g. Figure 3C). Thus, although similar to peptide-tag systems in the detection and isolation of proteins using antibodies (e.g. Flag or HA epitope tag), protein isolation using dsDNA oligos may provide an ideal mode of recombinant protein purification when contamination by antibody fragments is undesirable, such as during protein isolation for mass spectrometry analysis or therapeutic use.
Another advantage of nucleic acids as detection reagents over antibodies is that their synthesis is rapid and the position and stoichiometry of the chemical modification can be controlled. In addition, nucleic acids can be coupled with a variety of moieties during synthesis, including reactive thiols, amines, paramagnetic beads, fluorophores and biotin. Although we used fluorescent, biotinylated and gold-modified oligonucleotides for LM it would be possible to directly couple other moieties more useful for protein purification or ESI such as paramagnetic particles or metal complexes. In particular, the use of dsDNA oligos allows the detecting molecule to be easily functionalized at four independent sites during oligosynthesis (i.e. at the 3′ and 5′ of each DNA strand). In addition, further functional diversity could be accomplished by the direct coupling of peptides with desirable properties to the oligo, such as specific reactive groups or side chains (e.g. additional amine groups via poly-lysine). Thus, it should be possible to create pluri-functional oligos for use with our dsDNA oligo technology. For example, in the present study we created a pluri-functional dsDNA oligo by coupling a fluorophore on one DNA strand and a nanogold particle to the complementary DNA strand. The resulting dsDNA fluorogold oligo was then used for correlative LM (through the fluorophore) and EM (through post-detection silver enhancement of the gold moiety) (Figure 4).
Lac repressor is the prototype of a large family of prokaryotic helix–turn–helix (HTH) DNA binding proteins, the sequences of which are available for over 25 members including the fructose repressor (FruR), the purine repressor (PurR), and the galactose repressor (GalR) (35). In addition, the TetR, AraC, MerR, and MarR families of DNA binding proteins involved in multi-drug transport and resistance in prokaryotes provide additional examples of HTH-containing DNA binding proteins that bind specific operator sequences (36). Several of these proteins including TetR, BmrR, QacR, and EmrR have been studied extensively both biochemically and by X-ray crystallography.
Beyond the sheer number of examples, prokaryotic DNA binding proteins provide several advantages as peptide tags for in situ detection of epitope tagged-proteins in mammalian cells. First, many prokaryotic DNA binding proteins have evolved as components of regulatory operon systems that sense the presence of small molecules and metabolites. These small molecules modulate the binding of such proteins to their DNA operator sequences through binding to regulatory domains within these transcription factors and repressors. For example, IPTG can reduce the affinity of LacI for its operator in a dose-dependent manner (37). Similarly for TetR, the presence of tetracycline modulates the interaction of TetR with its operator (34). Thus, it may be possible to modulate the sensitivity of in situ detection by dsDNA oligos and as mentioned above, allow a gentle method of protein elution during purification of recombinant proteins using these oligos. Lastly, because of the evolutionary distance between prokaryotes and higher eukaryotes such as mammals, the existence of high affinity operator sequences within the mammalian genome would be unlikely, thus fusion proteins encoding these prokaryotic peptides are less likely to mislocalize by binding to genomic sequences. Taken together, these and other prokaryotic DNA binding proteins present a wealth of potential dsDNA oligo-peptide tag pairs for protein isolation and in situ detection using dsDNA oligos. Collectively, dsDNA oligo and aptamer in situ detection (Aptamer-ID) techniques represent a technology that could provide the means for massive parallel detection and localization of proteins in vitro or in situ by LM and EM. Using our knowledge of dsDNA oligo detection, we are currently evaluating modifications of the SELEX procedure to generate DNA aptamers that can be successfully used for Aptamer-ID of a wide range of cellular antigens.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr Michael Hendzel for plasmid reagents and Dr Amira Klip for valuable discussion regarding Aptamer-PI. G.D. is a senior postdoctoral fellow of the Canadian Institutes of Health Research (CIHR). This work is supported by an operating grant from the Natural Sciences and Engineering Research Council to D.P.B.-J. D.P.B.-J. holds a Canada Research Chair in Molecular and Cellular Imaging.
REFERENCES
- 1.Terpe K. (2003) Overview of tag protein fusions: from molecular and biochemical fundamentals to commercial systems. Appl. Microbiol. Biotechnol., 60, 523–533. [DOI] [PubMed] [Google Scholar]
- 2.Pines J. (1995) GFP in mammalian cells. Trends Genet., 11, 326–327. [DOI] [PubMed] [Google Scholar]
- 3.van Roessel P. and Brand,A.H. (2002) Imaging into the future: visualizing gene expression and protein interactions with fluorescent proteins. Nature Cell Biol., 4, E15–E20. [DOI] [PubMed] [Google Scholar]
- 4.Robinson J.M., Takizawa,T., Pombo,A. and Cook,P.R. (2001) Correlative fluorescence and electron microscopy on ultrathin cryosections: bridging the resolution gap. J. Histochem. Cytochem., 49, 803–808. [DOI] [PubMed] [Google Scholar]
- 5.Dellaire G., Nisman,R. and Bazett-Jones,D.P. (2004) Correlative light and electron spectroscopic imaging of chromatin in situ. Methods Enzymol., 375, 456–478. [DOI] [PubMed] [Google Scholar]
- 6.Brody E.N. and Gold,L. (2000) Aptamers as therapeutic and diagnostic agents. J. Biotechnol., 74, 5–13. [DOI] [PubMed] [Google Scholar]
- 7.Hoppe-Seyler F., Crnkovic-Mertens,I., Tomai,E. and Butz,K. (2004) Peptide aptamers: specific inhibitors of protein function. Curr. Mol. Med., 4:529–538. [DOI] [PubMed] [Google Scholar]
- 8.Tuerk C and Gold,L. (1990) Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science, 249, 505–510. [DOI] [PubMed] [Google Scholar]
- 9.Ellington A.D and Szostak,J.W. (1990) Selection in vitro of single-stranded DNA molecules that fold into specific ligand-binding structures. Nature, 346, 818–822. [DOI] [PubMed] [Google Scholar]
- 10.Bacher J.M. and Ellington,A.D. (1998) Nucleic acid selection as a tool for drug discovery. Drugs Discov. Today, 3, 265–273. [Google Scholar]
- 11.Jayasena S.D. (1999) Aptamers: an emerging class of molecules that rival antibodies in diagnostics. Clin. Chem., 45, 1628–1650. [PubMed] [Google Scholar]
- 12.Morris K.N., Jensen,K.B., Julin,C.M., Weil,M. and Gold,L. (1998) High affinity ligands from in vitro selection: complex targets. Proc. Natl Acad. Sci. USA, 95, 2902–2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roulet E., Busso,S., Camargo,A.A., Simpson,A.J., Mermod,N. and Bucher,P. (2002) High-throughput SELEX SAGE method for quantitative modeling of transcription-factor binding sites. Nat. Biotechnol., 20, 831–835. [DOI] [PubMed] [Google Scholar]
- 14.Bock L.C., Griffin,J.A.Latham,E.H.Vermaas and Toole,J.J. (1992) Selection of single-stranded DNA molecules that bind and inhibit human thrombin. Nature, 355, 564–566. [DOI] [PubMed] [Google Scholar]
- 15.Dougan H., Weitz,J.I., Stafford,A.R., Gillespie,K.D., Klement,P., Hobbs,J.B. and Lyster,D.M. (2003) Evaluation of DNA aptamers directed to thrombin as potential thrombus imaging agents. Nucl. Med. Biol., 30, 61–72. [DOI] [PubMed] [Google Scholar]
- 16.Stanlis K.K.H. and McIntosh,J.R. (2003) Single-strand DNA aptamers as probes for protein localization in cells. J. Histo. Cyto., 51, 797–808. [DOI] [PubMed] [Google Scholar]
- 17.Robinett C.C., Straight,A., Li,G., Willhelm,C., Sudlow,G., Murray,A. and Belmont,A.S. (1996) In vivo localization of DNA sequences and visualization of large-scale chromatin organization using lac operator/repressor recognition. J. Cell Biol., 135, 1685–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lewis M., Chang,G., Horton,N.C., Kercher,M.A., Pace,H.C., Schumacher,M.A., Brennan,R.G. and Lu,P. (1996) Crystal structure of the lactose operon repressor and its complexes with DNA and inducer. Science, 271, 1247–1254. [DOI] [PubMed] [Google Scholar]
- 19.Dellaire G., Makarov,E.M., Cowger,J.J, Longman,D., Sutherland,H.G.E., Luhrmann,R., Torchia,J. and Bickmore,W.A. (2002) Mammalian PRP4 kinase copurifies and interacts with components of both the U5 snRNP and the N-CoR deacetylase complexes. Mol. Cell. Biol., 22, 5141–5156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ren Y., Kruhlak,M.J. and Bazett-Jones,D.P. (2003) Same serial section correlative light and energy-filtered transmission electron microscopy. J Histochem. Cytochem., 51, 605–612. [DOI] [PubMed] [Google Scholar]
- 21.Spector D.L. (1993) Nuclear organization of pre-mRNA processing. Curr. Opin. Cell Biol., 5, 442–447. [DOI] [PubMed] [Google Scholar]
- 22.Salomoni P. and Pandolfi,P.P. (2002) The role of PML in tumor suppression. Cell, 108,150–170. [DOI] [PubMed] [Google Scholar]
- 23.Strudwick S. and Borden,K.L. (2002) Finding a role for PML in APL pathogenesis: a critical assessment of potential PML activities. Leukaemia, 16, 1906–1917. [DOI] [PubMed] [Google Scholar]
- 24.Dellaire G. and Bazett-Jones,D.P. (2004) PML nuclear bodies: Dynamic sensors of cellular stress and DNA damage. Bioessays, 26, 963–977. [DOI] [PubMed] [Google Scholar]
- 25.Roth M.B., Zahler,A.M. and Stolk,J.A. (1992) A conserved family of nuclear phosphoproteins localized to sites of polymerase II transcription. J. Cell. Biol., 115, 587–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blank M., Weinshenk,T., Priemer,M. and Schluesener,H. (2001) Systematic evolution of a DNA aptamer binding to rat brain tumor microvessels. Selective targeting of endothelial regulatory protein pigpen. J. Biol. Chem., 276, 16464–16468. [DOI] [PubMed] [Google Scholar]
- 27.Bianchini M., Radrizzani,M., Brocardo,M.G., Reyes,G.B., Solveyra,C.G. and Santa-Coloma,T.A. (2001) Specific oligobodies against ERK-2 that recognize both the native and the denatured state of the protein. J. Immunol. Methods, 252, 191–197. [DOI] [PubMed] [Google Scholar]
- 28.Hsieh M. and Brenowitz,M. (1997) Comparison of the DNA association kinetics of the Lac repressor tetramer, its dimeric mutant LacIadi, and the native dimeric Gal repressor. J. Biol. Chem., 272, 22092–22096. [DOI] [PubMed] [Google Scholar]
- 29.Hermann T. and Patel,D.J. (2000) Adaptive recognition by nucleic acid aptamers. Science, 287, 820–825. [DOI] [PubMed] [Google Scholar]
- 30.Gadgil H., Oak,S.A. and Jarrett,H.W. (2001). Affinity purification of DNA-binding proteins. J. Biochem. Biophys. Methods, 49, 607–624. [DOI] [PubMed] [Google Scholar]
- 31.Romig T.S., Bell,C. and Drolet,D.W. (1999) Aptamer affinity chromatography: combinatorial chemistry applied to protein purification. J. Chromatogr. B Biomed. Sci. Appl., 31, 275–284. [PubMed] [Google Scholar]
- 32.Puig O., Caspary,F., Rigaut,G., Rutz,B., Bouveret,E., Bragado-Nilsson,E., Wilm,M. and Seraphin,B. (2001) The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods, 24, 218–229. [DOI] [PubMed] [Google Scholar]
- 33.Gadgil H. and Jarrett,H.W. (1999) Heparin elution of transcription factors from DNA-Sepharose columns. J. Chromatogr. A., 848, 131–138. [DOI] [PubMed] [Google Scholar]
- 34.Hillen W. and Berens,C. (1994) Mechanisms underlying expression of Tn10 encoded tetracycline resistance. Annu. Rev. Microbiol., 48, 345–369. [DOI] [PubMed] [Google Scholar]
- 35.Nguyen C.C. and Saier,M.H., Jr. (1995) Phylogenetic, structural and functional analyses of the LacI-GalR family of bacterial transcription factors. FEBS Lett., 377, 98–102. [DOI] [PubMed] [Google Scholar]
- 36.Grkovic S., Brown,M.H. and Skurray,R.A. (2002) Regulation of bacterial drug export systems. Microbiol. Mol. Biol. Rev., 66, 671–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kercher M.A., Lu,P. and Lewis,M. (1997) Lac repressor–operator complex. Curr. Opin. Struct. Biol., 7, 76–85. [DOI] [PubMed] [Google Scholar]