

Abstract
The c-Jun NH2-terminal kinase (JNK) has been implicated in both cell death and survival responses to different stimuli. Here we reexamine the function of JNK in tumor necrosis factor (TNF)-stimulated cell death using fibroblasts isolated from wild-type, Mkk4-/- Mkk7-/-, and Jnk1-/- Jnk2-/- mice. We demonstrate that JNK can act to suppress TNF-stimulated apoptosis. However, we find that JNK can also potentiate TNF-stimulated necrosis by increasing the production of reactive oxygen species (ROS). Together, these data indicate that JNK can shift the balance of TNF-stimulated cell death from apoptosis to necrosis. Increased necrosis may represent a contributing factor in stress-induced inflammatory responses mediated by JNK.
Keywords: TNF, necrosis, apoptosis, JNK, reactive oxygen species
Tumor necrosis factor (TNF) is an important regulator of immune responses that also influences cell differentiation, survival, and apoptosis. The binding of TNF to the cell surface receptor TNF-R1 causes the recruitment of the adapter protein TRADD to the cytoplasmic domain of the receptor. TRADD then serves to recruit additional signaling molecules, including FADD, TRAF2, and RIP1 (Chen and Goeddel 2002). Apoptotic responses to TNF can be mediated by the activation of caspase-8 by FADD (Varfolomeev et al. 1998; Yeh et al. 1998), while anti-apoptotic responses can be mediated by activation of the NF-κB pathway by TRAF2 and RIP1 (Yeh et al. 1997; Kelliher et al. 1998). The cellular response to TNF, in part, represents a balance between these opposing pathways (Varfolomeev and Ashkenazi 2004).
TNF can also activate JNK (Kyriakis et al. 1994; Sluss et al. 1994) by a TRAF2-dependent mechanism (Yeh et al. 1997). It has been established that JNK activation is critical for TNF-stimulated AP-1-dependent gene expression (Ventura et al. 2003). However, the role of JNK in TNF-stimulated death signaling is unclear (Davis 2000). Some studies indicate that JNK is not essential for TNF-stimulated apoptosis (Liu et al. 1996; Natoli et al. 1997; Yeh et al. 1997; Lamb et al. 2003; Sakon et al. 2003). Indeed, JNK may antagonize TNF-stimulated apoptosis (Lee et al. 1997; Roulston et al. 1998; Reuther-Madrid et al. 2002; Lamb et al. 2003). In contrast, other studies indicate an essential role for JNK in TNF-induced cell death (Guo et al. 1998; De Smaele et al. 2001; Tang et al. 2001, 2002; Deng et al. 2003; Papa et al. 2004). Furthermore, genetic analysis of Drosophila demonstrates that JNK may mediate cell death caused by a TNF-related signaling pathway (Igaki et al. 2002; Moreno et al. 2002; Kauppila et al. 2003). Together, these studies suggest a complex relationship between JNK and TNF-stimulated cell death (Varfolomeev and Ashkenazi 2004).
The addition of TNF to many mammalian cells does not cause a cytotoxic response. The lack of cell death is most likely related to the ability of TNF to engage survival signaling pathways, including NF-κB (Beg and Baltimore 1996; Van Antwerp et al. 1996; Wang et al. 1996). Consequently, inactivation of survival signaling is required to study TNF-stimulated cell death and most studies have employed one of two different approaches: (1) partial inhibition of macromolecular synthesis using inhibitors of transcription or translation, and (2) inhibition of the NF-κB pathway. The use of these two different experimental strategies may contribute, in part, to the divergent conclusions reached in previous studies concerning the role of JNK in TNF-stimulated cell death. It is also possible that the role of JNK in TNF-stimulated cell death markedly differs between individual cell types.
The purpose of this study was to reexamine the role of JNK in TNF-induced cell death using comparative studies of fibroblasts isolated from wild-type, Mkk4-/- Mkk7-/-, and Jnk1-/- Jnk2-/- mice. We report that JNK has a dual role in the TNF-stimulated death of fibroblasts. JNK can suppress TNF-stimulated apoptosis, but can also potentiate TNF-stimulated necrosis. The increased necrosis is mediated, in part, by a requirement of JNK for TNF-induced production of cytotoxic reactive oxygen species (ROS).
Results
Wild-type and Jnk1-/- Jnk2-/- (Jnk-/-) fibroblasts were transduced with retroviruses to obtain populations of cells that either do or do not express an inhibitor of the NF-κB signaling pathway, a mutant form of IκBα that is not phosphorylated by the IκB kinase (ΔN-IκBα) (Fig. 1A). Control studies demonstrated that ΔN-IκBα inhibited the expression of an NF-κB target gene (IL-6) in TNF-stimulated cells (Fig. 1B). Furthermore, TNF-stimulated NF-κB DNA binding activity was detected in nuclear extracts prepared from wild-type and Jnk-/- fibroblasts, but not from wild-type (ΔN-IκBα) or Jnk-/- (ΔN-IκBα) fibroblasts, in an electrophoretic mobility shift assay using an oligonucleotide probe with an NF-κB-binding site (data not shown). Immunofluorescence analysis confirmed that TNF-stimulated nuclear translocation of NF-κB was inhibited in both wild-type and Jnk-/- cells expressing ΔN-IκBα (Fig. 1C). We conclude that these cells represent a defined group that can be used to test the role of JNK in both of the major paradigms of experimentally induced TNF-stimulated cell death (partial inhibition of macromolecular synthesis or inhibition of NF-κB).
Figure 1.
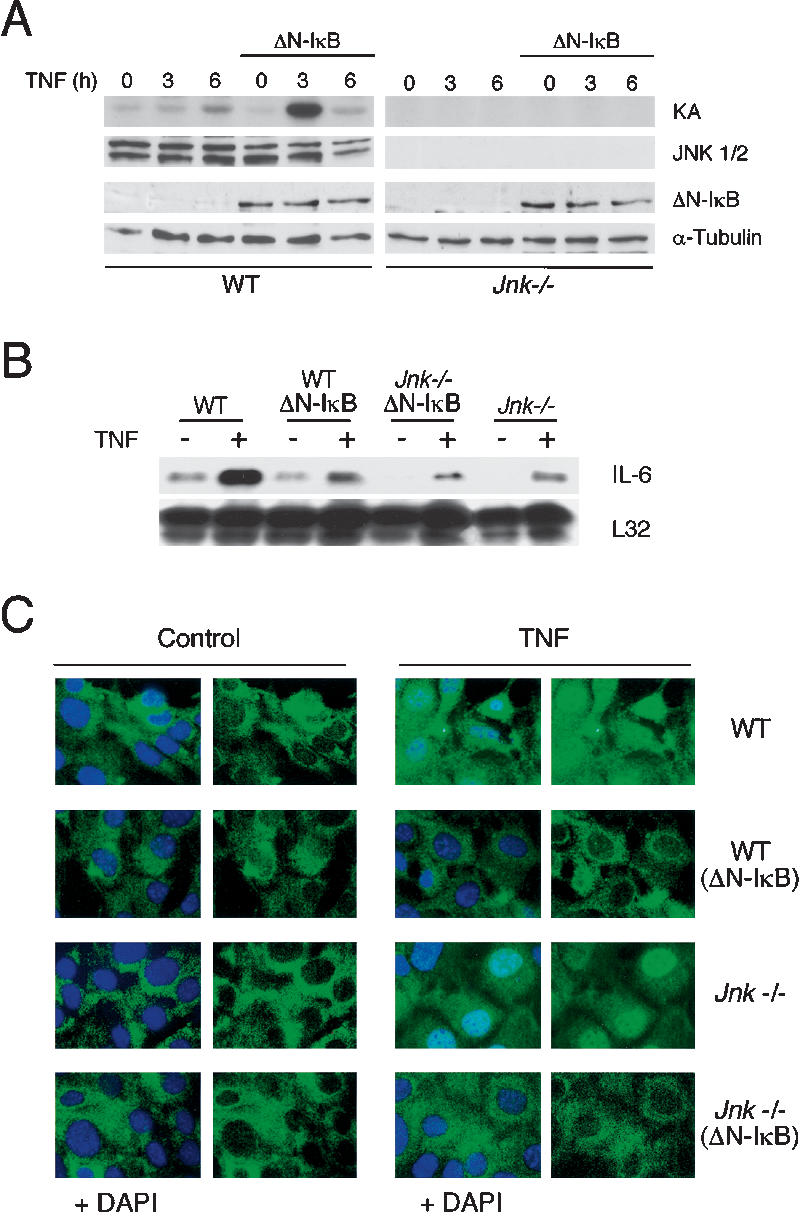
NF-κB inhibition increases TNF-stimulated JNK activation. (A) Wild-type (WT) and Jnk-/- fibroblasts were transduced with a retrovirus that expresses Flag-tagged ΔN-IκBα. The expression of ΔN-IκBα and JNK was detected by immunoblot analysis. JNK activity in cells treated with 10 ng/mL TNF was examined in an in vitro kinase assay (KA) using c-Jun as the substrate. (B) The expression of IL-6 and ribosomal protein L32 mRNA was examined in a ribonuclease protection assay. The cells were treated without and with 10 ng/mL TNF (45 min). (C) The subcellular distribution of p65 NF-κB was examined in wild-type (WT) and Jnk-/- fibroblasts. The cells were treated without or with 10 ng/mL TNF (30 min). The effect of expression of ΔN-IκBα was examined. The nuclear accumulation of p65 NF-κB (green) was examined by immunofluorescence analysis. Nuclei stained with DAPI (blue) are shown in merged images.
Cross-talk between the NF-κB and the JNK signaling pathways has been identified in previous studies (De Smaele et al. 2001; Tang et al. 2001; Reuther-Madrid et al. 2002). These studies have demonstrated that NF-κB functions to suppress sustained JNK activation in cells exposed to TNF. Increased expression of the NF-κB target genes GADD45β (De Smaele et al. 2001; Papa et al. 2004) and XIAP (Tang et al. 2001) has been implicated in this process and it appears that NF-κB functions to suppress the TNF-stimulated ROS production that causes sustained JNK activation (Sakon et al. 2003). We therefore performed control experiments to examine JNK activity using the cells isolated in this study.
Treatment with TNF caused a rapid and transient increase in JNK activation in wild-type cells that was maximal at 15 min and declined to basal levels within 45 min (data not shown). Expression of ΔN-IκBα caused no change in this early time course of JNK activation (data not shown). A second phase of JNK activation was detected during prolonged incubation of wild-type (ΔN-IκBα) cells with TNF (e.g., 3 h), but was not observed in experiments using wild-type cells (Fig. 1A). In contrast, no JNK protein or activity were detected in Jnk-/- or Jnk-/- (ΔN-IκBα) cells (Fig. 1A). Together, these data confirm the observation that NF-κB acts to suppress the second phase of TNF-stimulated JNK activity that is observed during the prolonged incubation of cells with TNF.
JNK can suppress TNF-stimulated apoptosis
We compared the apoptotic response of cells to TNF by measuring the fragmentation of genomic DNA. Treatment of wild-type and Jnk-/- cells with TNF did not cause significant changes in DNA fragmentation (Fig. 2A) or cell survival (Fig. 3A). In contrast, studies of wild-type (ΔN-IκBα) and Jnk-/- (ΔN-IκBα) cells demonstrated that TNF caused increased DNA fragmentation (Fig. 2A). No marked differences between wild-type (ΔN-IκBα) and Jnk-/- (ΔN-IκBα) cells were detected (Fig. 2A). These data demonstrate that JNK is not required for TNF-stimulated apoptosis in cells lacking a functional NF-κB pathway.
Figure 2.

JNK represses TNF-stimulated apoptosis. (A) Wild-type (WT) and Jnk-/- fibroblasts were incubated with 10 ng/mL TNF (6 h). The effect of the expression of ΔN-IκBα was examined. Apoptosis was investigated by measuring DNA fragmentation by ELISA. Wild-type fibroblasts were also incubated with TNF plus the protein synthesis inhibitor emetine (2 μM). The data shown are the mean ± SD of triplicate determinations. The data are representative of three independent experiments. (B) Wild-type (WT) and Jnk-/- fibroblasts incubated without and with 10 ng/mL TNF (6 h) in the presence of the protein synthesis inhibitor emetine (2 μM) were examined. The effect of the expression of ΔN-IκBα was examined. Apoptosis was investigated by measuring DNA fragmentation by ELISA. The data shown are the mean ± SD of triplicate determinations. The data are representative of three independent experiments. (C) Wild-type (WT) and Jnk-/- fibroblasts were incubated (6 h) without and with 10 ng/mL TNF plus 2 μM emetine. Nuclear DNA was stained with DAPI (blue). Apoptotic cells were detected by using a TUNEL assay (red).
Figure 3.

JNK promotes TNF-stimulated necrosis. (A) Wild-type (WT) and Jnk-/- fibroblasts were incubated without and with 10 ng/mL TNF. The effect of the addition of emetine (2 μM) was examined. Cell survival was measured by staining the cells with crystal violet. The data shown are the mean ± SD of triplicate determinations. The data are representative of three independent experiments. (B) Wild-type (WT) (ΔN-IκBα) and Jnk-/- (ΔN-IκBα) fibroblasts were incubated without and with 10 ng/mL TNF. The effect of the caspase inhibitor zVAD-fmk (10 μM) was examined. Cell survival was measured by staining with crystal violet. The data shown are the mean ± SD of triplicate determinations. The data are representative of three independent experiments. (C) Complementation analysis was performed using wild-type (WT) (ΔN-IκBα) and Jnk-/- (ΔN-IκBα) fibroblasts incubated without and with 10 ng/mL TNF. The effect of expression of JNK1 or JNK2 was examined. Cell survival was measured by staining with crystal violet. The data shown are the mean ± SD of triplicate determinations. The data are representative of three independent experiments. (D) Wild-type (WT) and Jnk-/- fibroblasts were incubated without and with 10 ng/mL TNF. The effect of ΔN-IκBα expression was examined. Cell survival was measured by counting cells using a Neubauer hemacytometer. The data shown are the mean ± SD of triplicate determinations. The data are representative of three independent experiments. (E) Wild-type (WT) (ΔN-IκBα) and Jnk-/- (ΔN-IκBα) fibroblasts were incubated without (Control) and with 10 ng/mL TNF (6 h). The cells were then replated in 10-cm dishes in fresh culture medium and incubated (5 d). Surviving colonies were stained with crystal violet and photographed. The data shown are representative of three independent experiments.
TNF-stimulated apoptosis can also be detected following partial inhibition of macromolecular synthesis. Thus, in the presence of the protein synthesis inhibitor emetine, TNF caused increased DNA fragmentation in both wild-type and Jnk-/- cells (Fig. 2B) and decreased cell survival (Fig. 3A). This apoptotic response was suppressed by caspase inhibition caused by the drug zVAD-fmk (data not shown). Interestingly, the apoptotic response of Jnk-/- cells under these conditions was increased compared to that of wild-type cells (Figs. 2B, 3A). TUNEL assays confirmed that treatment of Jnk-/- fibroblasts with TNF plus emetine caused increased apoptosis compared to wild-type cells (Fig. 2C). Time course studies demonstrated that this increased apoptosis represents an acceleration of the rate of Jnk-/- fibroblast death following treatment with TNF plus emetine compared to wild-type fibroblasts and does not represent an increase in the final extent of the apoptotic response (Lamb et al. 2003). Together, these data establish that JNK can suppress TNF-stimulated apoptosis.
It is interesting that the effect of JNK deficiency to accelerate TNF-stimulated apoptosis was observed following partial inhibition of macromolecular synthesis (Fig. 2B), but was not observed when the NF-κB pathway was inhibited (Fig. 2A). These data suggest that the role of JNK to suppress TNF-stimulated apoptosis requires a functional NF-κB pathway. This conclusion is consistent with previous studies that demonstrate that survival signaling by JNK in TNF-treated cells is mediated, in part, by cooperation between the JNK and NF-κB pathways (Lamb et al. 2003). Nevertheless, in the presence of emetine, Jnk-/- (ΔN-IκBα) cells exhibited increased TNF-stimulated apoptosis compared with wild-type (ΔN-IκBα) cells (Fig. 2B), indicating that this cooperative role of NF-κB may be redundant with other survival signaling pathways. Indeed, cooperative anti-apoptosis signaling by JNK together with the PI-3′ Kinase/AKT pathway has also been described (Lamb et al. 2003).
JNK can potentiate TNF-stimulated necrosis
Nonapoptotic (necrotic) cell death can be induced by TNF. Thus, the TNF-stimulated necrotic death of mouse L929 cells can be mediated by reactive oxygen species (ROS) and is increased by caspase inhibition (Vercammen et al. 1998; Fiers et al. 1999). Similarly, TNF-stimulated necrotic death of fibroblasts caused by ROS is increased by inhibition of the NF-κB pathway (Sakon et al. 2003). We therefore examined the potential role of JNK in TNF-stimulated necrotic death using wild-type and Jnk-/- fibroblasts incubated in the absence of the protein synthesis inhibitor emetine. As expected, measurement of cell survival indicated that wild-type cells were not killed by TNF (Fig. 3A), but were rapidly killed by TNF if the NF-κB pathway was inhibited by ΔN-IκBα (Fig. 3B). This TNF-stimulated cell death was not blocked by the caspase inhibitor zVAD-fmk and was attenuated in Jnk-/- cells (Fig. 3B). Time course analysis demonstrated that JNK deficiency delayed cell death for at least 24 h (Fig. 3B). Measurement of cell number confirmed that TNF caused decreased viability of wild-type (ΔN-IκBα) fibroblasts, but did not cause decreased viability of wild-type, Jnk-/-, or Jnk-/- (ΔN-IκBα) fibroblasts (Fig. 3D). Similarly, studies of cell viability following TNF treatment using colony formation assays demonstrated that TNF killed wild-type (ΔN-IκBα) fibroblasts, but not Jnk-/- (ΔN-IκBα) fibroblasts (Fig. 3E). Complementation analysis demonstrated that the expression of JNK1 or JNK2 increased the TNF-stimulated death of Jnk-/- (ΔN-IκBα) fibroblasts (Fig. 3C), indicating that the increased survival of TNF-stimulated Jnk-/- (ΔN-IκBα) cells compared to wild-type (ΔN-IκBα) cells was caused by JNK deficiency. Together, these data indicate that TNF can cause cell death that is dependent on JNK when the NF-κB pathway is inhibited.
To confirm that the JNK-dependent TNF-induced cell death was not apoptosis, we examined the morphology of the cells by electron microscopy (Fig. 4). TNF-treated wild-type (ΔN-IκBα) cells displayed altered morphology, including the presence of distended cytoplasmic organelles and rupture of nuclear and plasma membranes, in the absence of obvious apoptotic bodies. These data indicate that the JNK-dependent TNF-stimulated cell death observed in wild-type (ΔN-IκBα) cells is mediated, at least in part, by a nonapoptotic (necrotic) mechanism.
Figure 4.
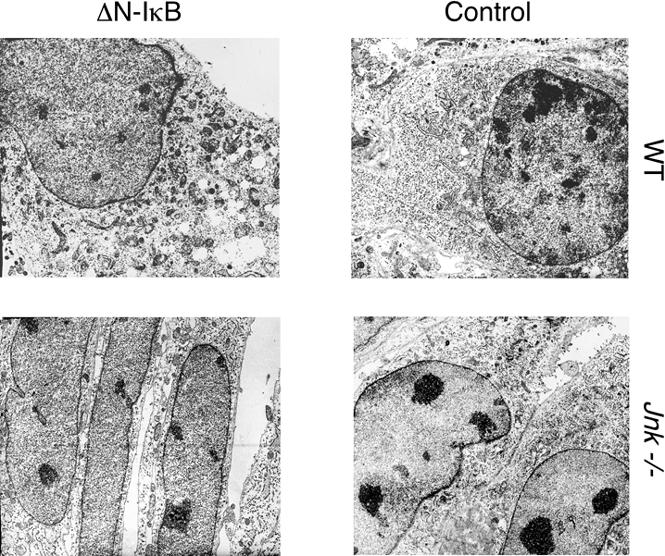
JNK-dependent cell death caused by TNF is associated with necrotic cellular morphology. TNF-treated wild-type (WT) and Jnk-/- fibroblasts were examined by transmission electron microscopy. The effect of the expression of ΔN-IκBα was examined. Representative images are presented.
JNK pathway mutations can suppress TNF-stimulated cell death
Complementation analysis indicates that the effect of JNK deficiency to cause reduced TNF-stimulated necrosis is most likely caused by the loss of JNK signaling (Fig. 3C). To obtain independent evidence for a role of JNK signaling in TNF-stimulated necrosis, we examined the effect of Mkk4 and Mkk7 gene disruption. It has been established in previous studies that the MAP kinase kinases MKK4 and MKK7 are required for JNK activation (Tournier et al. 2001). Compound mutation of the Mkk4 and Mkk7 genes prevents JNK activation (Tournier et al. 2001). Indeed, TNF-stimulated JNK activation was observed in wild-type fibroblasts, but not in Mkk4-/- Mkk7-/- fibroblasts (Fig. 5A). Previous studies of primary embryonic fibroblasts have demonstrated that MKK7 has an essential role while MKK4 plays a collaborative role in TNF-stimulated JNK activation because TNF selectively increases the activity of MKK7, but not MKK4 (Tournier et al. 2001). In this study, using 3T3 fibroblasts derived from primary embryonic fibroblasts, we found that TNF was able to activate both MKK4 and MKK7 (Fig. 5B). Consistent with this observation, we found a partial loss of TNF-stimulated JNK activation in both Mkk4-/- and Mkk7-/- fibroblasts (Fig. 5A). Thus, in 3T3 fibroblasts, the MKK4 and MKK7 protein kinases appear to serve partially redundant functions in TNF-stimulated JNK activation. These data (Fig. 5), together with previous studies (Tournier et al. 2001), suggest that the roles of MKK4 and MKK7 in TNF-stimulated JNK activation may differ between cell types.
Figure 5.

Effect of Mkk4 and Mkk7 gene disruption on TNF-stimulated JNK activation and necrosis. (A) Wild-type (WT), Mkk4-/-, Mkk7-/-, and Mkk4-/- Mkk7-/- fibroblasts were incubated (10 min) without and with 10 ng/mL TNF. The amount of tubulin, JNK, and phospho-JNK was examined by immunoblot analysis. The data shown are representative of three independent experiments. (B) Wild-type (WT) fibroblasts were incubated with 10 ng/mL TNF. The amount of tubulin, MKK4, phospho-MKK4, MKK7, and phospho-MKK7 was examined by immunoblot analysis. The data shown are representative of three independent experiments. (C) Wild-type (WT), Mkk4-/-, Mkk7-/-, and Mkk4-/- Mkk7-/- fibroblasts that express ΔN-IκBα were incubated without and with 10 ng/mL TNF. Cell survival after incubation for 6 and 24 h was examined by staining with crystal violet. The data shown are the mean ± SD of triplicate determinations. The data are representative of three independent experiments. (D) Wild-type (WT), Mkk4-/-, Mkk7-/-, and Mkk4-/- Mkk7-/- fibroblasts were incubated with 10 ng/mL TNF. The effect of ΔN-IκBα expression was examined. The amount of tubulin, ΔN-IκBα, and JNK was examined by immunoblot analysis. JNK activity was examined in an in vitro kinase assay (KA) using the substrate c-Jun. The data shown are representative of three independent experiments.
Measurement of JNK activity using an in vitro protein kinase assay demonstrated that TNF caused rapid and transient JNK activation in wild-type, Mkk4-/-, and Mkk7-/- fibroblasts (Fig. 5D). Expression of ΔN-IκBα did not markedly alter this early phase JNK activation, but did cause a second and more sustained phase of JNK activation at later times following treatment with TNF. As expected, JNK activity was not detected in the compound mutant Mkk4-/- Mkk7-/- fibroblasts (Fig. 5D). These fibroblasts, which lack JNK activity, provide the possibility for an alternative test of the role of JNK in TNF-stimulated necrosis.
Survival studies performed in the absence of emetine demonstrated that TNF did not cause decreased fibroblast viability. However, TNF-stimulated killing was observed if ΔN-IκBα was expressed in the fibroblasts. Survival assays demonstrated that the death response of wild-type, Mkk4-/-, and Mkk7-/- fibroblasts was similar (Fig. 5C). In contrast, the compound mutant Mkk4-/- Mkk7-/- fibroblasts that lack JNK activity (Fig. 5D) were found to be resistant to the cytotoxic effects of TNF (Fig. 5C). These data provide independent evidence in favor of the hypothesis that JNK activity contributes to TNF-stimulated death when the NF-κB pathway is inhibited.
ROS production contributes to TNF-stimulated necrosis
The production of ROS has been implicated as a mechanism of TNF-stimulated necrotic death (Vercammen et al. 1998; Fiers et al. 1999). To examine whether ROS may be relevant to TNF-stimulated JNK-dependent necrotic cell death, we investigated the effect of butylated hydroxyanisole (BHA), a scavenger of ROS. Measurement of cell survival demonstrated that BHA inhibited the TNF-induced death of wild-type (ΔN-IκBα) cells (Fig. 6A). Similar results were obtained when wild-type (ΔN-IκBα) and Jnk-/- (ΔN-IκBα) fibroblasts were examined by flow cytometry (data not shown). These data indicate that JNK-dependent TNF-stimulated necrosis may be mediated by ROS.
Figure 6.

TNF-stimulated necrosis requires ROS production. (A) Wild-type (WT) and Jnk-/- fibroblasts expressing ΔN-IκBα were treated with 10 ng/mL TNF (12 h). Cell survival was measured by staining with crystal violet. The effect of the addition of butylated hydroxyanisole (BHA; 100 μM) was examined. The data shown are the mean ± SD of triplicate determinations. The data are representative of three independent experiments. (B) Wild-type (WT) and Jnk-/- fibroblasts were incubated (4 h) with different concentrations of hydrogen peroxide. The effect of ΔN-IκBα expression was examined. Cell survival was measured by staining with crystal violet. The data shown are the mean ± SD of triplicate determinations. The data are representative of three independent experiments. (C) Wild-type (WT) (ΔN-IκBα) and Jnk-/- (ΔN-IκBα) fibroblasts were incubated with 50 μM hydrogen peroxide. Cell survival was measured by staining with crystal violet. The data shown are the mean ± SD of triplicate determinations. The data are representative of three independent experiments.
To test whether JNK may play a role in ROS-mediated cell death, we examined the effect of the exposure of wild-type and Jnk-/- cells to exogenous peroxide (Fig. 6B). Both wild-type and Jnk-/- cells were killed by peroxide. Dose response analysis indicated that a lower concentration of peroxide was required to kill Jnk-/- fibroblasts than wild-type fibroblasts (Fig. 6B). Time course analysis demonstrated that the increased sensitivity of Jnk-/- fibroblasts to peroxide was associated with an accelerated rate of cell death but not with an increase in the final extent of the cell death response (Fig. 6C). Together, these data indicate that JNK can play a protective role in cells exposed to redox stress.
JNK contributes to TNF-stimulated ROS production
The increased sensitivity of Jnk-/- fibroblasts to peroxide compared to wild-type fibroblasts (Fig. 6A,B) contrasts with resistance of Jnk-/- fibroblasts to ROS-dependent necrotic cell death caused by TNF (Fig. 6A). These findings suggest that the resistance of Jnk-/- fibroblasts to TNF-stimulated necrosis was not caused by differences in the response of wild-type and Jnk-/- fibroblasts to ROS. Instead these data suggest that wild-type and Jnk-/- fibroblasts may differ in their ability to produce ROS following treatment with TNF.
To test whether JNK is relevant to TNF-induced ROS production, we used the probe CM-H2DCFDA that exhibits increased fluorescence following exposure to peroxide and hydroxyl radicals. ROS were not detected in studies of wild-type and Jnk-/- cells treated with TNF. In contrast, studies of wild-type (ΔN-IκBα) cells indicated that TNF caused a marked increase in ROS production (Fig. 7A), consistent with previous observations (Sakon et al. 2003). However, TNF did not cause ROS production in Jnk-/- (ΔN-IκBα) cells (Fig. 7A). To confirm the requirement of JNK, we examined TNF-stimulated ROS production in mixed cultures of wild-type (ΔN-IκBα) and Jnk-/- (ΔN-IκBα) fibroblasts; this analysis demonstrated that ROS production was only detected in wild-type (ΔN-IκBα) fibroblasts (Fig. 7B). Interestingly, new gene expression was not required for TNF-stimulated ROS production since treatment of wild-type (ΔN-IκBα) fibroblasts with emetine did not prevent TNF-stimulated ROS production (Fig. 7A).
Figure 7.

JNK-dependent ROS production contributes to TNF-stimulated necrosis. (A) Wild-type (WT) and Jnk-/- fibroblasts expressing ΔN-IκBα were incubated (4 h) without or with 10 ng/mL TNF or 10 ng/mL TNF plus 2 μM emetine. The cells were stained with 1 μM CM-H2DCFDA to examine ROS production (upper panels; green) and with DAPI to detect nuclear DNA (lower panels; blue). The data shown are representative of three independent experiments. (B) Jnk-/- (ΔN-IκBα) fibroblasts were stained with CellTracker Red CMTPX, cocultured with wild-type (WT) (ΔN-IκBα) fibroblasts, and incubated (4 h) with 10 ng/mL TNF. The cells were stained with 1 μM CM-H2DCFDA to examine ROS production (green) and with DAPI to detect nuclear DNA (blue). The data shown are representative of three independent experiments.
Together, these data indicate that JNK is essential for TNF-induced ROS production by fibroblasts when the NF-κB pathway is inhibited (Fig. 7). In contrast, JNK is not required for cell death caused by ROS (Fig. 6). Together, these data indicate that JNK-dependent ROS production may account for the requirement of JNK for efficient TNF-stimulated necrosis.
Discussion
It is known that TNF can cause cell death by both apoptotic and nonapoptotic (necrotic) mechanisms (Tracey and Cerami 1993). TNF-stimulated apoptotic cell death is mediated by caspase activation (Chen and Goeddel 2002). Indeed Caspase 8-/- fibroblasts are resistant to TNF-stimulated apoptosis when protein synthesis is partially inhibited (Varfolomeev et al. 1998). In contrast, it is established that ROS play a critical role in TNF-stimulated necrotic death (Vercammen et al. 1998; Fiers et al. 1999). The results of the present study demonstrate that JNK can have opposing actions on these two mechanisms of cell death: inhibition of TNF-stimulated apoptosis and potentiation of TNF-stimulated necrosis.
It is interesting that both the timing and the mechanism of JNK activation associated with these effects of JNK on TNF-stimulated cell death are different. Thus, anti-apoptotic JNK signaling is associated with the rapid and transient activation of JNK mediated by TRAF2 (Lamb et al. 2003), while pro-necrotic JNK signaling is associated with the later and more sustained phase of JNK activation that is mediated by ROS (Sakon et al. 2003).
TNF-stimulated ROS production requires JNK and is suppressed by NF-κB
Necrotic cell death caused by TNF is promoted by inhibition of NF-κB and is mediated, in part, by increased ROS production (Lin et al. 2003; Sakon et al. 2003). These data indicate that NF-κB acts to suppress ROS production in cells exposed to TNF. This function of NF-κB may be mediated by increased expression of NF-κB target genes, including manganese-dependent superoxide dismutase (Tanaka et al. 2002). Endogenously produced ROS can be scavenged by superoxide dismutases, glutathione peroxidases, and peroxiredoxins (Thannickal and Fanburg 2000). TNF-stimulated ROS production may therefore not be detected in normal cells, but can be observed following inhibition of the NF-κB pathway (Sakon et al. 2003).
It is established that TRADD, RIP1, and TRAF2 are required for TNF-stimulated ROS production (Chan et al. 2003; Lin et al. 2003). Here we demonstrate that JNK is also required for TNF-stimulated ROS production. Since TRADD and TRAF2 are required for rapid TNF-stimulated JNK activation (Chen and Goeddel 2002), it is possible that TRADD, TRAF2, and JNK function within a signaling module that can initiate ROS production. Nevertheless, the mechanism that accounts for the requirement of JNK for TNF-stimulated ROS production is unclear. Since JNK has been implicated in the regulation of mitochondria (an important source of endogenous cellular ROS), it is likely that JNK may regulate ROS production by mitochondria (Davis 2000).
Cells deficient in NF-κB signaling exhibit greatly increased sustained TNF-stimulated JNK activity (De Smaele et al. 2001; Tang et al. 2001; Reuther-Madrid et al. 2002). This sustained JNK activation is mediated by increased ROS production in NF-κB-deficient cells (Sakon et al. 2003). It is therefore interesting that TNF-stimulated ROS production is JNK dependent (Fig. 7). These data suggest that a positive feedback cycle operates in NF-κB-deficient cells in which TNF-stimulated ROS production causes increased JNK activity that leads to a further increase in ROS production (Fig. 8). This type of positive feedback involving JNK may generate sufficient amounts of ROS to cause necrosis (Fig. 8).
Figure 8.

Schematic illustration of the role of JNK in fibroblasts during TNF-stimulated cell death. TNF causes transient JNK activation that is mediated by a TRAF2-dependent mechanism. This transient activation of JNK acts to inhibit TNF-stimulated apoptosis observed following partial inhibition of mRNA or protein synthesis. This effect of JNK is mediated, in part, by cooperation between JNK and the NF-κB and PI-3′ kinase survival signaling pathways (Lamb et al. 2003). TNF also causes activation of the NF-κB pathway. One function of NF-κB is to suppress TNF-stimulated ROS production, in part, by increased expression of manganese-dependent superoxide dismutase. However, when NF-κB signaling is reduced, TNF causes ROS production. The TNF-stimulated ROS acts in a positive feedback cycle that activates JNK, which then leads to more ROS production, and ultimately to necrotic cell death. It is possible that the transient activation of JNK can initiate this postitive feedback cycle (dashed line), but other signaling pathways are likely to play an important role. Nevertheless, JNK is required for the sustained phase of ROS production. Consequently, JNK can potentiate TNF-stimulated necrosis in fibroblasts.
JNK can suppress TNF-stimulated apoptosis
Treatment of cells with TNF causes rapid and transient activation of JNK by a TRAF2-dependent mechanism (Yeh et al. 1997). A second and more sustained phase of JNK activation occurs at later times, is mediated by TNF-stimulated ROS production, and is suppressed by NF-κB (Sakon et al. 2003). The TRAF2-mediated early phase of JNK activation is not required for TNF-stimulated apoptosis (Yeh et al. 1997). Indeed, the rapid and transient activation of JNK caused by TNF appears to mediate an anti-apoptotic response (Lee et al. 1997; Roulston et al. 1998; Reuther-Madrid et al. 2002; Lamb et al. 2003). Thus, JNK-deficient fibroblasts exhibit accelerated TNF-stimulated apoptosis following partial inhibition of protein synthesis. Complementation analysis indicates that this anti-apoptotic action of JNK involves JunD and is mediated, in part, by functional cooperation of JNK with survival signaling pathways (including NF-κB and PI-3′ kinase) to cause increased expression of anti-apoptotic genes (Lamb et al. 2003). The observation that NF-κB inhibition does not prevent the increased TNF-stimulated apoptosis of Jnk-/- cells following partial inhibition of protein synthesis (Fig. 2B) suggests that there is some functional redundancy between the survival signaling pathways that can cooperate with JNK.
Signaling cell death by TNF-R1 and TNF-R2
Gene disruption studies have demonstrated that TNF-R1, but not TNF-R2, is essential for TNF-stimulated necrosis (Chan et al. 2003; Lin et al. 2003). Nevertheless, an important role for TNF-R2 in death signaling has been established by the observation that TNF-R2 can cause degradation of the adapter protein TRAF2 (Chan and Lenardo 2000; Fotin-Mleczek et al. 2002; Li et al. 2002). Since TRAF2 activates the NF-κB and JNK anti-apoptosis signaling pathways, TRAF2 degradation caused by TNF-R2 may enhance death signaling by TNF-R1. Studies of wild-type and Jnk-/- fibroblasts indicate that the level of TNF-R1 and TNF-R2 expression is similar, but TNF-induced TRAF2 degradation was not detected (Lamb et al. 2003). However, TNF-R2-stimulated TRAF2 degradation may be critical for TNF-stimulated cell death under different experimental conditions or in other cell types. Thus, disruption of the IκB kinase gene Ikkβ in murine liver causes reduced (but not eliminated) NF-κB activation and results in increased liver damage in response to membrane-bound TNF (that binds both TNF-R1 and TNF-R2), but not in response to soluble TNF (that primarily binds TNF-R1) (Maeda et al. 2003). Interestingly, the liver damage caused by membrane-bound TNF is dependent upon JNK. Since TRAF2 is degraded, it is possible that the mechanism of JNK activation in this model is mediated by ROS (rather than by TRAF2). This form of liver damage may therefore be mediated by necrosis in addition to apoptosis. The ability of TNF-R2 to cause TRAF2 degradation, and thus suppress TNF-R1 stimulated NF-κB activation, may represent an important mechanism in vivo that promotes JNK-dependent TNF-stimulated cell death. The relative expression of TNF-R1 and TNF-R2 by different types of cells may therefore be an important factor that influences the role of JNK in death signaling. These considerations suggest that the role of JNK in TNF-induced cell death may be cell type dependent. Our study of fibroblasts reported here indicates that JNK can inhibit TNF-induced apoptosis and potentiate necrosis. It will be important to extend our analysis of fibroblasts in future studies to examine the role of JNK in other cell types.
Increased necrosis and reduced apoptosis may contribute to JNK-induced inflammation
Cell death in vivo can be mediated by apoptotic and nonapoptotic (necrotic) responses. Both of these mechanisms can contribute to cell death in response to one stimulus. Thus, brain ischemia can cause necrosis at the site of injury followed by a delayed apoptotic response in surrounding tissues. Similarly, cell death in model systems in vitro can be a mixture of both apoptotic and necrotic processes. For example, treatment of NF-κB-deficient fibroblasts with TNF causes both an apoptotic response (Fig. 2) and a necrotic response (Figs. 3, 4). The presence of both types of cell death is significant for inflammatory responses in vivo. It is established that apoptosis represents an anti-inflammatory form of cell death, whereas necrosis leads to marked inflammation. Since JNK can suppress apoptosis and potentiate necrosis caused by TNF, it is possible that this shift in the balance of cell death from apoptosis to necrosis may be a contributing factor in JNK-induced inflammation.
Conclusions
The JNK signaling pathway has a complex relationship with TNF-stimulated cell death. The results of the present study of fibroblasts indicate that JNK may have no role in TNF-stimulated apoptosis (Fig. 2A), may provide an anti-apoptotic signal (Fig. 2B,C), or may potentiate TNF-stimulated necrosis (Fig. 3B), depending on the experimental conditions of the analysis. It is likely that this complexity of JNK function may account for the divergent conclusions reached in previous studies concerning the role of JNK in TNF-stimulated cell death. An important future goal will be to extend our analysis of fibroblasts to other cell types.
Materials and methods
Tissue culture
Wild-type, Jnk1-/- Jnk2-/- (Jnk-/-), Mkk4-/-, Mkk7-/-, and Mkk4-/- Mkk7-/- fibroblasts were established in culture from mouse strain 129svJ E13.5 embryos (Tournier et al. 2000, 2001; Kennedy et al. 2003) using a standard 3T3 protocol. All animals were housed in a facility accredited by the American Association for Laboratory Animal Care and the animal studies were approved by the Institutional Animal Care and Use Committee of the University of Massachusetts. The fibroblasts were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat inactivated fetal bovine serum (FBS), 2 mM glutamine, 100 units/mL of penicillin, 100 μg/mL of streptomycin (Invitrogen) at 37°C in a humidified atmosphere with 5% CO2. The fibroblasts were transduced with the retroviral vectors pBABE-IRES- PuroR or pBABE-Flag-ΔN-IκBα-IRES-PuroR (Wisdom et al. 1999) and selected by incubation in medium containing 2 μg/mL puromycin. The fibroblasts were also transduced with the retroviral vectors pMSCV-Flag-JNK1-IRES-eGFP or pMSCV-Flag-JNK2-IRES-eGFP (Lamb et al. 2003) and selected by flow cytometry. All experiments were performed using pools of clones (>104) to prevent problems of interpretation caused by clonal variability.
Cell number in proliferation assays was assessed using a Neubauer hemacytometer. Relative cell number was also assessed by staining with crystal violet (Tournier et al. 2000). Colony survival assays were performed by incubating cells in 35-mm dishes with 10 ng/mL TNF (6 h) and then replating the cells in 10-cm dishes in fresh culture medium; surviving cells growing in colonies were detected after incubation for 5 d using crystal violet.
Protein kinase studies
Immunoblot analysis was performed by probing with antibodies to JNK (Pharmingen), phospho-JNK1/2 (Cell Signaling), MKK4 (SantaCruz), phospho-MKK4 (Cell Signaling), MKK7 (Pharmingen), phospho-MKK7 (Biosource), α-Tubulin (Sigma), or Flag epitope tag (M2) (Sigma) and immune complexes were detected by enhanced chemiluminescence (NEN). JNK activity was measured by an in vitro kinase assay using c-Jun as the substrate (Whitmarsh and Davis 2001).
Apoptosis assays
TUNEL assays and DNA fragmentation assays were performed using the In Situ Cell Death Detection Kit (TMR red) and the Cell Death Detection Elisaplus kit, respectively, following the manufacturer's recommedations (Roche). The fibroblasts on coverslips were mounted in Vectashield with 4′-6′-diamino-2-phenylindole (DAPI; Vector Laboratories) and examined by fluorescence microscopy with a Zeiss Axioplan microscope.
RNase protection assays
Total RNA (5 μg) was examined using the “Multi-probe RNase protection assay” (Pharmingen) with the template set mCK2b following the manufacturer's recommendations. The products were separated on a 5% sequencing gel, detected by autoradiography, and quantitated by PhosphorImager analysis (Molecular Dynamics).
Measurement of ROS
Exponentially growing fibroblasts were incubated with TNF (4 h) and with 1 μM 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescence diacetate (CM-H2DCFDA; Molecular Probes) in PBS (15 min) (Sakon et al. 2003). Fluorescence was detected using 488-nm laser illumination with a Leica confocal microscope or by flow cytometry. In some experiments, wild-type and Jnk-/- cells were examined in mixed culture; the wild-type and Jnk-/- cells were distinguished by selective labeling of one cell population with CellTracker Red CMTPX (Molecular Probes). Nuclei were stained with DAPI (1:10,000; Molecular Probes, Inc.).
Electron microscopy
Fibroblasts were incubated without and with TNF (4 h), fixed with 2.5% glutaraldehyde, and processed for electron microscopy using standard techniques.
Immunofluorescence microscopy
Cells were grown on glass coverslips and fixed with 4% paraformaldehyde (Ted Pella, Inc.) in PBS (10 min), washed with PBS, incubated with 50 mM NH4Cl in PBS (10 min), and permeabilized with 0.1% Triton X-100 in PBS. After incubation (1 h) with 5% (w/v) bovine serum albumin (BSA) in PBS, the coverslips were incubated (1 h) with an antibody to p65 NF-κB (Santa Cruz) in PBS with 3% BSA. Immune complexes were detected with fluorescein-conjugated anti-rabbit Ig secondary antibody (Jackson ImmunoResearch Inc.) in PBS with 1% BSA. Nuclei were stained with DAPI (1:10,000; Molecular Probes, Inc.). Coverslips were mounted with polyvinylalcohol 30,000-70,000 (Sigma). Fluorescence microscopy was performed with a conventional Zeiss Axioplan microscope.
Acknowledgments
We thank Dr. R. Wisdom for providing essential reagents, J. Nunnari for assistance with electron microscopy, and K. Gemme for expert administrative assistance. R.J.D. and R.A.F. are investigators of the Howard Hughes Medical Institute. This study was supported, in part, by research grants CA65861 (to R.J.D.), AI35098 (to A.S.B.), CA73756 (to A.S.B.), and by a Diabetes and Endocrinology Center grant (DK32520) from the National Institutes of Health.
Article published online ahead of print. Article and publication date are at http://www.genesdev.org/cgi/doi/10.1101/gad.1223004.
References
- Beg A.A. and Baltimore, D. 1996. An essential role for NF-κB in preventing TNF-α-induced cell death. Science 274: 782-784. [DOI] [PubMed] [Google Scholar]
- Chan F.K. and Lenardo, M.J. 2000. A crucial role for p80 TNF-R2 in amplifying p60 TNF-R1 apoptosis signals in T lymphocytes. Eur. J. Immunol. 30: 652-660. [DOI] [PubMed] [Google Scholar]
- Chan F.K., Shisler, J., Bixby, J.G., Felices, M., Zheng, L., Appel, M., Orenstein, J., Moss, B., and Lenardo, M.J. 2003. A role for tumor necrosis factor receptor-2 and receptor-interacting protein in programmed necrosis and antiviral responses. J. Biol. Chem. 278: 51613-51621. [DOI] [PubMed] [Google Scholar]
- Chen G. and Goeddel, D.V. 2002. TNF-R1 signaling: A beautiful pathway. Science 296: 1634-1635. [DOI] [PubMed] [Google Scholar]
- Davis R.J. 2000. Signal transduction by the JNK group of MAP kinases. Cell 103: 239-252. [DOI] [PubMed] [Google Scholar]
- Deng Y., Ren, X., Yang, L., Lin, Y., and Wu, X. 2003. A JNK-dependent pathway is required for TNFα-induced apoptosis. Cell 115: 61-70. [DOI] [PubMed] [Google Scholar]
- De Smaele E., Zazzeroni, F., Papa, S., Nguyen, D.U., Jin, R., Jones, J., Cong, R., and Franzoso, G. 2001. Induction of gadd45β by NF-κB downregulates pro-apoptotic JNK signalling. Nature 414: 308-313. [DOI] [PubMed] [Google Scholar]
- Fiers W., Beyaert, R., Declercq, W., and Vandenabeele, P. 1999. More than one way to die: Apoptosis, necrosis and reactive oxygen damage. Oncogene 18: 7719-7730. [DOI] [PubMed] [Google Scholar]
- Fotin-Mleczek M., Henkler, F., Samel, D., Reichwein, M., Hausser, A., Parmryd, I., Scheurich, P., Schmid, J.A., and Wajant, H. 2002. Apoptotic crosstalk of TNF receptors: TNF-R2-induces depletion of TRAF2 and IAP proteins and accelerates TNF-R1-dependent activation of caspase-8. J. Cell Sci. 115: 2757-2770. [DOI] [PubMed] [Google Scholar]
- Guo Y.L., Baysal, K., Kang, B., Yang, L.J., and Williamson, J.R. 1998. Correlation between sustained c-Jun N-terminal protein kinase activation and apoptosis induced by tumor necrosis factor-α in rat mesangial cells. J. Biol. Chem. 273: 4027-4034. [DOI] [PubMed] [Google Scholar]
- Igaki T., Kanda, H., Yamamoto-Goto, Y., Kanuka, H., Kuranaga, E., Aigaki, T., and Miura, M. 2002. Eiger, a TNF superfamily ligand that triggers the Drosophila JNK pathway. EMBO J. 21: 3009-3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauppila S., Maaty, W.S., Chen, P., Tomar, R.S., Eby, M.T., Chapo, J., Chew, S., Rathore, N., Zachariah, S., Sinha, S.K., et al. 2003. Eiger and its receptor, Wengen, comprise a TNF-like system in Drosophila. Oncogene 22: 4860-4867. [DOI] [PubMed] [Google Scholar]
- Kelliher M.A., Grimm, S., Ishida, Y., Kuo, F., Stanger, B.Z., and Leder, P. 1998. The death domain kinase RIP mediates the TNF-induced NF-κB signal. Immunity 8: 297-303. [DOI] [PubMed] [Google Scholar]
- Kennedy N.J., Sluss, H.K., Jones, S.N., Bar-Sagi, D., Flavell, R.A., and Davis, R.J. 2003. Suppression of Ras-stimulated transformation by the JNK signal transduction pathway. Genes & Dev. 17: 629-637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyriakis J.M., Banerjee, P., Nikolakaki, E., Dai, T., Rubie, E.A., Ahmad, M.F., Avruch, J., and Woodgett, J.R. 1994. The stress-activated protein kinase subfamily of c-Jun kinases. Nature 369: 156-160. [DOI] [PubMed] [Google Scholar]
- Lamb J.A., Ventura, J.J., Hess, P., Flavell, R.A., and Davis, R.J. 2003. JunD mediates survival signaling by the JNK signal transduction pathway. Mol. Cell 11: 1479-1489. [DOI] [PubMed] [Google Scholar]
- Lee S.Y., Reichlin, A., Santana, A., Sokol, K.A., Nussenzweig, M.C., and Choi, Y. 1997. TRAF2 is essential for JNK but not NF-κB activation and regulates lymphocyte proliferation and survival. Immunity 7: 703-713. [DOI] [PubMed] [Google Scholar]
- Li X., Yang, Y., and Ashwell, J.D. 2002. TNF-RII and c-IAP1 mediate ubiquitination and degradation of TRAF2. Nature 416: 345-347. [DOI] [PubMed] [Google Scholar]
- Lin Y., Choksi, S., Shen, H.M., Yang, Q.F., Hur, G.M., Kim, Y.S., Tran, J.H., Nedospasov, S.A., and Liu, Z.G. 2004. Tumor necrosis factor-induced nonapoptotic cell death requires receptor-interacting protein-mediated cellular reactive oxygen species accumulation. J. Biol. Chem. 279: 10822-10828. Epub 2003 Dec 29. [DOI] [PubMed] [Google Scholar]
- Liu Z.G., Hsu, H., Goeddel, D.V., and Karin, M. 1996. Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-κB activation prevents cell death. Cell 87: 565-576. [DOI] [PubMed] [Google Scholar]
- Maeda S., Chang, L., Li, Z.W., Luo, J.L., Leffert, H., and Karin, M. 2003. IKKβ is required for prevention of apoptosis mediated by cell-bound but not by circulating TNFα. Immunity 19: 725-737. [DOI] [PubMed] [Google Scholar]
- Moreno E., Yan, M., and Basler, K. 2002. Evolution of TNF signaling mechanisms: JNK-dependent apoptosis triggered by Eiger, the Drosophila homolog of the TNF superfamily. Curr. Biol. 12: 1263-1268. [DOI] [PubMed] [Google Scholar]
- Natoli G., Costanzo, A., Ianni, A., Templeton, D.J., Woodgett, J.R., Balsano, C., and Levrero, M. 1997. Activation of SAPK/JNK by TNF receptor 1 through a noncytotoxic TRAF2-dependent pathway. Science 275: 200-203. [DOI] [PubMed] [Google Scholar]
- Papa S., Zazzeroni, F., Bubici, C., Jayawardena, S., Alvarez, K., Matsuda, S., Nguyen, D.U., Pham, C.G., Nelsbach, A.H., Melis, T., et al. 2004. Gadd45β mediates the NF-κB suppression of JNK signalling by targeting MKK7/JNKK2. Nat. Cell Biol. 6: 146-153. [DOI] [PubMed] [Google Scholar]
- Reuther-Madrid J.Y., Kashatus, D., Chen, S., Li, X., Westwick, J., Davis, R.J., Earp, H.S., Wang, C.Y., and Baldwin Jr., A.S. 2002. The p65/RelA subunit of NF-κB suppresses the sustained, antiapoptotic activity of Jun kinase induced by tumor necrosis factor. Mol. Cell. Biol. 22: 8175-8183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roulston A., Reinhard, C., Amiri, P., and Williams, L.T. 1998. Early activation of c-Jun N-terminal kinase and p38 kinase regulate cell survival in response to tumor necrosis factor α. J. Biol. Chem. 273: 10232-10239. [DOI] [PubMed] [Google Scholar]
- Sakon S., Xue, X., Takekawa, M., Sasazuki, T., Okazaki, T., Kojima, Y., Piao, J.H., Yagita, H., Okumura, K., Doi, T., et al. 2003. NF-κB inhibits TNF-induced accumulation of ROS that mediate prolonged MAPK activation and necrotic cell death. EMBO J. 22: 3898-3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sluss H.K., Barrett, T., Derijard, B., and Davis, R.J. 1994. Signal transduction by tumor necrosis factor mediated by JNK protein kinases. Mol. Cell. Biol. 14: 8376-884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka H., Matsumura, I., Ezoe, S., Satoh, Y., Sakamaki, T., Albanese, C., Machii, T., Pestell, R.G., and Kanakura, Y. 2002. E2F1 and c-Myc potentiate apoptosis through inhibition of NF-κB activity that facilitates MnSOD-mediated ROS elimination. Mol. Cell 9: 1017-1029. [DOI] [PubMed] [Google Scholar]
- Tang G., Minemoto, Y., Dibling, B., Purcell, N.H., Li, Z., Karin, M., and Lin, A. 2001. Inhibition of JNK activation through NF-κB target genes. Nature 414: 313-317. [DOI] [PubMed] [Google Scholar]
- Tang F., Tang, G., Xiang, J., Dai, Q., Rosner, M.R., and Lin, A. 2002. The absence of NF-κB-mediated inhibition of c-Jun N-terminal kinase activation contributes to tumor necrosis factor α-induced apoptosis. Mol. Cell. Biol. 22: 8571-8579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thannickal V.J. and Fanburg, B.L. 2000. Reactive oxygen species in cell signaling. Am. J. Physiol. Lung Cell Mol. Physiol. 279: L1005-L1028. [DOI] [PubMed] [Google Scholar]
- Tournier C., Hess, P., Yang, D.D., Xu, J., Turner, T.K., Nimnual, A., Bar-Sagi, D., Jones, S.N., Flavell, R.A., and Davis, R.J. 2000. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science 288: 870-874. [DOI] [PubMed] [Google Scholar]
- Tournier C., Dong, C., Turner, T.K., Jones, S.N., Flavell, R.A., and Davis, R.J. 2001. MKK7 is an essential component of the JNK signal transduction pathway activated by proinflammatory cytokines. Genes & Dev. 15: 1419-1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracey K.J. and Cerami, A. 1993. Tumor necrosis factor, other cytokines and disease. Annu. Rev. Cell Biol. 9: 317-343. [DOI] [PubMed] [Google Scholar]
- Van Antwerp D.J., Martin, S.J., Kafri, T., Green, D.R., and Verma, I.M. 1996. Suppression of TNF-α-induced apoptosis by NF-κB. Science 274: 787-789. [DOI] [PubMed] [Google Scholar]
- Varfolomeev E. and Ashkenazi, A. 2004. Tumor necrosis factor: An apoptosis JunKie. Cell 116: 491-497. [DOI] [PubMed] [Google Scholar]
- Varfolomeev E.E., Schuchmann, M., Luria, V., Chiannilkulchai, N., Beckmann, J.S., Mett, I.L., Rebrikov, D., Brodianski, V.M., Kemper, O.C., Kollet, O., et al. 1998. Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity 9: 267-276. [DOI] [PubMed] [Google Scholar]
- Ventura J.J., Kennedy, N.J., Lamb, J.A., Flavell, R.A., and Davis, R.J. 2003. c-Jun NH(2)-terminal kinase is essential for the regulation of AP-1 by tumor necrosis factor. Mol. Cell. Biol. 23: 2871-2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vercammen D., Beyaert, R., Denecker, G., Goossens, V., Van Loo, G., Declercq, W., Grooten, J., Fiers, W., and Vandenabeele, P. 1998. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J. Exp. Med. 187: 1477-1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.Y., Mayo, M.W., and Baldwin Jr, A.S. 1996. TNF- and cancer therapy-induced apoptosis: Potentiation by inhibition of NF-κB. Science 274: 784-787. [DOI] [PubMed] [Google Scholar]
- Whitmarsh A.J. and Davis, R.J. 2001. Analyzing JNK and p38 mitogen-activated protein kinase activity. Methods Enzymol. 332: 319-336. [DOI] [PubMed] [Google Scholar]
- Wisdom R., Johnson, R.S., and Moore, C. 1999. c-Jun regulates cell cycle progression and apoptosis by distinct mechanisms. EMBO J. 18: 188-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh W.C., Shahinian, A., Speiser, D., Kraunus, J., Billia, F., Wakeham, A., de la Pompa, J.L., Ferrick, D., Hum, B., Iscove, N., et al. 1997. Early lethality, functional NF-κB activation, and increased sensitivity to TNF-induced cell death in TRAF2-deficient mice. Immunity 7: 715-725. [DOI] [PubMed] [Google Scholar]
- Yeh W.C., Pompa, J.L., McCurrach, M.E., Shu, H.B., Elia, A.J., Shahinian, A., Ng, M., Wakeham, A., Khoo, W., Mitchell, K., et al. 1998. FADD: Essential for embryo development and signaling from some, but not all, inducers of apoptosis. Science 279: 1954-1958. [DOI] [PubMed] [Google Scholar]