

Abstract
Exercise is known to have numerous beneficial effects. Recent studies indicate that exercise improves mitochondrial energetics not only in skeletal muscle but also in other tissues. While exercise elicits positive effects on memory, neurogenesis, and synaptic plasticity, the effects of exercise on brain mitochondrial energetics remain relatively unknown. Herein, we studied the effects of exercise training in old and young mice on brain mitochondrial energetics, in comparison to known effects on peripheral tissues that utilize fatty acid oxidation. Exercise improved the capacity for mucle and liver to oxidize palmitate in old mice, but not young mice. In the brain, exercise increased rates of respiration and reactive oxygen species (ROS) production in the old group only while utilizing complex I substrates, effects that were not seen in the young group. Coupled complex I to III enzymatic activity was significantly increased in old trained versus untrained mice with no effect on coupled II to III enzymatic activity. Mitochondrial protein content and markers of mitochondrial biogenesis (PGC-1α and TFAM) were not affected by exercise training in the brain, in contrast to the skeletal muscle of old mice. Brain levels of the autophagy marker LC3-II and protein levels of other signaling proteins that regulate metabolism or transport (BDNF, HSP60, phosphorylated mTOR, FNDC5, SIRT3) were not significantly altered. Old exercised mice showed a significant increase in DRP1 protein levels in the brain without changes in phosphorylation, while MFN2 and OPA1 protein levels were unchanged. Our results suggest that exercise training in old mice can improve brain mitochondrial function through effects on electron transport chain function and mitochondrial dynamics without increasing mitochondrial biogenesis.
Keywords: mitochondria, exercise, brain, cortex, complex I, DRP1
1. Introduction
Mitochondria have taken a central role in several hypotheses of aging for decades (Harman, 1956). Mitochondrial function, in particular respiration and the activities of electron transport chain (ETC) complexes I and IV, decline with aging in the brain, liver, heart and kidney, which also show changes in mitochondrial morphology (Manczak et al., 2005; Shigenaga et al., 1994). Mitochondrial DNA (mtDNA) mutations accumulate with age in the brain (Corral-Debrinski et al., 1992; Joseph et al., 2016), and altered brain energetics results in fission arrest and formation of abnormally fused presynaptic mitochondria in the prefrontal cortex (Hara et al., 2014; Zhang et al., 2016). Impaired mitochondrial function is critical in the pathogenesis of age related neurodegenerative disorders (Esteves et al., 2010; Lin and Beal, 2006), making it pivotal to develop strategies aimed at improving mitochondrial function.
Recent work indicates that exercise training increases mitochondrial biogenesis in skeletal muscle and improves lipid oxidation through activation of upstream signaling pathways such as peroxisome proliferation-activated receptor gamma co-activator 1α (PGC-1α) and mitochondrial transcription factor A (TFAM) (Joseph et al., 2016; Yan et al., 2012). However, β-oxidation is not favored in the brain as neurons exhibit a greater dependence on aerobic glucose metabolism than peripheral cells (Schönfeld and Reiser, 2013). Thus, the effects of exercise on brain mitochondrial function may differ from its effects in muscle and other peripheral tissues.
In addition to its cardiovascular benefits, exercise improves brain health and neuronal plasticity (Voss et al., 2013). In mouse models, exercise increases neurogenesis and synaptic plasticity (Cotman and Berchtold, 2002; van Praag et al., 2005). Some of these effects may be due to upregulation of brain-derived neurotrophic factor (BDNF) (Marosi and Mattson, 2014), which occurs in the hippocampus, cortex, and cerebellum in response to exercise (Neeper et al., 1996). The effects of exercise are particularly pronounced in the hippocampus with exercise improving hippocampus-dependent spatial memory in rodent models (Erickson et al., 2011). Exercise may also have protective effects against the development of neurodegenerative conditions (Paillard et al., 2015). Upregulation of BDNF, increased mitochondrial biogenesis, and modulation of autophagy have been suggested as beneficial effects in this context (reviewed in (Mattson, 2014)), particularly as BDNF stimulates PGC-1α expression in hippocampal neurons (E et al., 2014; Khabour et al., 2013). However, relatively little is known about the effects of exercise on brain mitochondrial function with aging.
Here, we analyzed mitochondrial function and pathways implicated in regulating mitochondrial biogenesis in young and old mice after three weeks of exercise. We demonstrate that in concert with improvements in oxidative capacity of liver and muscle, complex I linked mitochondrial function is improved in old mice after exercise training without affecting complex II linked mitochondrial function. We found no evidence of increased brain mitochondrial protein expression or mitochondrial biogenesis, however protein levels of DRP1 were significantly elevated by exercise in brains of old mice, implicating enhanced mitochondrial transport or turnover.
2. Methods
2.1 Animal care and maintenance
C57BL/6J mice, both 4-week and 24-month old males, were obtained from the National Institute on Aging aged rodent colony (Charles River Laboratories, Madison, WI). Prior to the initiation of each experiment, mice were maintained on a constant 12-h light and 12-h dark cycle with free access to water, were fed ad libitum with a standard chow diet, and were allowed to acclimate for a week prior to the experimental protocol. All procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Pittsburgh, and were in accordance with the National Research Council’s Guide for the Care and Use of Laboratory Animals.
2.2 Treadmill running protocol
Exercise training was performed similarly to previously published treadmill running protocols (Perrino et al., 2011). The 24-month old mice were randomly divided between control (old control, OC) and treadmill trained groups (old trained, OT). The 4-week old mice were divided into a young control group (YC) and treadmill trained group (YT). The OT and YT mice ran on a motor-driven treadmill (AccuScan Instruments, Columbus, OH) 5 days a week for 3 weeks and two days, for a total of 17 days of training. Training always started with 5 min of warm-up running at a velocity of 10 m/min. On the first day, and following 5 min warm-up, mice ran for 20 min at a 0° incline, at a velocity of 15 m/min. Over 4 days, the time of training progressively increased to 60 min at a 10° incline and a velocity of 15 m/min. During the second week, the mice ran for 60 min at 10° incline and a velocity of 15 m/min. During the third week the velocity increased to 17 m/min, and for the final two days of training the velocity was increased to 19 m/min. To encourage animals to move onto the treadmill, a mild electric shock was applied from a grid behind the treadmill. All mice in both groups were able to complete the training regimen without developing behavioral signs of fatigue. For control purposes, the YC and OC mice were handled in exactly the same way, but the treadmill was not turned on.
2.3 Whole body physiology and tissue harvest
Forty-four hours following the final bout of exercise, oxygen consumption (VO2), expired CO2, and respiratory quotient (RQ) were determined over a 24 hour period using a Comprehensive Laboratory Animal Monitoring System (CLAMS) (Columbus Instruments, Columbus, OH). Following CLAMS, mice were fasted for 6-hours and then euthanized. Liver, gastrocnemius, and brain were harvested immediately and flash frozen in liquid nitrogen and stored at −80°C for Western blot, or processed for mitochondrial or fatty acid oxidation assays. Each group (OC, OT, YC, YT) consisted of 7–8 biological replicates
2.4 Experiments with isolated mitochondria
2.4.1 Mitochondrial Isolation
After being euthanized, the brains were promptly removed, a portion of the cortex and striatum were dissected from one side and snap frozen in liquid nitrogen for Western blot analysis, with the remainder of the brain used for mitochondrial isolation as previously described (Gusdon et al., 2009). Briefly, dilutions of Percoll were prepared in isolation buffer I (IB I, containing 225 mM mannitol, 75 mM sucrose, 10 mM HEPES Potassium, 1 mM EDTA, 0.1% fatty acid free BSA, pH 7.4). All steps were performed at 4°C. Brain tissues were homogenized in 12% Percoll, layered on top of a gradient of 24% and 42% Percoll, and centrifuged at 27,000 × g for 10 minutes. The mitochondrial fraction was located between the 24% and 42% layers and was removed with a syringe, diluted in IB I, and centrifuged at 10,000 × g for 10 minutes. The supernatant was discarded, and the pellet was re-suspended in isolation buffer II (IB II, containing 225 mM mannitol, 75 mM sucrose, 10 mM HEPES potassium, 0.1 mM EDTA, pH 7.4) and centrifuged at 10,000 × g for 10 minutes. The supernatant was again discarded, and mitochondria were cryopreserved as described (Nukala et al., 2006) by re-suspending the pellet in 1.5 mL of 10% dimethyl sulfoxide (DMSO) in IB II. This solution was then placed in a Nalgene freezing container for cooling to −80°C at a uniform rate of about 1 °C/min. Protein concentrations for cortical and striatal mitochondrial preparations were determined using the bicinchoninic acid (BCA) protein assay (Thermo Fisher Scientific, Rockford, IL).
2.4.2 Mitochondrial respiration
Mitochondrial respiration was determined by measuring oxygen consumption using an Oxygraph-2k (Oroboros Instruments, Innsburck, Austria). Incubation media was supplemented with 5 mM L-malate and 5 mM L-glutamate to support complex I respiration or 5 mM L-succinate to support complex II respiration. For each measurement, a concentration of 0.2 mg/mL mitochondrial protein was added. State 4 respiration was recorded for two minutes after the addition of mitochondria. State 3 respiration was induced by the addition of 0.5 mM ADP. The rate of state 3 respiration was measured for at least three minutes after maximum phosphorylating respiration was reached.
2.4.3 Mitochondria ROS production
Mitochondrial ROS production was assessed as previously described (Votyakova and Reynolds, 2005). Briefly, 300 μg of mitochondrial protein was added to incubation media supplemented with either 5 mM L-glutamate and 5 mM L-malate or 5 mM succinate. ROS production was measured by recording the real-time oxidation of 2 μM Amplex Red dye (Molecular Probes, Eugene, OR) to the fluorescent molecule resorufin as catalyzed by 1 U/mL horseradish peroxidase. Fluorescence was measured at an excitation wavelength of 560 nm and an emission wavelength of 590 nm. Rates of ROS production were measured over 5 minutes for each treatment, and all rates were linear over the time intervals used. Slopes were converted into units of pmol H2O2/min/mg.
2.4.4 ETC enzymatic activity
The electron shuttling activity between complex I and complex III was determined as described (Gusdon et al., 2008) by monitoring the reduction of ferricytochrome c to ferrocytochrome c at 550 nm using a Spectramax E5 spectrofluorophotometer (Molecular Devices). The assay medium contained 25 mM potassium phosphate (pH 7.2 at 20°C), 2.5 mM MgCl2, 2.5 mg/mL bovine serum albumin, and 2 mM KCN. KCN was included in order to inhibit cytochrome c oxidase and prevent the reoxidation of ferrocytochrome c. A baseline was recorded for 1 minute at 550 nm after the addition of alamethacin (30 μg/mL), ferricytochrome c (15 μM), NADH (0.13 mM), and malonate (20 mM). In parallel wells, rotenone (2 μM) was added. Reactions were initiated by the addition of coupled mitochondria (200 μg/mL), and the rate of reduction of ferricytochrome c to ferrocytochrome c was recorded for three minutes. Complex I-III activity was determined by subtracting the rotenone insensitive activity from the total activity.
2.4.5 Complex II-III Activity
Enzymatic activity was determined as described (Kennaway et al., 1984) by monitoring the reduction of ferricytochrome c to ferrocytochrome c at 550 nm. The same assay medium as for complex I-III activity was used. A baseline was recorded for 1 minute at 550 nm after the addition of alamethacin (30 μg/mL), ferricytochrome c (15 μM), and rotenone (2 μM). In parallel wells, malonate (20 mM) was added. Reactions were initiated with the addition of mitochondria (200 μg/mL) preincubated with 20 mM succinate at 30°C for 20 minutes in potassium phosphate buffer. The rate of reduction of ferricytochrome c was recorded for three minutes. Coupled complex II-III activity was determined by subtracting the malonate insensitive activity from the total activity.
2.5 Western blotting
Western blotting was performed as described (Zhu et al., 2007). Protein concentration was determined using a BCA protein assay (Thermo Fisher Scientific, Rockford, IL), and equal amounts of protein (25 μg) were added to each well. The following primary antibody dilutions were used: 1:5,000 mouse monoclonal MitoProfile Total OXPHOS antibody cocktail (MitoSciences, Eugene, OR), 1:1,000 rabbit anti-FNDC5 (Abcam, Cambridge, MA), 1:1,000 rabbit anti-PGC-1α (Santa Cruz, Dallas, TX), 1:10,000 rabbit anti-TFAM (courtesy of Dr. Craig Cameron, Penn State University), 1:1,1000 rabbit anti-HSP60 (Abcam), 1:1,000 rabbit anti-BDNF (Abcam), 1:500 mouse monoclonal anti-LC3 (5F10 clone, Nanotools, Teningen, Germany), 1:1,000 rabbit anti-Phospho-mTOR (Ser2448, Cell Signaling, Danvers, MA), 1:1,000 rabbit anti-mTOR (Cell Signaling, Danvers, MA), 1:1000 anti-DRP1 (BD Biosciences, Franklin Lakes, NJ), 1:1000 anti-SIRT3 (courtesy of Dr. Eric Verdin, UCSF, CA), 1:2,000 anti-MFN2 (Abcam), 1:5,000 anti-TOM20 (Santa Cruz), and 1:1,000 anti-MnSOD (Abcam) followed by ECL detection (GE Healthcare, Little Chalfont, UK). Membranes were stripped and reprobed with 1:10,000 rabbit anti-GAPDH (Abcam). Densitometry was performed using ImageJ’s Gel analyzer (NIH, Bathesda, MD).
2.6 Muscle and liver palmitate oxidation assay
The assay was carried out using gastrocnemius and liver homogenates as described previously (Huynh et al., 2014). Following tissue harvesting, 60 mg of the specimen was placed into ice-cold sucrose-EDTA medium (SETH buffer; 250 nm sucrose, 1 mM EDTA, and 10 mM Tris-HCl, pH 7.4). The sample was then blotted dry, and placed in 200 μl of SETH buffer, minced thoroughly with scissors (~200 snips) and diluted 20-fold with additional SETH buffer. The minced tissue was then homogenized (12 passes) on ice using a Teflon coated pestle and glass homogenizer (Kontes Duall, Kimble Chase, Vineland, NJ). Forty microliters of homogenate were then added to the incubation well of a modified 48-well plate with a channel cut between the adjacent trap wells (Nunc, Thermo Fisher Scientific, Rochestrer, NY). The trap wells contained 200 μL of 1N sodium hydroxide for the collection of liberated 14-CO2. To the homogenate, 160 μl of incubation buffer (0.2 mM palmitate ([1-14-C] palmitate at 0.5 Ci/ml), 100 mM sucrose, 10 mM Tris-HCL, 5 mM potassium phosphate, 80 mM potassium chloride, 1 mM magnesium chloride, 0.1 mM malate, 2 mM ATP, 1 mM dithiothreitol, 0.2 mM EDTA, 1 mM L-Carnitine, 0.05 mM coenzyme A, and 0.5% fatty acid free bovine serum albumin, pH 7.4) was added to initiate the reaction. The plate was quickly sealed and incubated in a shaking water bath at 37°C for 30 minutes. Following the incubation, 100 μL of 70% perchloric acid was added to terminate the reaction. One hundred and fifty microliters of the sodium hydroxide from the trap wells was counted for label incorporation into trapped 14-CO2 as an index of complete palmitate oxidation. This was determined by scintillation counting using 4ml of EcoScint (National Diagnostics, Atlanta, GA).
2.7 Statistical analysis
Statistical significance among groups was determined using one-way ANOVA analysis and protected Student’s t-test. Results were considered statistically significant when P < 0.05. All figures were created with and statistical analysis was performed using R (R Foundation for Statistical Computing).
3. Results
3.1 Whole body physiology
Analysis of whole body physiology by CLAMS in the four groups revealed diminished systemic metabolism in aged mice that was reversed by exercise training. Both VO2 and VCO2 were lower in the OC group and were restored with exercise training in the OT group (Figure 1A and B, respectively). Similarly, RQ was decreased in the OC and was restored after exercise training in the OT mice (Figure 1C). The rates of fatty acid oxidation (FAO) were determined in homogenates from the gastrocnemius and liver as described (Huynh et al., 2014) by measuring the oxidation of radiolabeled palmitate. Rates of FAO were decreased in the OC group in both gastrocnemius and liver and were restored with exercise training in OT mice (Figure 1D).
Figure 1.

Exercise training affects whole body physiology and fatty acid oxidation. Forty-four hours after the final round of exercise training, total body oxygen consumption (VO2), expired CO2 (VCO2), and respiratory quotient (RQ) were determine over a 24-hour period using a Comprehensive Laboratory Animal Monitoring System (CLAMS). Rates of fatty acid oxidation were determined using tissues homogenates from the gastrocnemius and liver. Rates of oxygen consumption were decreased in OC mice which were restored after exercise training in OT mice (A). Rates of CO2 production were decreased in OC mice which were restored after exercise training in OT mice (B). The respiratory quotient was decreased in OC mice which was restored with exercise training in OT mice (C). Rates of palmitate oxidation were decreased in OC mice in tissue homogenates from both gastrocnemius and liver, which was restored with exercise training in OT mice (D). Data are presented as means ± SD. In Panels A–C, bar graphs at the top left represent areas under the curve (AUC) from 8 to 24 hr. In Panel A: *P < 0.05 in OC compared with YC, YT, and OT. In Panel B: *P < 0.05 in OC compared with YC, YT, and OT. In Panel C: *P < 0.05 in OC compared with YC, YT, and OT and †P < 0.05 in OT compared with YC, YT, and OC. In Panel D: *P < 0.05 in OC compared with YC, YT, and OT. YC, N=8; YT, N=8; OC, N=7; OT, N=7.
3.2 Effects of exercise on mitochondrial function
3.2.1 Respiration
To investigate the effects of exercise training on brain mitochondrial function, a tissue that is dependent on glucose metabolism rather than FAO, the rates of mitochondrial oxygen consumption were measured using glutamate and malate to support complex I mediated respiration and succinate to support complex II mediated respiration. State 4 respiration was assessed by quantifying mitochondrial oxygen consumption in the presence of complex I or complex II substrates without adding ADP. State 4 respiration was not significantly different among the groups utilizing complex I substrates (Figure 2A). However, when utilizing complex II substrates, respiration in old control OC and OT was significantly elevated compared with YC and YT (Figure 2B), but there was no effect of exercise training. State 3 respiration was assessed after the addition of ADP. Utilizing either complex I or complex II substrates, both OC and OT exhibited increased rates of respiration compared with YC and YT. Additionally, exercise training significantly increased complex I, but not complex II state 3 respiration in the OT group compared with OC (Figure 2A,B).
Figure 2.

Exercise training increases complex I substrate driven state 3 respiration, rotenone-induced ROS production, and complex I–III enzymatic activity in the brains of old mice. Utilizing complex I substrates, rates of state 3 respiration were increased in both old groups compared to the young groups, and in old mice, exercise training further increased the rate of state 3 respiration (A). Utilizing complex II substrates, rates of state 4 and 3 respiration were increased in old mice compared with young, however exercise training had no significant effect on succinate-supported state 3 respiration in the old mice (B). Utilizing complex I substrates, rates of basal ROS production were characteristically low (C). After inhibition with rotenone, rates of ROS production were significantly increased in the OT group (C). As expected, rates of ROS production utilizing succinate were basally elevated due to reverse electron transport, which was ablated after elimination of membrane potential with the uncoupling agent FCCP (D). There was no difference in ROS production utilizing complex II substrates among the four mouse groups (D). Complex I–III coupled activity was significantly increased in old mice with exercise training (E). Complex II–III coupled activity showed no significant difference among the four groups (F). Data are presented as means ± SD. In Panel A: *P < 0.05 vs YC and YT in state 3 respiration; †P < 0.05 vs OC in state 3 respiration. In Panel B: *P < 0.05 vs YC and YT in state 4 respiration; †P < 0.05 vs YC and YT in state 3 respiration. In Panel C: *P < 0.05 vs OC after rotenone treatment. In Panel E: *P < 0.05 vs YC. †P<0.05 vs OC. YC, N=8; YT, N=8; OC, N=7; OT, N=7.
3.2.2 ROS production
Mitochondrial ROS production was assessed by measuring the rate of oxidation of Amplex red by hydrogen peroxide in the presence of superoxide dismutase. Utilizing complex I substrates, brain mitochondria produce characteristically low levels of ROS, and there was no significant difference among the groups (Figure 2C). Inhibiting distally within complex I with rotenone markedly increased ROS production from complex I redox centers, and after rotenone treatment, the rate of ROS production was increased in OT compared with OC (Figure 2C). Utilizing complex II substrates, brain mitochondria produce higher basal rates of ROS when membrane potential is maximum due to reverse electron transport (Votyakova and Reynolds, 2001), and there was no significant difference among the groups (Figure 2D). Dissipating membrane potential with FCCP decreases ROS production, and again there was no significant difference among the groups (Figure 2D).
3.2.3 Enzymatic activities
Changes in enzymatic activity could account for the increased rates of respiration observed in mitochondria from the OT group in the presence of glutamate, malate and ADP (Figure 2A). ETC activity was assessed by determining the rates of coupled enzymatic activity. Complex I-III activity was determined by following the reduction of cytochrome c in the presence of NADH, as a source of electrons for complex I, and malonate to inhibit complex II. Complex II-III activity was determined by following the reduction of cytochrome c by mitochondria, which had been preincubated with succinate, in the presence of rotenone to inhibit complex I. Complex I-III activity was significantly decreased in OC compared to YC, and this decrease was reversed by exercise training (Figure 2E). Complex II-III activity did not differ among the four groups (Figure 2F).
3.2.4 Mitochondrial protein content and biogenesis
To investigate whether the increase in respiration and coupled I-III activity observed in old trained mice could be due to increased mitochondrial content, cortical and striatal lysates were immunoblotted for components of each of the ETC complexes. Consistent with previous reports (Haase et al., 2011; Menshikova et al., 2006), mitochondrial protein content was significantly increased with exercise training in skeletal muscle (Supplementary Figure 1). In contrast, there were no significant differences in protein content of complex I, II, III, IV, or V subunits in the cortex or striatum in old or young mice after exercise training (Figure 3A–J; Supplementary Figure 2A). Additionally, we observed no significant difference in the protein content of TOM20 or MnSOD (Supplementary Figure 2B). PGC-1α and TFAM each play important roles in controlling mitochondrial biogenesis and their expression in the hippocampus has been shown to be affected by exercise training (Yan et al., 2012). In the cortex and striatum, exercise training did not alter expression of PGC-1 α or TFAM (Figure 4; Supplementary Figure 3). There were also no effects on levels of the mitochondrial chaperonin heat shock protein 60 (Hsp60) (Figure 4; Supplementary Figure 3) or the mitochondrial deacetylase SIRT3 (Figure 4; Supplementary Figure 3), a regulator of mitochondrial biogenesis, ATP generation, ROS production, and the uncoupled protein response (McDonnell et al., 2015).
Figure 3.
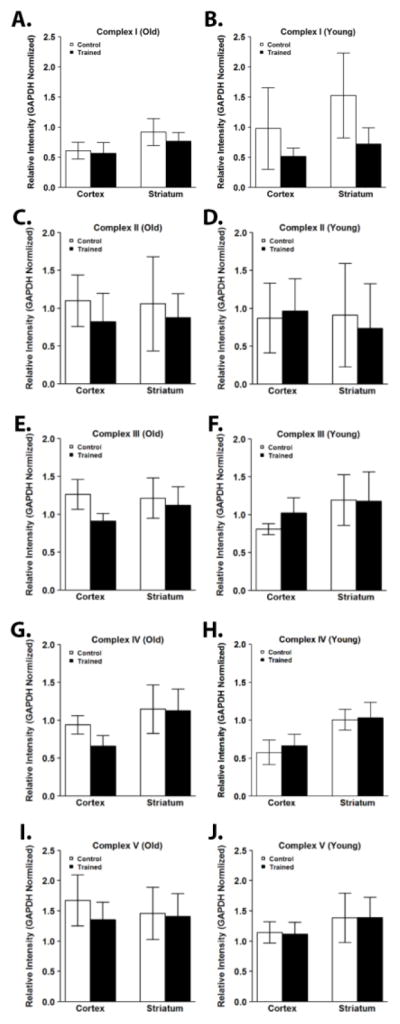
Exercise training resulted in no significant difference in brain mitochondrial content. Mitochondrial content was determined by Western blotting for a component of each of the ETC complexes. There were no significant differences after exercise training in the protein content of mitochondrial complex I (A), complex II (B), complex III (C), complex IV (D), or complex V (E) subunits in the cortex and striatum of young and old mice. Data are presented as means ± SD. YC, N=8; YT, N=8; OC, N=7; OT, N=7.
Figure 4.

Exercise training does not affect markers of mitochondrial biogenesis, HSP60, or SIRT3 in the brain. Mitochondrial biogenesis was assessed by Western blotting for PGC-1α and TFAM. Western blotting was also performed for HSP60 and SIRT3. Protein levels of PGC-1α were unchanged after exercise training in the cortex or striatum of old (A) or young (B) mice. Protein levels of TFAM were also not significantly different in the cortex or striatum of old (C) or (D) mice. Protein levels of HSP60 (E, old; F, young) and SIRT3 (G, old; H, young) were unchanged after exercise training in the cortex or striatum of old or young mice. Data are presented as means ± SD. YC, N=8; YT, N=8; OC, N=7; OT, N=7.
3.2.5 Upstream regulation of mitochondrial biogenesis
BDNF has been shown to be increased in the brain in response to exercise (Neeper et al., 1996), where it drives PGC-1α expression. However, we found no difference in protein levels of BDNF after exercise training in the cortex or striatum of old or young mice (Figure 5; Supplementary Figure 4). The membrane protein fibronectin type III domain containing 5 (FNDC5) is induced by PGC-1α, and can be cleaved in exercised muscle to form the hormone irisin (Boström et al., 2012). We found no significant difference in protein levels of FNDC5 after exercise training in the cortex or striatum of old or young mice (Figure 5; Supplementary Figure 4).
Figure 5.

Exercise training does not affect brain levels of BDNF and FNDC5, markers of autophagy, or phosphorylation of mTOR. Expression levels of BDNF, FNDC5, LC3-II, phosphorylated mTOR (P-mTOR) were assessed by Western blotting. P-mTOR levels were normalized to total mTOR levels. Protein levels of BDNF (A,B), FNDC5 (C,D), LC3-II (E,F), and P-mTOR (G,H) were not significantly different after exercise training in the cortex or striatum of old (A,C,E,G) or young (B,D,F,H) mice. Data are presented as means ± SD. YC, N=8; YT, N=8; OC, N=7; OT, N=7.
3.2.6 Effect of exercise on autophagy
Autophagy is critical for the response of muscle tissue to exercise and has also been shown to be increased in the brain in response to exercise (He et al., 2012). However, we found no difference in the amount of LC3-II after exercise training in the cortex or striatum of old or young mice (Figure 5; Supplementary Figure 4). The mammalian target of rapamycin (mTOR) plays an important role in suppressing autophagy, and activity of mTOR has been shown to be increased with exercise (Watson and Baar, 2014). We found that the ratio of phosphorylated to unphosphorylated mTOR was not changed with exercise training in the cortex or striatum of old or young mice (Figure 5; Supplementary Figure 4). Additionally, absolute levels of phosphorylated and total mTOR did not differ with exercise training in the cortex or striatum between old or young mice (Supplementary Figure 4).
3.2.6 Mitochondrial dynamics
Mitochondria are dynamic organelles and constantly undergo fission and fusion. Alterations in mitochondrial homeostasis and dynamics have been implicated in aging and age-related disorders (Hara et al., 2014; Zhang et al., 2016) and may also be influenced by exercise training (Santos-alves et al., 2015). Protein levels of DRP1, a protein involved in mitochondrial fission, were significantly increased after exercise training in the cortex of old mice (Figure 6A), while levels of DRP1 were not affected after exercise training in the brains of young mice (Figure 6B). DRP1 protein levels in old trained mice were significantly higher than in young control and exercised mice (P<0.05). Protein lvel phosphorylated DRP1 to total DRP1 was not changed with exercise training (Supplementary Figure 5A). Exercise training did not affect the levels of the mitochondrial fusion proteins MFN2 (Figure 6C,D) or OPA1 (Supplementary Figure 5B).
Figure 6.

Exercise training affects markers of mitochondrial dynamics in the brain. Mitochondrial dynamics was assessed by Western blotting for markers of mitochondrial fission and fusion, DRP1 and MFN2, respectively. Protein levels of DRP1 were significantly elevated in the cortex of old, exercised mice (A), however levels were unchanged in young mice in response to exercise training (B). *P < 0.05 vs. OC, YC and YT. Protein levels of MFN2 were unchanged after exercise in either striatum or cortex of old (C) or young (D) mice. Data are presented as means ± SD. YC, N=8; YT, N=8; OC, N=7; OT, N=7.
4. Discussion
Mitochondria play a key role in aging (Bratic and Larsson, 2013; Joseph et al., 2016), and interventions that improve mitochondrial function may ameliorate age-associated pathophysiological changes. Exercise has long been known to have beneficial effects on whole body metabolism, skeletal muscle oxidative capacity, and mitochondrial function, in part through stimulating mitochondrial biogenesis (Yan et al., 2012). Aging has been shown to result in mitochondrial dysfunction as well as reduced FAO in skeletal muscle (Hebert et al., 2010; Picard et al., 2011). We show that exercise training improved rates of FAO in old mice (Figure 1D) confirming the ability of exercise in our model to result in positive physiological adaptations. This is in line with studies in rodents and humans showing that exercise has a powerful effect on skeletal muscle FAO in older individuals (Chow et al., 2007; Lanza et al., 2008). This is in contrast to some reports suggesting that aged animals have lost the capacity to adapt to exercise stimuli (Ballak et al., 2015). These tissue specific changes were reflected in positive adaptations at the whole body level as demonstrated by improved VO2 and VCO2, (Figure 1A–C). While exercise yields a number of benefits in the central nervous system, the ability of exercise to affect mitochondrial function in the brain with aging has not been systematically studied.
4.1 Improvements in complex I substrate linked mitochondrial function
Our results demonstrate for the first time, that brain mitochondria from aged animals respond to exercise training by selectively improving the activity of complex I of the ETC. While rates of state 3 respiration were increased in old mice, the overall quality of electron transport appers to decrease with aging, as shown by the lower complex I-III coupled activity in old control mice (Figure 2E) and increased rates of state 4 respiration (Figure 2A,B), leading to a lower RQ. After exercise training, mitochondria isolated from the old group exhibited further increases in respiration (Figure 2A) accompanied by slight increases in rotenone-driven ROS production (Figure 2C) while utilizing complex I substrates. The increased ROS production in the old group after exercising training likely occurred as a result of increased electron flux through the ETC, and is in accord with increased rates of respiration. Inhibiting distal to complex I redox centers with rotenone results in supra-physiological increases in ROS production, which can make slight differences in ROS production more apparent. In situ ROS production during exercise is likely much lower given the presence of ADP and reduced membrane potential during active ATP generation (Goncalves et al., 2014). Notably, the decrease in complex I-III coupled activity observed in old mice relative to young mice was fully reversed by exercise training (Fig. 2E). The RQ was also improved by exercise training due to the increase in state 3 respiratory rates (Fig. 2A).
4.2 Role of mitochondrial biogenesis
While increased mitochondrial mass could result in increased rates of respiration, we did not observe an increase in the brain expression of the two most important factors involved in mitochondrial biogenesis: PGC-1α and TFAM (Figure 4A–D), the latter of which is tightly correlated with mtDNA content (Ekstrand et al., 2004; Ikeda et al., 2015; Lu et al., 2013; Mei et al., 2015). Nor were there changes in HSP60, which is responsible for transport of proteins into the matrix (Kaufman et al., 2003), or in SIRT3 (Figure 4E–H), which also regulates mitochondrial biogenesis (McDonnell et al., 2015). Likewise, there were no significant changes in levels of either nuclear DNA-encoded or mtDNA-encoded respiratory complex subunits (Figure 3, Supplementary Figure 2A) or of mitochondrial outer membrane or matrix proteins (Supplementary Figure 2B), although we did confirm that mitochondrial protein content was increased in liver and muscle after exercise training in our experimental model (Supplementary Figure 1), consistent with previous reports (Haase et al., 2011; Menshikova et al., 2006).
The fact that we did not observe changes in mitochondrial protein content or markers of biogenesis is not unexpected when considering the pattern of exercise-induced changes in respiration and ROS production. As shown in Figure 2, increased mitochondrial respiration and ROS production was only found when mitochondria were utilizing complex I substrates. If mitochondrial content were increased as a result of increased mitochondrial biogenesis, respiration and ROS production would also be increased while utilizing complex II substrates. Consistently, the activity of both complexes I and II were elevated in the studies that demonstrated increased mitochondrial biogenesis and content in skeletal muscle (Menshikova et al., 2006). The different physiology of muscle and brain cells may contribute, although a recent study showed that complex I linked mitochondrial function was also affected by aging in muscle (Kruse et al., 2016). Local augmentation of mitochondrial mass would allow skeletal muscle to perform a larger proportion of work by aerobic rather than anaerobic metabolism, as less ADP would be required to stimulate the same degree of mitochondrial respiration (Constable et al., 1987). Because neurons function exclusively through aerobic metabolism, these adaptive mechanisms may be less relevant to brain mitochondrial function.
One recent study indirectly suggested that six weeks of exercise training could increase mitochondrial biogenesis in the hippocampus, as evidenced by increased mRNA expression of TFAM and an accessory subunit of complex I (Aguiar et al., 2014). This study also showed that exercise training increased the activity of complex I and made complex I more resistant to inhibition by rotenone, consistent with increased mitochondrial content, as both rotenone-sensitive and insensitive activity would be expected to increase in proportion to increased mitochondrial mass. The fact that increased brain mitochondrial biogenesis or mass was not observed in our study might be due to the shorter duration of exercise (3 weeks) compared with other studies (6–8 weeks) (Aguiar et al., 2014; E et al., 2014; Steiner et al., 2011). However, another study showed that 24 weeks of exercise training increased complex I activity without affecting complex II activity, similar to our results (Navarro et al., 2004). Alternatively, increased biogenesis may be more pronounced in the hippocampus, which is capable of undergoing exercise-induced neurogenesis, a process that requires a net gain in mitochondrial mass (van Praag et al., 2014). In any case, the data presented here indicate that a shorter duration of peripheral exercise in old animals is sufficient to enhance brain mitochondrial function through mechanisms other than upregulation of mitochondrial mass.
4.3 Effect of exercise on upstream signaling pathways
We also evaluated the contribution of other upstream signaling pathways. BNDF is implicated in the beneficial effects of exercise on memory, neurogenesis, and synaptic plasticity (Voss et al., 2013), acting upstream of PGC-1α in stimulating mitochondrial biogenesis (Cheng et al., 2012). However, we did not detect a significant difference in the expression of BNDF after exercise (Figure 5A,B). Recently, the myokine irisin (generated by the cleavage of the membrane protein FNDC5) was shown to be induced by PGC-1α, playing an important role for energy expenditure by stimulating UCP1 expression (Boström et al., 2012). We did not find a difference in the expression of FNDC5 (Figure 5C,D) in the cortex or striatum. Another study showed that seven days of running upregulated SIRT3 expression in the hippocampus, and led to a pattern of protein acetylation contributing to resistance against celluar stress (Cheng et al., 2015). We did not find an increase in SIRT3 expression after exercise (Figure 4. The effects of exercise on hippocampal BDNF and SIRT3 were observed after only 2–7 days of exercise (Neeper et al., 1996) and may not be sustained. Alternatively, there may be brain region selective effects of exercise.
Induction of autophagy during exercise is believed to be important to supply amino acids to skeletal muscles during exercise (Jamart et al., 2013) and may also protect against oxidative stress (Dutta et al., 2013). Furthermore, some studies have suggested that a brief duration of exercise induces autophagy in the brain (He et al., 2012). However, we did not observe a difference in levels of the autophagosome marker LC3-II after exercise (Figure 5E,F). Activation of mTOR down-regulates autophagy and up-regulates protein synthesis in skeletal muscle, and its activation may be caused by increased levels of BDNF in response to exercise (Watson and Baar, 2014). The fact that we did not observe changes in phosphorylated mTOR in the cortex or striatum after exercise (Figure 5G,H) is consistent with the lack of alteration in LC3-II and in BDNF. It should be noted, however, that a lack of change in steady state levels of LC3-II may be observed in the context of increased autophagic flux, if autophagy and lysosomal degradation are upregulated in parallel. Further studies, as new methods to monitor autophagy activity in vivo are developed, will be necessary to determine the effects of transient or sustained exercise on levels of brain autophagy.
4.4 A role for mitochondrial dynamics
Interestingly, our data demonstrated that improved brain complex I activity due to exercise training in old mice was correlated with a significant increase in the protein levels of the mitochondrial fission protein DRP1 in the cortex (Figure 6A,B). We found no change in the protein levels of the key fusion proteins MFN2 (Figure 6) or OPA1 (Supplementary Figure 5). Additionally, there was no change in DRP1 that is phosphorylated at the inactivating PKA phosphorylation site (Supplementary Figure 5). Taken together, these data suggest that a shift in the mitochondrial fission-fusion balance toward fission contributes to improved mitochondrial function in the cortex after exercise. As altered brain energetics induces mitochondrial fission arrest (Zhang et al., 2016) in Alzheimer’s disease, upregulation of the DRP1 may act to reverse age-related alterations in the brain.
There are several mechanisms by which upregulation of DRP1 may be beneficial to neurons. Mitochondrial fission is necessary for effective autophagic clearance of damaged mitochondria in both PINK1-Parkin dependent and independent mitophagy pathways (Dagda et al., 2009; Kageyama et al., 2014). Moreover, mitochondrial fission is required for the development and maintenance of proper mitochondrial and synaptic function in neurons (Fukumitsu et al., 2016; Ishihara et al., 2009), and improves neuronal survival by decreasing oxidative stress (Kageyama et al., 2012). DRP1 has also been shown to be important for exercise-induced hippocampal neurogenesis and plasticity (Steib et al., 2014). This may be mediated in part by a requirement for fission in order to achieve optimal delivery of mitochondria to both presynaptic and postsynaptic terminals (Fukumitsu et al., 2016). Indeed, the requirement for local ATP production and calcium buffering at synapses results in more mitochondrial traffic toward synapses and increased synaptic mitochondrial content, while impaired mitochondrial transport precedes neurodegeneration (Chang et al., 2006; MacAskill et al., 2010; Yamada et al., 2016; Yu et al., 2016). Alternatively, fission serves to limit propagation of mitochondrial calcium, with dysregulation of this process implicated in excitotoxicity and Parkinsonian neurodegeneration (Cherra et al., 2013). Modulation of mitochondral dynamics and calcium buffering have also been implicated in synaptic plasticity in the aging brain (Cheng et al., 2010).
The increased activity-dependent synaptic plasticity and glutaminergic synaptic activity in the cortex compared with the striatum, as well as brain regional differences in metabolism (Pandya et al., 2016; Sauerbeck et al., 2011), may help to explain different effects of exercise training noted in different brain regions. The relative levels of fission-fusion proteins are known to vary according to body region (Santos-alves et al., 2015). Of note, fission-fusion proteins also varied with duration of exercise, with one study in skeletal muscle showing a decrease in MFN2 levels in skeletal muscle after 45 minutes of exercise with subsequent recovery to levels higher than the pre-exercise baseline (Ding et al., 2010). Additionally, abnormally fused mitochondria are observed specifically in presynaptic regions of the aging primate cortex (Hara et al., 2014), suggesting that increased levels of fission proteins induced by exercise may serve to normalize these aging effects.
Since we did not observe a difference with exercise training on a number of signaling pathways that could affect mitochondrial homeostasis (Figures 4 and 5), we can only speculate as to the mechanism linking exercise to mitochondrial homeostasis in the brains of old, but not young, animals. Recently, the role of microRNAs in aging has been demonstrated, including those that affect mitochondrial homeostasis. The miR-30 family members are upregulated during aging and cellular senescence, but are reduced by caloric restriction in mice with improved longevity (Smith-Vikos and Slack, 2012). As miR-30 family microRNAs act to suppress expression of DRP1 by inhibiting the p53 mediated activation of the DRP1 promoter (Li et al., 2010), as well as a number of autophagy mediators, it is possible that exercise training resulted in increased DRP1 expression and improved mitochondrial quality control and function by alleviating aging related miRNA changes in old mice.
Our results are consistent with prior reports demonstrating that preventing mitochondrial fission leads to decreased mitochondrial respiration and coupling with increased susceptibility to oxidative stress (Kageyama et al., 2012; Parone et al., 2008). However, this does not necessarily explain the complex I specificity of our findings. One explanation could be that exercise drives structural alterations in the ETC. Exercise was shown to be able to alter the membrane-dependent interactions among mitochondria in mice (Picard et al., 2013), which could affect mitochondrial morphology and superstructure. The redox status of the cell, which could be modified by exercise, has also been shown to modify mitochondrial morphology (Shutt et al., 2012). In addition, mitochondrial function is governed by supercomplexes formed between complexes I and III and also among complexes I, III, and IV (Genova and Lenaz, 2014). Thus, exercise may improve the stability of the complex I/III/IV supercomplex, increasing electron flow through complex I while having little effect on the flow of electrons delivered through complex II. This mechanism would be consistent with our finding that complex I linked enzymatic activity was increased after exercise training (Figure 2E) without having an effect on complex II linked enzymatic activity (Figure 2F).
5. Conclusions
Our results indicate that exercise training improves muscle and liver FAO in aged mice, changes that are reflected in improved whole body metabolism. In brain mitochondria from aged mice, exercise selectively improved mitochondrial complex I function. Moreover, exercise increased levels of DRP1 without affecting indices of mitochondrial biogenesis or other stress response pathways. These data indicate that changes in mitochondrial dynamics and possibly structural alterations to complex I occur in response to exercise in the absence of, or prior to, changes in overall brain mitochondrial content. Importantly, the specific improvements elicited by exercise for brain complex I activity was observed in the old age group, suggesting that beneficial effects of exercise observed with aging and neurodegenerative diseases may be mediated in part by improved brain mitochondrial function.
Supplementary Material
Exercise training increases mitochondrial protein expression in the gastrocnemius. Mitochondrial content was determined by Western blotting for components of each of ETC complexes. Protein content of complexes II, III, IV, and V were significantly increased in old mice after exercise but not significantly different in young mice after exercise in the gastrocnemius. Data are presented as means ± SD. YC, N=8; YT, N=8; OC, N=7; OT, N=7. *P<0.05.
Exercise training does not affect mitochochondrial mass in the brain. Protein components of mitochondrial complexes I-V were assessed by Western blotting (A). Protein levels of mitochondrial complexes I-V were not significantly different in the cortex or striatum of young mice. Protein levels of the mitochondrial proteins TOM20 and MnSOD were determined by Western blotting (B). There were no significant differences in the protein expression of TOM20 and MnSOD after exercise in old or young mice. Data are presented as means ± SD. YC, N=8; YT, N=8; OC, N=7; OT, N=7.
Exercise training does not increase markers of mitochondrial biogenesis in the brain. Protein levels of PGC-1α, TFAM, HSP60, and SIRT3 were assessed by Western blotting. There was no significant difference in protein levels of PGC-1α, TFAM, HSP60, and SIRT3 in the cortex or striatum of old or young mice after exercise training. YC, N=8; YT, N=8; OC, N=7; OT, N=7.
Exercise training does not affect expression of BDNF, FNDC5, LC3-II, or mTOR. Protien levels of BNDF, FNDC5, LC3-II, and phosphorylated and total mTOR were assessed by Western blotting. There was no significant change in the protein expression of BDNF, FNDC5, LC3-II, or phospho-mTOR after exercise in the cortex or striatum of young or old mice. YC, N=8; YT, N=8; OC, N=7; OT, N=7.
Exercise training does not affect phosphorylated DRP1 or OPA1 levels. Phosph-DRP1 (P-DRP1, phosphorylated at Ser616) and OPA1 levels were assessed by Western blotting. Protein levels of P-DRP1 were normalized to GAPDH and were not significantly different in the cortex or striatum of old or young mice (A). OPA1 levels were normalized to GAPDH and were not significantly different in the cortex of old or young mice (B). Data are presented as means ± SD. YC, N=8; YT, N=8; OC, N=7; OT, N=7.
Highlights.
Exercise training increased whole body oxygen consumption and fatty acid oxidation.
Complex I-III coupled mitochondrial activity was decreased in the brains of old mice.
Exercise increased complex I linked respiration and complex I-III coupled ativity in old mice.
Exercise training did not affect mitochondrial protein content or biogenesis in the brain.
Expression of the fission protein DRP1 was increased in the brains of old exercised mice.
Acknowledgments
We would like to thank Dr. Emine Koc and Dr. Craig Cameron for providing the anti-TFAM antibody. We would like to thank Dr. Eric Verdin for providing the anti-SIRT3 antibody.
Funding
This work was supported in part by the National Institutes of Health (AG026389 and NS065789 to C.T.C; AG044437 to PMC), the A. Julio Martinez Endowment (C.T.C.), and pilot funding from the University of Pittsburgh Claude D. Pepper Center (to PMC) and the University of Pittsburgh Institute on Aging (to BHG).
Abbreviations
- PGC-1α
peroxisome proliferator-activated receptor gamma co-activator 1α
- TFAM
mitochondrial transcription factor A
- BDNF
brain-derived neurotrophic factor
- ETC
electron transport chain
- OC
old control group
- OT
old trained group
- YC
young control group
- YT
young trained group
- DMSO
dimethyl sulfoxide
- IB I
isolation buffer I
- IB II
isolation buffer II
- BCA
bicinchronic acid
- ROS
reactive oxygen species
- DRP1
dynamin-related protein 1
- MFN2
mitofusin-2
- SIRT3
sirtuin 3
- mTOR
mammalian target of rapamycin
- FCCP
carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone
- FNDC5
fibronectin type III domain containing 5
- HSP60
heat shock protein 60
- COX7RP
cytochrome c oxidase subunit 7a-related protein
- TOM20
translocase of the outer membrane 20
- MnSOD
manganese superoxide dismutase
Footnotes
Conflict of interest
The authors declare no conflicts of interest.
Contributions
A.M.G. designed and carried out the experiments, performed data analysis, and wrote the manuscript. J.C. performed experiments and approved the manuscript. G.D. performed experiments and edited the manuscript. R.M.O. designed the CLAMS experiments and approved the manuscript. B.H.G. contributed to the experimental design and edited the manuscript. P.M.C. designed the experiments, performed data analysis, and edited the manuscript. C.T.C. supervised the project, designed experiments, performed data analysis, and helped write the manuscript. The authors declare that they have no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aguiar aS, Stragier E, da Luz Scheffer D, Remor aP, Oliveira Pa, Prediger RD, Latini a, Raisman-Vozari R, Mongeau R, Lanfumey L. Effects of exercise on mitochondrial function, neuroplasticity and anxio-depressive behavior of mice. Neuroscience. 2014;271:56–63. doi: 10.1016/j.neuroscience.2014.04.027. [DOI] [PubMed] [Google Scholar]
- Ballak SB, Jaspers RT, Deldicque L, Chalil S, Peters EL, de Haan A, Degens H. Blunted hypertrophic response in old mouse muscle is associated with a lower satellite cell density and is not alleviated by resveratrol. Exp Gerontol. 2015;62:23–31. doi: 10.1016/j.exger.2014.12.020. [DOI] [PubMed] [Google Scholar]
- Boström P, Wu J, Jedrychowski MP, Korde A, Ye L, Lo JC, Rasbach Ka, Boström EA, Choi JH, Long JZ, Kajimura S, Zingaretti MC, Vind BF, Tu H, Cinti S, Højlund K, Gygi SP, Spiegelman BM. A PGC1-α-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature. 2012;481:463–8. doi: 10.1038/nature10777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bratic A, Larsson N. The role of mitochondria in aging. J Clin Invest. 2013;123:951–957. doi: 10.1172/JCI64125.Mitochondrial. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang DTW, Honick AS, Reynolds IJ. Mitochondrial trafficking to synapses in cultured primary cortical neurons. J Neurosci. 2006;26:7035–7045. doi: 10.1523/JNEUROSCI.1012-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng A, Hou Y, Mattson MP. Mitochondria and neuroplasticity. ASN Neuro. 2010;2:e00045. doi: 10.1042/AN20100019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng A, Wan R, Yang JL, Kamimura N, Son TG, Ouyang X, Luo Y, Okun E, Mattson MP. Involvement of PGC-1α in the formation and maintenance of neuronal dendritic spines. Nat Commun. 2012;3:1250. doi: 10.1038/ncomms2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng A, Yang Y, Zhou Y, Maharana C, Lu D, Peng W, Liu Y, Wan R, Marosi K, Misiak M, Bohr VA, Mattson MP. Mitochondrial SIRT3 Mediates Adaptive Responses of Neurons to Exercise and Metabolic and Excitatory Challenges. Cell Metab. 2015;23:128–142. doi: 10.1016/j.cmet.2015.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherra SJ, Steer E, Gusdon AM, Kiselyov K, Chu CT. Mutant LRRK2 elicits calcium imbalance and depletion of dendritic mitochondria in neurons. Am J Pathol. 2013;182:474–84. doi: 10.1016/j.ajpath.2012.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow LS, Greenlund LJ, Asmann YW, Short KR, McCrady SK, Levine JA, Nair KS. Impact of endurance training on murine spontaneous activity, muscle mitochondrial DNA abundance, gene transcripts, and function. J Appl Physiol (Bethesda, Md 1985) 2007;102:1078–1089. doi: 10.1152/japplphysiol.00791.2006. [DOI] [PubMed] [Google Scholar]
- Constable SH, Favier RJ, Fell RD, Chen M, Holloszy J. Energy metabolism in contracting adaptation to exercise training rat skeletal muscle: Am. J Physiol. 1987;253:C316–322. doi: 10.1152/ajpcell.1987.253.2.C316. [DOI] [PubMed] [Google Scholar]
- Corral-Debrinski M, Horton T, Lott MT, Shoffner JM, Beal MF, Wallace DC. Mitochondrial DNA deletions in human brain: regional variability and increase with advanced age. Nat Genet. 1992;2:324–329. doi: 10.1038/ng1292-324. [DOI] [PubMed] [Google Scholar]
- Cotman CW, Berchtold NC. Exercise: a behavioral intervention to enhance brain health and plasticity. TRENDS Neurosci. 2002;25:295–301. doi: 10.1016/s0166-2236(02)02143-4. [DOI] [PubMed] [Google Scholar]
- Dagda RK, Cherra SJ, Kulich SM, Tandon A, Park D, Chu CT. Loss of PINK1 Function Promotes Mitophagy through Effects on Oxidative Stress and Mitochondrial Fission. J Biol Chem. 2009;284:13843–13855. doi: 10.1074/jbc.M808515200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding H, Jiang N, Liu H, Liu X, Liu D, Zhao F, Wen L, Liu S, Ji LL, Zhang Y. Response of mitochondrial fusion and fission protein gene expression to exercise in rat skeletal muscle. Biochim Biophys Acta - Gen Subj. 2010;1800:250–256. doi: 10.1016/j.bbagen.2009.08.007. [DOI] [PubMed] [Google Scholar]
- Dutta D, Xu J, Kim JS, Dunn WA, Leeuwenburgh C. Upregulated autophagy protects cardiomyocytes from oxidative stress-induced toxicity. Autophagy. 2013;9:328–344. doi: 10.4161/auto.22971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EL, Burns JM, Swerdlow RH. Effect of high-intensity exercise on aged mouse brain mitochondria, neurogenesis, and inflammation. Neurobiol Aging. 2014;35:2574–2583. doi: 10.1016/j.neurobiolaging.2014.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekstrand MI, Falkenberg M, Rantanen A, Park CB, Gaspari M, Hultenby K, Rustin P, Gustafsson CM, Larsson NG. Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum Mol Genet. 2004;13:935–944. doi: 10.1093/hmg/ddh109. [DOI] [PubMed] [Google Scholar]
- Erickson KI, Voss MW, Prakash RS, Basak C, Szabo A, Chaddock L, Kim JS, Heo S, Alves H, White SM, Wojcicki TR, Mailey E, Vieira VJ, Martin Sa, Pence BD, Woods Ja, McAuley E, Kramer AF. Exercise training increases size of hippocampus and improves memory. Proc Natl Acad Sci U S A. 2011;108:3017–22. doi: 10.1073/pnas.1015950108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteves AR, Lu J, Rodova M, Onyango I, Lezi E, Dubinsky R, Lyons KE, Pahwa R, Burns JM, Cardoso SM, Swerdlow RH. Mitochondrial respiration and respiration-associated proteins in cell lines created through Parkinson’s subject mitochondrial transfer. J Neurochem. 2010;113:674–682. doi: 10.1111/j.1471-4159.2010.06631.x. [DOI] [PubMed] [Google Scholar]
- Fukumitsu K, Hatsukano T, Yoshimura A, Heuser J, Fujishima K, Kengaku M. Mitochondrial fission protein Drp1 regulates mitochondrial transport and dendritic arborization in cerebellar Purkinje cells. Mol Cell Neurosci. 2016;71:56–65. doi: 10.1016/j.mcn.2015.12.006. [DOI] [PubMed] [Google Scholar]
- Genova ML, Lenaz G. Functional role of mitochondrial respiratory supercomplexes. Biochim Biophys Acta. 2014;1837:427–43. doi: 10.1016/j.bbabio.2013.11.002. [DOI] [PubMed] [Google Scholar]
- Goncalves RLS, Quinlan CL, Perevoshchikova IV, Hey-Mogensen M, Brand MD. Sites of Superoxide and Hydrogen Peroxide Production by Muscle Mitochondria Assessed ex vivo Under Conditions Mimicking Rest and Exercise. J Biol Chem. 2014:0–44. doi: 10.1074/jbc.M114.619072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusdon AM, Chen J, Votyakova TV, Mathews CE. Methods in enzymology. 1. Elsevier Inc; 2009. Chapter 24 Quantification, localization, and tissue specificities of mouse mitochondrial reactive oxygen species production. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusdon AM, Votyakova TV, Mathews CE. mt-Nd2a suppresses reactive oxygen species production by mitochondrial complexes I and III. J Biol Chem. 2008;283:10690–7. doi: 10.1074/jbc.M708801200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haase TN, Ringholm S, Leick L, Bienso RS, Kiilerich K, Johansen S, Nielsen MM, Wojtaszewski JF, Hidalgo J, Pedersen Pa, Pilegaard H. Role of PGC-1 in exercise and fasting-induced adaptations in mouse liver. AJP Regul Integr Comp Physiol. 2011;301:R1501–R1509. doi: 10.1152/ajpregu.00775.2010. [DOI] [PubMed] [Google Scholar]
- Hara Y, Yuk F, Puri R, Janssen WGM, Rapp PR, Morrison JH. Presynaptic mitochondrial morphology in monkey prefrontal cortex correlates with working memory and is improved with estrogen treatment. Proc Natl Acad Sci U S A. 2014;111:486–91. doi: 10.1073/pnas.1311310110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harman D. Aging: a Theory Based on Free Radical and Radiation Chemistry. J Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- He C, Sumpter R, Levine B. Exercise induces autophagy in peripheral tissues and in the brain. Autophagy. 2012;8:1548–51. doi: 10.4161/auto.21327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert SL, Lanza IR, Nair KS. Mitochondrial DNA alterations and reduced mitochondrial function in aging. Mech Ageing Dev. 2010;131:451–462. doi: 10.1016/j.mad.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh FK, Green MF, Koves TR, Hirschey MD. Measurement of fatty acid oxidation rates in animal tissues and cell lines. Methods Enzymol. 2014;542:391–405. doi: 10.1016/B978-0-12-416618-9.00020-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda M, Ide T, Fujino T, Arai S, Saku K, Kakino T, Tyynismaa H, Yamasaki T, Yamada KI, Kang D, Suomalainen A, Sunagawa K. Overexpression of TFAM or twinkle increases mtDNA copy number and facilitates cardioprotection associated with limited mitochondrial oxidative stress. PLoS One. 2015;10:1–19. doi: 10.1371/journal.pone.0119687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara N, Nomura M, Jofuku A, Kato H, Suzuki SO, Masuda K, Otera H, Nakanishi Y, Nonaka I, Goto YI, Taguchi N, Morinaga H, Maeda M, Takayanagi R, Yokota S, Mihara K. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat Cell Biol. 2009;11:958–966. doi: 10.1038/ncb1907. [DOI] [PubMed] [Google Scholar]
- Jamart C, Naslain D, Gilson H, Francaux M. Higher activation of autophagy in skeletal muscle of mice during endurance exercise in the fasted state. Am J Physiol Endocrinol Metab. 2013;305:E964–74. doi: 10.1152/ajpendo.00270.2013. [DOI] [PubMed] [Google Scholar]
- Joseph AM, Adhihetty PJ, Leeuwenburgh C. Beneficial effects of exercise on age-related mitochondrial dysfunction and oxidative stress in skeletal muscle. J Physiol. 2016;594:5105–5123. doi: 10.1113/JP270659.This. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kageyama Y, Hoshijima M, Seo K, Bedja D, Sysa-Shah P, Andrabi Sa, Chen W, Hoke A, Dawson VL, Dawson TM, Gabrielson K, Kass Da, Iijima M, Sesaki H. Parkin-independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain. EMBO J. 2014;33:1–16. doi: 10.15252/embj.201488658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kageyama Y, Zhang Z, Roda R, Fukaya M, Wakabayashi J, Wakabayashi N, Kensler TW, Hemachandra Reddy P, Iijima M, Sesaki H. Mitochondrial division ensures the survival of postmitotic neurons by suppressing oxidative damage. J Cell Biol. 2012;197:535–551. doi: 10.1083/jcb.201110034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman Ba, Kolesar JE, Perlman PS, Butow Ra. A function for the mitochondrial chaperonin Hsp60 in the structure and transmission of mitochondrial DNA nucleoids in Saccharomyces cerevisiae. J Cell Biol. 2003;163:457–61. doi: 10.1083/jcb.200306132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennaway NG, Buist NRM, Darley-Usmar VM, Papadimitriou A, DiMauro S, Kelley RI, Capaldi RA, Blank NK, D’Agostino A. Lactic Acidosis and Mitochondrial Myopathy Associatd with Deficiency of Several Components of Complex III of the Respiratory Chain. Pediatr Res. 1984;18:991–9. doi: 10.1203/00006450-198410000-00017. [DOI] [PubMed] [Google Scholar]
- Khabour OF, Alzoubi KH, Alomari Ma, Alzubi Ma. Changes in spatial memory and BDNF expression to simultaneous dietary restriction and forced exercise. Brain Res Bull. 2013;90:19–24. doi: 10.1016/j.brainresbull.2012.08.005. [DOI] [PubMed] [Google Scholar]
- Kruse SE, Karunadharma PP, Basisty N, Johnson R, Beyer RP, Maccoss MJ, Rabinovitch PS, Marcinek DJ. Age modifies respiratory complex I and protein homeostasis in a muscle type-specific manner. Aging Cell. 2016;15:89–99. doi: 10.1111/acel.12412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanza IR, Short DK, Short KR, Raghavakaimal S, Basu R, Joyner MJ, McConnell JP, Nair KS. Endurance exercise as a countermeasure for aging. Diabetes. 2008;57:2933–2942. doi: 10.2337/db08-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Donath S, Li Y, Qin D, Prabhakar BS, Li P. miR-30 regulates mitochondrial fission through targeting p53 and the dynamin-related protein-1 pathway. PLoS Genet. 2010:6. doi: 10.1371/journal.pgen.1000795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. nature05292[pii]\r. [DOI] [PubMed] [Google Scholar]
- Lu B, Lee J, Nie X, Li M, Morozov YI, Venkatesh S, Bogenhagen DF, Temiakov D, Suzuki CK. Phosphorylation of Human TFAM in Mitochondria Impairs DNA Binding and Promotes Degradation by the AAA+ Lon Protease. Mol Cell. 2013;49:121–132. doi: 10.1016/j.molcel.2012.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacAskill AF, Atkin TA, Kittler JT. Mitochondrial tracking and the provision of energy and calcium buffering at excitatory synapses. Eur J Neurosci. 2010;32:231–240. doi: 10.1111/j.1460-9568.2010.07345.x. [DOI] [PubMed] [Google Scholar]
- Manczak M, Jung Y, Park BS, Partovi D, Reddy PH. Time-course of mitochondrial gene expressions in mice brains: Implications for mitochondrial dysfunction, oxidative damage, and cytochrome c in aging. J Neurochem. 2005;92:494–504. doi: 10.1111/j.1471-4159.2004.02884.x. [DOI] [PubMed] [Google Scholar]
- Marosi K, Mattson MP. BDNF mediates adaptive brain and body responses to energetic challenges. Trends Endocrinol Metab. 2014;25:89–98. doi: 10.1016/j.tem.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP. Interventions that improve body and brain bioenergetics for parkinson’s disease risk reduction and therapy. J Parkinsons Dis. 2014;4:1–13. doi: 10.3233/JPD-130335. [DOI] [PubMed] [Google Scholar]
- McDonnell E, Peterson BS, Bomze HM, Hirschey MD. SIRT3 regulates progression and development of diseases of aging. Trends Endocrinol Metab. 2015;26:1–7. doi: 10.1016/j.tem.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei H, Sun S, Bai Y, Chen Y, Chai R, Li H. Reduced mtDNA copy number increases the sensitivity of tumor cells to chemotherapeutic drugs. Cell death {&} Dis. 2015;6:e1710. doi: 10.1038/cddis.2015.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menshikova EV, Ritov VB, Fairfull L, Ferrell RE, Kelley DE, Goodpaster BH. Effects of Exercise on Mitochondrial Content and Function in Aging Human Skeletal Muscle. Journals Gerontol Ser A Biol Sci Med Sci. 2006;61:534–540. doi: 10.1093/gerona/61.6.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro A, Gomez C, Lo M, Boveris A. Beneficial effects of moderate exercise on mice aging: survival, behavior, oxidative stress, and mitochondrial electron transfer. Am J Physi. 2004:505–511. doi: 10.1152/ajpregu.00208.2003. [DOI] [PubMed] [Google Scholar]
- Neeper SA, Gdmez-pinilla F, Choi J, Cotman CW. Physical activity increases mRNA for brain-derived neurotrophic factor and nerve growth factor in rat brain. Brain Res. 1996;726:49–56. [PubMed] [Google Scholar]
- Nukala VN, Singh IN, Davis LM, Sullivan PG. Cryopreservation of brain mitochondria: A novel methodology for functional studies. J Neurosci Methods. 2006;152:48–54. doi: 10.1016/j.jneumeth.2005.08.017. [DOI] [PubMed] [Google Scholar]
- Paillard T, Rolland Y, de Philipe SB. Protective Effects of Physical Exercise in Alzheimer’s Disease and Parkinson’s Disease: A Narrative Review. J Clin Neurol. 2015;11:212–219. doi: 10.3988/jcn.2015.11.3.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandya JD, Royland JE, MacPhail RC, Sullivan PG, Kodavanti PRS. Age- and brain region-specific differences in mitochondrial bioenergetics in Brown Norway rats. Neurobiol Aging. 2016;42:25–34. doi: 10.1016/j.neurobiolaging.2016.02.027. [DOI] [PubMed] [Google Scholar]
- Parone PA, Da Druz S, Tondera D, Mattenberger Y, James DI, Maechler P, Barja F, Martinou JC. Preventing mitochondrial fission impairs mitochondrial function and leads to loss of mitochondrial DNA. PLoS One. 2008;3:1–10. doi: 10.1371/journal.pone.0003257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrino C, Gargiulo G, Pironti G, Franzone A, Scudiero L, De Laurentis M, Magliulo F, Ilardi F, Carotenuto G, Schiattarella GG, Esposito G. Cardiovascular effects of treadmill exercise in physiological and pathological preclinical settings. Am J Physiol Heart Circ Physiol. 2011;300:H1983–H1989. doi: 10.1152/ajpheart.00784.2010. [DOI] [PubMed] [Google Scholar]
- Picard M, Gentil BJ, McManus MJ, White K, St Louis K, Gartside SE, Wallace DC, Turnbull DM. Acute exercise remodels mitochondrial membrane interactions in mouse skeletal muscle. J Appl Physiol. 2013;115:1562–71. doi: 10.1152/japplphysiol.00819.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard M, Ritchie D, Thomas MM, Wright KJ, Hepple RT. Alterations in intrinsic mitochondrial function with aging are fiber type-specific and do not explain differential atrophy between muscles. Aging Cell. 2011;10:1047–1055. doi: 10.1111/j.1474-9726.2011.00745.x. [DOI] [PubMed] [Google Scholar]
- Santos-alves E, Marques-aleixo I, Rizo-roca D, Torrella JR, Oliveira PJ, Magalhães J, Ascensão A. Exercise modulates liver cellular and mitochondrial proteins related to quality control signaling. Life Sci. 2015;135:124–130. doi: 10.1016/j.lfs.2015.06.007. [DOI] [PubMed] [Google Scholar]
- Sauerbeck A, Pandya J, Singh I, Bittman K, Readnower R, Bing G, Sullivan P. Analysis of regional brain mitochondrial bioenergetics and susceptibility to mitochondrial inhibition utilizing a microplate based system. J Neurosci Methods. 2011;198:36–43. doi: 10.1016/j.jneumeth.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schönfeld P, Reiser G. Why does brain metabolism not favor burning of fatty acids to provide energy? J Cereb blood flow Metab. 2013;33:1493–9. doi: 10.1038/jcbfm.2013.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigenaga MK, Hagen TM, Ames BN. Oxidative damage and mitochondrial decay in aging. Proc Natl Acad Sci U S A. 1994;91:10771–8. doi: 10.1073/pnas.91.23.10771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shutt T, Geoffrion M, Milne R, McBride HM. The intracellular redox state is a core determinant of mitochondrial fusion. EMBO Rep. 2012;13:909–15. doi: 10.1038/embor.2012.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith-Vikos T, Slack FJ. MicroRNAs and their roles in aging. J Cell Sci. 2012;125:7–17. doi: 10.1242/jcs.099200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steib K, Schaffner I, Jagasia R, Ebert B, Lie DC. Mitochondria Modify Exercise-Induced Development of Stem Cell-Derived Neurons in the Adult Brain. J Neurosci. 2014;34:6624–6633. doi: 10.1523/JNEUROSCI.4972-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner JL, Murphy EA, McClellan JL, Carmichael MD, Davis JM. Exercise training increases mitochondrial biogenesis in the brain. J Appl Physiol. 2011;111:1066–71. doi: 10.1152/japplphysiol.00343.2011. [DOI] [PubMed] [Google Scholar]
- van Praag H, Fleshner M, Schwartz MW, Mattson MP. Exercise, Energy Intake, Glucose Homeostasis, and the Brain. J Neurosci. 2014;34:15139–15149. doi: 10.1523/JNEUROSCI.2814-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Praag H, Shubert T, Zhao C, Gage FH. Exercise enhances learning and hippocampal neurogenesis in aged mice. J Neurosci. 2005;25:8680–5. doi: 10.1523/JNEUROSCI.1731-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss MW, Vivar C, Kramer AF, van Praag H. Bridging animal and human models of exercise-induced brain plasticity. Trends Cogn Sci. 2013;17:525–44. doi: 10.1016/j.tics.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Votyakova TV, Reynolds IJ. Ca2+-induced permeabilization promotes free radical release from rat brain mitochondria with partially inhibited complex I. J Neurochem. 2005;93:526–37. doi: 10.1111/j.1471-4159.2005.03042.x. [DOI] [PubMed] [Google Scholar]
- Votyakova TV, Reynolds IJ. DeltaPsi(m)-Dependent and -independent production of reactive oxygen species by rat brain mitochondria. J Neurochem. 2001;79:266–277. doi: 10.1046/j.1471-4159.2001.00548.x. [DOI] [PubMed] [Google Scholar]
- Watson K, Baar K. mTOR and the health benefits of exercise. Semin Cell Dev Biol. 2014;36:130–9. doi: 10.1016/j.semcdb.2014.08.013. [DOI] [PubMed] [Google Scholar]
- Yamada T, Adachi Y, Fukaya M, Iijima M, Sesaki H. Dynamin-Related Protein 1 Deficiency Leads to Receptor-Interacting Protein Kinase 3 Mediated Necroptotic Neurodegeneration. Am J Pathol. 2016;186:2798–2802. doi: 10.1016/j.ajpath.2016.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Lira Va, Greene NP. Exercise training-induced regulation of mitochondrial quality. Exerc Sport Sci Rev. 2012;40:159–64. doi: 10.1097/JES.0b013e3182575599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Q, Fang D, Swerdlow RH, Yu H, Chen JX, Yan SS. Antioxidants Rescue Mitochondrial Transport in Differentiated Alzheimer’s Disease Trans-Mitochondrial Cybrid Cells. J Alzheimer’s Dis. 2016;54:679–690. doi: 10.3233/JAD-160532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Trushin S, Christensen TA, Bachmeier BV, Gateno B, Schroeder A, Yao J, Itoh K, Sesaki H, Poon WW, Gylys KH, Patterson ER, Parisi JE, Diaz Brinton R, Salisbury JL, Trushina E. Altered brain energetics induces mitochondrial fission arrest in Alzheimer’s Disease. Sci Rep. 2016;6:18725. doi: 10.1038/srep18725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu JH, Horbinski C, Guo F, Watkins S, Uchiyama Y, Chu CT. Regulation of autophagy by extracellular signal-regulated protein kinases during 1-methyl-4-phenylpyridinium-induced cell death. Am J Pathol. 2007;170:75–86. doi: 10.2353/ajpath.2007.060524. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Exercise training increases mitochondrial protein expression in the gastrocnemius. Mitochondrial content was determined by Western blotting for components of each of ETC complexes. Protein content of complexes II, III, IV, and V were significantly increased in old mice after exercise but not significantly different in young mice after exercise in the gastrocnemius. Data are presented as means ± SD. YC, N=8; YT, N=8; OC, N=7; OT, N=7. *P<0.05.
Exercise training does not affect mitochochondrial mass in the brain. Protein components of mitochondrial complexes I-V were assessed by Western blotting (A). Protein levels of mitochondrial complexes I-V were not significantly different in the cortex or striatum of young mice. Protein levels of the mitochondrial proteins TOM20 and MnSOD were determined by Western blotting (B). There were no significant differences in the protein expression of TOM20 and MnSOD after exercise in old or young mice. Data are presented as means ± SD. YC, N=8; YT, N=8; OC, N=7; OT, N=7.
Exercise training does not increase markers of mitochondrial biogenesis in the brain. Protein levels of PGC-1α, TFAM, HSP60, and SIRT3 were assessed by Western blotting. There was no significant difference in protein levels of PGC-1α, TFAM, HSP60, and SIRT3 in the cortex or striatum of old or young mice after exercise training. YC, N=8; YT, N=8; OC, N=7; OT, N=7.
Exercise training does not affect expression of BDNF, FNDC5, LC3-II, or mTOR. Protien levels of BNDF, FNDC5, LC3-II, and phosphorylated and total mTOR were assessed by Western blotting. There was no significant change in the protein expression of BDNF, FNDC5, LC3-II, or phospho-mTOR after exercise in the cortex or striatum of young or old mice. YC, N=8; YT, N=8; OC, N=7; OT, N=7.
Exercise training does not affect phosphorylated DRP1 or OPA1 levels. Phosph-DRP1 (P-DRP1, phosphorylated at Ser616) and OPA1 levels were assessed by Western blotting. Protein levels of P-DRP1 were normalized to GAPDH and were not significantly different in the cortex or striatum of old or young mice (A). OPA1 levels were normalized to GAPDH and were not significantly different in the cortex of old or young mice (B). Data are presented as means ± SD. YC, N=8; YT, N=8; OC, N=7; OT, N=7.