

Abstract
It is believed that most modern mammalian lineages arose from a series of rapid speciation events near the Cretaceous-Tertiary boundary. It is shown that such a phylogeny makes the common ancestral genome sequence an ideal target for reconstruction. Simulations suggest that with methods currently available, we can expect to get 98% of the bases correct in reconstructing megabase-scale euchromatic regions of an eutherian ancestral genome from the genomes of ∼20 optimally chosen modern mammals. Using actual genomic sequences from 19 extant mammals, we reconstruct 1.1 Mb of ancient genome sequence around the CFTR locus. Detailed examination suggests the reconstruction is accurate and that it allows us to identify features in modern species, such as remnants of ancient transposon insertions, that were not identified by direct analysis. Tracing the predicted evolutionary history of the bases in the reconstructed region, estimates are made of the amount of DNA turnover due to insertion, deletion, and substitution in the different placental mammalian lineages since the common eutherian ancestor, showing considerable variation between lineages. In coming years, such reconstructions may help in identifying and understanding the genetic features common to eutherian mammals and may shed light on the evolution of human or primate-specific traits.
Following completion of the human genome sequence, there is now considerable interest in obtaining a more comprehensive understanding of its evolution (International Human Genome Sequencing Consortium [IHGSC] 2001; International Mouse Genome Sequencing Consortium [IMGSC] 2002; Rat Genome Sequencing Project Consortium [RGSPC] 2004). Patterns of evolutionary conservation are used to screen human DNA mutations to predict those that will be deleterious to protein function (Sunyaev et al. 2001; Ng and Henikoff 2002) and to identify noncoding sequences that are under negative selection, and hence, may perform regulatory or structural functions (Hardison 2000; Boffelli et al. 2003; Cooper et al. 2003; Margulies et al. 2003; Bejerano et al. 2004). Long periods of conservation followed by sudden change may provide clues to the evolution of new human traits (Goodman et al. 1971; Challem 1997; Enard et al. 2002). All of these efforts depend, directly or indirectly, on reconstructing the evolutionary history of the bases in the human genome, and hence, on reconstructing the genomes of our distant ancestors.
The hope of learning about long extinct species by recovering and cloning their DNA has engaged the popular as well as the scientific imagination, but the reality of such endeavors falls short of expectations on two grounds. The first is lack of information; there is not enough intact DNA in the modern remains of species that have been extinct for many millions of years to infer ancestral genome sequences (Austin et al. 1997; Marota and Rollo 2002). The second is lack of the necessary biotechnology to synthesize large genomic regions from many small pieces. While there is recent progress in overcoming the second obstacle (Smith et al. 2003), the problem of loss of information appears to be insurmountable for species from, say, the Jurassic or Cretaceous periods that have left behind few modern descendants. However, for ancient species with many different modern descendants, there is still the possibility that large regions of their genomes can be approximately inferred from the genomes of modern species using a model of molecular evolution. On a smaller scale, such ancestral reconstructions have been performed for protein families including rhodopsin (Chang et al. 2002), ultraviolet vision gene SWS1 (Shi and Yokoyama 2003), ribonucleases (Jermann et al. 1995; Zhang and Rosenberg 2002), Tu elongation factors (Gaucher et al. 2003), steroid receptors (Thornton et al. 2003) (for review, see Chang and Donoghue 2000; Thornton 2004), for transposons (Adey et al. 1994; Smit and Riggs 1996; Ivics et al. 1997; Jurka 2000), and for small genomes like HIV (Hillis et al. 1994), in which case the predicted ancestral sequences were compared with the known ones. However, studies of large-scale computational genome reconstruction, an undertaking that might be termed computational “paleogenomics” (Birnbaum et al. 2000), have been limited to higher-level genome properties such as gene order (Blanchette et al. 1999; El-Mabrouk and Sankoff 1999; Pevzner and Tesler 2003; Bourque et al. 2004) or karyotype (Graphodatsky et al. 2002; Yang et al. 2003).
Maximum likelihood algorithms for the reconstruction of ancestral amino acids or DNA bases have been developed and used by several groups (Yang et al. 1995; Koshi and Goldstein 1996; Cunningham et al. 1998; Schultz et al. 1996; Pupko et al. 2000, 2002). The maximal likelihood approach appears to work better than parsimony methods (Zhang and Nei 1997). Bayesian methods that take into account uncertainties in the tree, branch lengths, and model parameters have also been explored (Schultz and Churchill 1999; Huelsenbeck and Bollback 2001), although these involve more computationally expensive Markov Chain Monte Carlo sampling methods. With few exceptions (Hein 1989; Fredslund et al. 2003), algorithms have been limited to pure substitution models, and have not considered reconstruction in the presence of insertions and deletions.
We argue that a good target species for a genomic reconstruction is one that has generated a large number of independent, successful descendant lineages through a rapid series of ancestral speciation events. In this case, the problem can be viewed as attempting to reconstruct an original from many independent noisy copies. In the limit of an instantaneous radiation, the accuracy of the reconstruction approaches 100% exponentially fast as the number of copies increases (see Discussion). From the Cretaceous period, a good choice for reconstruction would be the genome of the eutherian ancestor, as this species is believed to have spawned the relatively rapid radiation of the different lineages of modern placental mammals (see Eizirik et al. 2001 for the radiation model used in this study, and Springer et al. 2003 for alternate hypotheses about the pace of the mammalian radiation). This ancient species also has the added advantage of being a human ancestor, so its reconstruction, however speculative, may shed additional light on our own evolution, perhaps helping to explain features of the human and other modern mammalian genomes. This study uses computational simulations to show that large parts of the euchromatic genome of that early eutherian, including many of its noncoding regions, could be accurately reconstructed if sufficiently many well-chosen extant mammalian genomes were available.
Results
Simulations
To assess the reconstructability of ancestral mammalian genomic sequences, we performed a series of computational simulations of the neutral evolution of a hypothetical ∼50 Kb ancestral genomic region into orthologous regions in 20 modern mammals (Fig. 1). Simulation parameters for substitution, deletion, and insertion were based on the analysis of ∼1.8 Mb of data from nine mammals in the regions orthologous to the human CFTR locus (Margulies et al. 2003; Thomas et al. 2003), as well as on a genome-wide comparison of the human and mouse genomes (Kent et al. 2003), and on a recently derived phylogenetic tree (Eizirik et al. 2001). The simulations included insertion of lineage-specific transposons and increased rates of substitution in CpG dinucleotides. For each pair of orthologous sequences generated, we verified that the average number of substitutions, insertions, and deletions are close to those observed in the neutrally evolving regions of the greater CFTR region. We also verified that the distribution of the sizes of insertions and deletions, as well as the frequency and age distribution of each type of repetitive element are close to those previously reported (IHGSC 2001; IMGSC 2002). Further details of the simulation process and its validation are given in Methods and in Blanchette et al. (2004).
Figure 1.

Estimated reconstructability of ancestral mammalian sequences. Average base-by-base error rate in the reconstruction of each simulated ancestral sequence. The error rate shown is the sum of the percentages of bases that are missing, added, or mismatched as a result of errors in the reconstruction, averaged over 100 simulations of sets of orthologous sequences of length ∼50 kb. Error rates are given first for all regions, and in parentheses for nonrepetitive regions only. The Boreoeutherian ancestor, which is the ancestor that can best be reconstructed, is indicated by the arrow. Branches completely located inside the box are called “early branches” (see text). The species names at the leaves only indicate what organisms we simulated; no actual biological sequences were used here. The tree topology and branch lengths are derived directly from Eizirik et al. (2001).
A crucial first step toward reconstructing ancestral sequences is to build an accurate multiple alignment of the extant sequences, thus establishing orthology relationships among the nucleotides of each sequence. To this end, we used a multiple-sequence alignment tool called TBA (Blanchette et al. 2004) based on the well-established pair-wise alignment program BLASTZ (Schwartz et al. 2003). Given TBA's multiple sequence alignment of the soft-repeat-masked extant sequences and a phylogenetic tree relating these sequences, whose topology is assumed to be known, but whose branch lengths are inferred using the HKY model (Hasegawa et al. 1985) and the PHYML program (Guindon and Gascuel 2003), we predicted which positions of the alignment correspond to ancestral bases and which correspond to nucleotides inserted after the ancestor. Here, we used a greedy algorithm that seeks to explain the observed alignment using a set of insertions and deletions of maximum likelihood (see Methods). The identity of the nucleotide at each ancestral position was then predicted using a context-dependent maximum-likelihood estimation. The only data available to the alignment and reconstruction procedure were the sequences of extant species. No information about the simulation process (neither its parameters nor its realization) was used to inform or set the parameters of the reconstruction process apart from the assumed common knowledge of the phylogenetic tree topology, the parameters of the HKY substitution model, and the known classes of transposons.
We compared the actual ancestral sequence used in our simulations with the predicted ancestral sequence by aligning them and counting the number of missing bases (those present in the actual ancestor, but not in the reconstruction), added bases (present in the reconstruction, but not in the actual ancestor), and mismatch errors (positions in the reconstruction assigned the incorrect nucleotide). The sum of the rates of all three types of errors was calculated separately at each ancestral node in the phylogenetic tree (Fig. 1). The results showed that under this phylogenetic tree with a relatively rapid placental mammalian radiation, the neutral nonrepetitive regions of the Boreoeutherian ancestral genome that have evolved like those in our simulations can be reconstructed with about 99% base-by-base accuracy from the genomes of 20 present-day mammals. Repetitive regions are not reconstructed as accurately, because they are more often involved in misalignments, which can result in incorrect predictions. Nonetheless, even counting errors in repetitive regions, the total accuracy is >98%. If a reconstructed base is chosen at random, chances are it lies at least within a 343-bp error-free sequence, showing that reconstruction errors are often clustered together, leaving large error-free regions. The simulated and reconstructed sequences, as well as statistics validating the simulation process, are available at http://genome.ucsc.edu/ancestors. The simulations suggest that even in the nonrepetitive regions, much of the difficulty of the reconstruction problem lies in the computation of the multiple alignment, as a reconstruction based on the correct multiple alignment derived from the simulation itself (and thus unavailable for actual sequences) had less than half the number of reconstruction errors.
Looking at the reconstructability in other ancestral species in the tree, a strong “local tree topology effect” is seen, whereby ancestral sequences at the center of rapid radiations are much more reconstructable than those with longer incident branches. This effect is so strong that sequences of early eutherians living in times of rapid radiation can be reconstructed more accurately than those of most of the more recent ancestors.
Examining reconstructions made using smaller subsets of this set of 20 species, it was found that, including repetitive regions, an accuracy of about 97% can be achieved using only 10 species chosen to sample most major mammalian lineages (Fig. 2). Sampling only five of the most slowly evolving lineages yields an accuracy of about 94%. Little is gained with our current reconstruction procedures by adding more than 10 species, because the risk of misalignment increases, while the unavoidable loss of information in the early branches persists (dashed box, Fig. 1; also see Discussion). However, further improvements to the multiple alignment methodology might change this.
Figure 2.

Estimated reconstructability of the Boreoeutherian ancestor. Fraction of the simulated Boreoeutherian ancestral sequence reconstructed incorrectly as a function of the number of extant species used for the reconstruction. For each number of species used, results are given counting all bases (left columns) and only nonrepetitive bases (right columns). Species are added in the following order: human, cat, chipmunk, sloth, manatee, rousette bat, mole, pig, beaver, tree shrew, horse, pangolin, mouse, armadillo, aardvark, okapi, dog, mole-rat, rabbit, and lemur.
The accuracy of the reconstruction depends crucially on the length of the early branches. Additional simulations (Supplemental Fig. S1) revealed that if the major placental lineages had diverged instantaneously (early branches of length zero, see Fig. 1), we would be able to reconstruct the simulated Boreoeutherian ancestral sequence, including repetitive regions, with <1% error. In contrast, if the early branch lengths inferred by Eizirik et al. (2001) turned out to underestimate the actual lengths by a factor of two, the error rate would jump to 3%, and to 6% if they were underestimated by a factor of four.
The accuracy of the reconstruction is less dependent on the overall branch length, within reasonable limits. If the neutral substitution and indel rates used in the model are increased by 25%, which is considerably more than the typical 10% regional neutral rate fluctuations observed in different genomic regions in human–mouse genome comparisons (Hardison et al. 2003), the accuracy of reconstruction only decreases to 97.5%. On the other hand, if the rates are uniformly half of the neutral rate, which corresponds roughly to the rates observed for coding regions (Eizirik et al. 2001), the reconstructed bases are >99.8% correct, with most errors due to incorrect alignment in the vicinity of repetitive elements. If the true evolutionary rates vary from site to site between these extremes, we would thus expect the overall average reconstruction accuracy of a region to be >97.5%, with significantly higher local accuracy for the more evolutionarily constrained subregions.
An important assumption in our reconstruction procedure is that the topology of the phylogenetic tree is known in advance. Since the early branches of the eutherian tree are very short, there remains some uncertainty about the precise branching order of the main mammalian phyla. Moreover, in situations of rapid speciation, different regions of the genome may actually have different phylogenetic trees because of incomplete lineage sorting due to different recombination histories (Shedlock et al. 2000). To assess the consequences of using an incorrect tree as input to the reconstruction procedure, we repeated our simulation using the original tree to generate the sequences, but using the incorrect tree (Xenartha, Laurasiatheria, Primates), (Rodents, Afrotheria) for the reconstruction of the ancestor. We found that the pseudo-“Xenartha-Laurasiatheria-Primates” ancestor inferred was an approximation of the true Boreoeutherian ancestor that was still 98.4% accurate. The robustness of the reconstruction with respect to changes in early branching order may be due to the relatively small number of mutational events on these short branches of the tree. However, similar robustness of ancestral reconstruction to minor tree-topology changes has also been observed in simulations of amino acid evolution for more general kinds of trees (Zhang and Nei 1997).
Finally, in addition to estimates of the overall accuracy of the reconstruction, the simulations also suggest how we may estimate the confidence in the reconstruction of the ancestral base at a given site based on properties of the local alignment containing that site. In a situation where the phylogenetic tree and sequence alignment are known to be correct and there are no insertions or deletions, the posterior probabilities of each of the four possible ancestral nucleotides can be explicitly computed using standard substitution models (Yang et al. 1995), which readily provides the probability of reconstruction error. However, in the presence of indels or with an uncertain alignment, the analogous error calculation becomes problematic, even for a fixed tree (Hein et al. 2000; Huelsenbeck 2001; Lunter et al. 2003).
Here, we take a heuristic approach to estimating the confidence of the reconstructed base at a given site. The probability that an individual reconstructed base is a mismatch error or an added base is empirically estimated based on local properties of the alignment at and around that position (see Methods). Testing this approach in our simulations, we find that about 98.5% of the nucleotides of our simulated Boreoeutherian ancestral sequence can be reconstructed with at least 90% confidence that they are not mismatches or added bases, and about 95%, with at least 99%, confidence. An additional 1% of the bases of the ancestral sequence are missing from the reconstructed sequence, but the locations of these omissions cannot be accurately predicted.
Reconstruction of an ancestral region in the CFTR locus
Following our simulations, we applied the reconstruction method to actual high-quality sequence data from a 1.87-Mb region containing the human CFTR locus, using 18 additional orthologous mammalian genomic regions (Table 2, below) generated by the NISC Comparative Sequencing Program (Thomas et al. 2003) (see www.nisc.nih.gov). We reconstructed an approximation to all ancestral sequences of the CFTR locus for which orthologous sequence was available in at least 16 of the 19 species listed in Table 2, below. In human, this corresponds to several discontinuous segments covering a total of 1.274 Mb. Simulations on synthetic data like those described above indicate that for the topology and set of branch lengths for these 19 species, the ancestral sequence that can be most accurately reconstructed based on the sequences available is the Boreoeutherian ancestor, and that neutrally evolving regions of this ancestral genome can be reconstructed with an accuracy of about 96%. Notice that although we are using sequences from 19 mammals, the predicted accuracies obtained are lower than those reported in Figure 2, because not as many major lineages or outgroups are sampled. On a site-specific basis, simulations suggest that >90% of the bases of the predicted ancestor can be assigned confidence values >99%. The reconstructed ancestor and site-specific confidence estimates are available at http://genome.ucsc.edu/ancestors.
Table 2.
Comparison of modern sequences to predicted ancestor
|
Deletions
|
Insertions
|
Substitutions
|
|||
|---|---|---|---|---|---|
| Species | Size of region (kb) (a) | Nonrepetitive %GC-content (b) | % of ancestor lost (c) | % of extant species' bases acquired (nonrepetitive only) (d) | % of extant species' bases changed (expected # substitutions per site) (e) |
| Reconstructed Boreoeutherian ancestor | 1124 | 37.0 | N/A | N/A | N/A |
| Human | 1274 | 37.1 | 11.3 | 21.7 (2.0) | 8.6 (11.1) |
| Chimpanzee | 1278 | 37.1 | 11.5 | 21.8 (1.8) | 8.7 (11.1) |
| Gorilla | 1247 | 37.1 | 12.9 | 21.6 (1.9) | 8.7 (11.1) |
| Baboon | 1260 | 37.3 | 12.6 | 21.2 (2.1) | 9.1 (10.7) |
| Orangutan | 1268 | 37.1 | 11.7 | 21.2 (1.8) | 8.6 (11.2) |
| Vervet | 1229 | 37.2 | 13.5 | 20.7 (2.0) | 9.1 (11.8) |
| Macaque | 1255 | 36.4 | 12.2 | 21.0 (2.0) | 9.1 (11.7) |
| Lemur | 1071 | 37.7 | 19.1 | 11.6 (2.8) | 9.0 (10.9) |
| Mouse-lemur | 1085 | 37.5 | 18.0 | 14.5 (3.8) | 9.3 (11.6) |
| Mouse | 1110 | 39.2 | 39.1 | 38.3 (12.0) | 17.5 (34.3) |
| Rat | 1239 | 39.5 | 38.8 | 44.4 (10.1) | 15.9 (35.1) |
| Rabbit | 1348 | 42.7 | 29.4 | 37.9 (28.9) | 10.5 (21.3) |
| Cat | 1206 | 37.2 | 24.5 | 29.6 (6.9) | 11.3 (16.5) |
| Dog | 1122 | 39.4 | 26.4 | 22.5 (6.4) | 13.5 (19.2) |
| Cow | 1324 | 37.1 | 30.9 | 41.5 (7.7) | 11.1 (20.9) |
| Pig | 1158 | 36.8 | 33.7 | 29.6 (7.5) | 10.9 (19.7) |
| Horse | 1102 | 38.5 | 20.2 | 17.5 (8.0) | 12.1 (13.3) |
| Hedgehog | 1379 | 39.7 | 50.0 | 48.9 (38.6) | 8.9 (28.5) |
| Armadillo | 1339 | 39.4 | 28.9 | 34.2 (18.1) | 9.9 (20.2) |
Listed are some properties of sequences of the extant species in the greater-CFTR locus and the predicted changes they incurred during evolution from the Boreoeutherian ancestral sequence. (a) Length of sequence. (b) Fraction of nonrepetitive bases that are G or C. (c) Deletions: percentage of the ancestral sequence lost in each species. (d) Insertions: percentage of extant species' sequence that was inserted since the reconstructed ancestor (in parentheses, percentage of extant species' sequence that resulted from insertions of nonrepetitive sequences, using RepeatMasker to identify repetitive sequences.) The high fraction of nonrepetitive inserted bases in rabbit and hedgehog is most likely due to lack of complete RepeatMasker libraries for the transposons specific to these species. (e) Substitutions: percentage of extant species' bases that were derived from an ancestral base but differ from that base (this is different from the standard percentage identity measure, where only aligned bases are considered). In parentheses, the expected number of substitutions per site under a Kimura 2-parameter model (Kimura 1980) is given, here using only the aligned bases.
We confirmed that the 96% accuracy estimate is reasonable by analysis of transposable elements whose insertion predated the Boreoeutherian ancestor (“ancestral repeats”) (Fig. 3A). For each family of ancestral repeats, a consensus sequence is available, obtained from the many copies of these elements scattered in the genome. The consensus sequence is thought to represent the transposon sequence at the time of its insertion into this and other regions of the ancestral genome (Jurka 2000). We aligned the extant sequence H of each transposon relic identified in the human CFTR region by RepeatMasker (Smit and Green 1999) to the consensus sequence C for its ancestral repeat family, and estimated the expected number of substitutions per site between consensus and human relic, d(C,H), using a Kimura 2-parameter model (Kimura 1980). Let A be the true (unknown) ancestral Boreoeutherian sequence for this transposon relic and let A* be the reconstructed sequence. Since A stands on the evolutionary path between C and H, we would expect to have d(C, H) ≈ d(C, A) + d(A, H), where d(C, A) and d(A, H) are the expected substitutions per site between C and A, and between A and H, respectively. Reconstruction errors in A* would be expected to take this sequence away from the true evolutionary path, resulting in d(C, H) < d(C, A*) + d(A*, H). Figure 3A shows the average distances observed for ancestral repeats of the CFTR region. It indicates that d(C,A*) + d(A*,H) exceeds d(C,H) by 0.04 substitutions per site, which can be verified to correspond to a mismatch error rate in the reconstructed sequence A* of about 2.6%. This roughly confirms our estimate of 96% overall accuracy, since mismatch errors are expected to account for about half of the base-by-base errors made by our method in this case and errors are concentrated in repetitive regions.
Figure 3.

(A) Estimates of the expected number of substitutions per site between a repeat consensus C, it human descendent H, and the reconstructed ancestor A*, based on a Kimura 2-parameter model and averaged over all human ancestral repeats of the region considered. The true ancestor A cannot be observed, but a distance of 0.026 substitutions per site between it and A* is estimated from the three other distances. (B) Star phylogeny with n independent descendants. (C) A tree with bifurcating root. Irrevocable information loss occurs between R and its descendants A and B.
Figure 4 illustrates the reconstruction in a noncoding region of the CFTR locus that exhibits a typical level of sequence conservation. This region is located in a 32-Kb intron of the CAV1 gene, about 13 Kb from the 5′ exon. The bases in this region are relics left over from the insertion of a MER20 transposon sometime prior to the mammalian radiation, and are thus unlikely to be under selective pressure.
Figure 4.

Example of reconstruction of an ancestral Boreoeutherian sequence based on actual orthologous sequences derived from a MER20 retrotransposon. Arrows indicate positions where the reconstructed ancestor differs from the MER20 consensus. Longer arrows indicate the positions where the knowledge of the MER20 consensus sequence would have changed the ancestral base prediction. The position of the human sequence displayed is chr7:115,739,755-115,739,899 (NCBI build 34). The alignment of the flanking nonrepetitive DNA (data not shown) verifies that the sequences from the different species are, in fact,orthologous. The tree and branches are derived directly from Eizirik et al.(2001).
Notice that despite the fact that the alignment of certain species (in particular, mouse, rat, and hedgehog) appears somewhat unreliable, the inference of the presence or absence of a Boreoeutherian ancestral base at a given position is quite straightforward given the alignment, and to a lesser extent, so is the prediction of the actual ancestral base itself. The MER20 consensus is shown for comparison. Most positions where the reconstructed Boreoeutherian ancestral base disagrees with the MER20 consensus are likely due to substitutions in this MER20 relic that predated the Boreoeutherian ancestor, since the support of the reconstructed base is very strong in the extant species. If the MER20 consensus sequence is used as an outgroup in the reconstruction procedure, only two bases (indicated by a longer arrow) are reconstructed differently, indicating that the reconstructed ancestral sequence is very stable and most of it is likely to be correct.
Because the reconstructed Boreoeutherian ancestral sequence is evolutionarily closer to the older mammalian ancestral genomes that existed at the time of the insertions of ancestral transposons, it is superior to the human genome sequence for the recognition of these elements. In essence, it acts as an observatory that allows us to see even farther back in time. When RepeatMasker is run on the inferred Boreoeutherian ancestor, ancient repeat families such as L2 LINES and MIRs are detected in significantly larger fraction than when RepeatMasker is run on the human sequence, because they are much less decayed [Table 1, column (b)]. This improved ability to detect very old repeats results in an increase of 2.7% in the estimated total fraction of the human CFTR region that derives from a transposon insertion (from 37.7% to 40.4%).
Table 1.
Detected repetitive content of the reconstructed Boreoeutherian ancestor and of human
|
PreBoreoeutherian ancestral repeats
|
||
|---|---|---|
| Detectable in human and ancestor (kb)a | Detectable in ancestor only (kb)b | |
| Alu | 0 | 0 |
| LINE L1 | 83.5 | 9.1 |
| LINE L2 | 61.5 | 15.3 |
| LINE L3 | 2.4 | 0.7 |
| DNA | 23.7 | 2.3 |
| MIR | 40.3 | 5.6 |
| LTR | 38.3 | 1.8 |
| Others | 5.0 | 0.4 |
| Total | 254.7 | 35.2 |
Number of human kilobases labeled by RepeatMasker as belonging to the given family and present in the Boreoeutherian ancestor
Number of human kilobases that are not detected as repetitive in human, but that are detected as such in the corresponding ancestral region. All numbers were calculated using the sensitive mode of RepeatMasker
More importantly, reconstructed ancestral genome sequences allow us to make inferences about the specific evolutionary path of functional elements such as protein-coding regions (Jermann et al. 1995; Sunyaev et al. 2001; Chang et al. 2002; Ng and Henikoff 2002; Zhang and Rosenberg 2002; Gaucher et al. 2003; Shi and Yokoyama 2003; Thornton et al. 2003; Thornton 2004). About 5995 of the 6026 codons from the known human genes in the region used for this reconstruction are also clearly coding in the other extant species. All of these 5995 codons were reconstructed without introducing an in-frame stop codon or frame shift, despite the fact that the reconstruction algorithm used neither prior knowledge about exon positions nor model of codon evolution. This indirectly suggests that the accuracy of the reconstruction is quite high for elements of the genome that have been under purifying selection.
The accuracy of the inferred ancestral CFTR protein sequence was verified by comparing it to outgroups like chicken and the marsupial Didelphis virginiana (opposum). Of the 1481 amino acids of the ancestral CFTR protein, 1276 are most likely correct by virtue of a quasi-unanimity within eutherian mammals. Of the remaining 205 amino acids where the reconstruction is not completely obvious, 137 amino acids are strongly confirmed by a match in either chicken or opposum, and 29 others could only be weakly confirmed by a match in either frog or Fugu. On the other hand, 15 amino acids could be incorrectly reconstructed as indicated by the failure of the two tests above and by a match between one of the eutherian amino acids and either Didelphis or chicken. Overall, this gives an estimated accuracy of ∼99% at the amino acid level for the reconstruction of the ancestral CFTR protein. This corresponds to an ∼99.5% accuracy at the base level, because roughly 2/3 of random base changes are nonsynonymous, and there is a 3:1 ratio of bases to amino acids. This is not as good as the 99.8% accuracy expected, based on simulations of regions evolving at half the neutral rate on the more optimally chosen set of 20 species, but is consistent with what we would expect from the suboptimal set of 19 species used in this reconstruction. Interestingly, at two of the positions where the reconstructed ancestral CFTR protein differs from human CFTR, the reconstructed ancestral amino acid is associated with cystic fibrosis when it occurs as a human mutation (http://www.genet.sickkids.on.ca/cftr/): Phe → Leu at amino acid position 87 (Bienvenu et al. 1994) and Met → Ile at position 1028 (Onay et al 1998). These disease-causing human variants are the wild-type amino acids in several other species, as has been observed for other human disease proteins as well (Schaner et al. 2001). That the disease-causing amino acid variant was wild type in our eutherian ancestor is very likely in the former case, but the reconstruction is less clear in the latter case, because so many different substitutions occured in different lineages.
Sensible reconstruction of hypothesized structural RNAs was also obtained. Two regions of the CFTR locus in introns of the ST7 gene that appear to form stable RNA secondary structures (Margulies et al. 2003) are predicted to fold in a nearly identical fashion in the reconstructed ancestor.
The reconstructed ancestral sequence can also be used to gather statistics on the rates of gain and loss of DNA in different eutherian lineages, and the shifts in substitution spectra. After reconstruction of the Boreoeutherian ancestral sequence from the 19 present-day genomic sequences, we compared it with those sequences to derive these statistics (Table 2). The reconstructed ancestral sequence had a size (1124 Kb) about 10% smaller than those of extant old-world monkeys (1260 Kb on average, with most of the difference due to Alu insertions) and also smaller than those of most other species, with the exception of the two lemurs. The number of inserted and deleted bases in primates is low compared with many other mammals (Thomas et al. 2003), while those of rodents (but not rabbit) are high. Substitution rates follow a similar pattern (Cooper et al. 2003). Overall, the ancestral sequence is most closely related to that of primates, and perhaps, surprisingly, to that of horse.
It is predicted that the human sequence differs from that of the Boreoeutherian ancestor in 30.3% of its bases, 21.7% resulting from insertions, and thus not present in the ancestor, and 8.6% resulting from substitutions. In addition, the human sequence has lost about 11.3% of the ancestral bases. Most differences between the human and ancestral sequences derive from primate lineage insertions of transposons, in agreement with other recent studies (IMGSC 2002). In contrast, rodents differ in about 55%–60% of their bases and have lost about 39% of the ancestral bases, while hedgehog differs from the ancestor in 58% of its bases and has lost 50%. Though this high mutation rate makes these species very useful for detecting functional regions through comparative genomics (Margulies et al. 2003), it makes them of less use for reconstructing ancestral sequences. Because of the difficulty of aligning such rapidly evolving sequences, the accuracy of these estimates for rodents and hedgehog remains uncertain.
The set of 19 species we used is not a uniform sampling of the eutherian phylogenetic tree, but rather is biased toward close human relatives, containing seven old-world monkeys. To ensure that the number of closely related species does not unduly affect the reconstructed ancestor by biasing it toward the human sequence, we repeated the reconstruction procedure, removing all primates but human and lemur. The new reconstructed ancestor was not significantly farther from the human sequence, with 0.113 expected substitutions per site (compared with 0.111 previously), 10.8% deletions (compared with 11.3% previously), and 23.4% insertions (compared with 21.7% previously).
The availability of predicted ancestral sequences at every internal node of the tree offers a unique perspective on the deletion and insertion processes at work along each branch of the tree. Focusing on a 280-kb region where sequences from all 19 mammals were available, the number of microdeletions and microinsertions (of length at most 10 bp) along each branch of the tree was estimated (Fig. 5). We did not attempt to estimate the indel rates along the four deepest branches of the tree because (1) for the two deepest branches of the tree, deletions cannot be distinguished from insertions, and (2) for the two branches incident upon the Boreoeutherian ancestor, deletions and insertions are crucially determined by the presence or absence of aligned bases in armadillo, which is often unreliably aligned. Among the branches where indels can be accurately counted, the rate of deletions is consistently two to three times higher than the rate of insertions, with the lowest deletion/insertion ratios found in the dog and the prosimian lineages, and the highest ones found in the pre-mouse–rat-split rodents, horse, and cow lineages. Deletion and insertion rates are definitely not following a molecular clock, with rates in primates ∼2.5 times lower than those in rodents and 1.3–1.5 times lower than those in artiodactyls and carnivores. The results for human versus rodents are in relatively close agreement with those obtained from a study of the whole human, mouse, and rat genomes (RGSPC 2004). Deletion and insertion rates are closely correlated with substitution rates, with the expected number of substitutions per site between 15 and 20 times higher than the deletion rate (Supplemental Fig. S3), with outliers hedgehog (28 times higher) and pre-mouse–rat-split rodents (26 times higher).
Figure 5.

Frequency of microdeletions (1–10 bp) (left) and microinsertions (right) during eutherian evolution. Indel rates for the branches shown with dashed lines cannot be accurately estimated. Estimates are based on a set of regions totaling about 280 kb, for which sequence data is available for all 19 mammals.
Discussion
One of the nonintuitive results of this study is the observation that more ancient ancestral genomes can often be reconstructed more accurately than those of their more recent descendants. Why exactly is this so? For simplicity, consider the case of reconstructing a single binary ancestral character state in the root species (e.g., purine vs. pyrimidine at a given site) under a simple model in which the prior probability distribution on the ancestral character is uniform, substitution rates are known, symmetric, homogeneous, and not too high, and the total branch length in the phylogenetic tree from the root ancestor to each of the modern species is the same (i.e., assume a molecular clock). Here, each of n modern species has a state that differs from the ancestral one with the same probability p <1/2. If the tree exhibits a star topology (Fig. 3B), in which each of the modern species derives directly from the ancestor on an independent branch, then it is clear that the maximum likelihood and Bayesian maximum a posteriori reconstructions of the ancestral character agree, and the reconstructed state is the one that is most often observed in the n modern species. The probability of an error in reconstruction is:
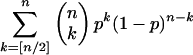 |
which is at most [4p(1 - p)]n/2 (Hoeffding 1963; Le Cam [Lemma 5 p.479] 1986). This error approaches zero exponentially fast as n increases. When n is too small, the ancestor is probably not reconstructable (Mossel 2003).
In contrast, a non-star topology (Fig. 3C) such as a binary tree that has the same total root-to-leaf branch length and the same number n of modern species at the leaves has two nonzero length branches from the root ancestor R leading to intermediate ancestors A and B, and information is irrevocably lost along these two branches. No matter how large the number n of modern descendant species derived from A and B, one can do no better at reconstructing the state at R than if one knew for certain the state in its immediate descendants A and B. Even with this knowledge, the accuracy of reconstruction of R from A and B will be strictly <100% for all reasonable models and nonzero branch lengths. The reconstruction gets poorer the longer the branch lengths are to A and B. This extends to the case where the ancestor R being reconstructed has a bounded number of independent immediate descendants and to the case where descendants of an earlier ancestor of R (outgroups) are also available. The long branches connecting them to the rest of the tree are why some more recent ancestral sequences in the tree of Figure 1 are less reconstructable than the Boreoeutherian ancestor, which acts almost like the root of a star topology.
The above analysis shows that the star tree is always the best topology for reconstruction in the limit as the number n of observed species becomes large, while the time to the common ancestor remains fixed. A stronger claim is that for every n and every time to the common ancestor, the star tree with n leaves is always more favorable for ancestral reconstruction than any branching tree that has internal “shared” nodes (but the same time to the common ancestor), because the star topology maximizes the mutual information between the residues at the leaves and at the root (Schultz et al. 1996; Schultz and Churchill 1999). This has been rigorously proven for a symmetric substitution model in the case of binary characters (Evans et al. 2000, Theorem 6.1). However, there are counterexamples with many-valued characters, e.g., amino acids, where for sufficiently long branches, the star topology does not provide the best ancestral reconstruction, i.e., the highest mutual information (B. Lucena and D. Haussler, in prep.). Thus, the precise relationship between tree topology and reconstructability of the ancestral state appears to be rather subtle in the general case.
While suggestive that reconstruction of a reasonable approximation to an eutherian ancestral euchromatic genome may be within our reach, our simulation results have a number of important limitations as follows: (1) The rates of substitutions, deletions, and small insertions are assumed to be constant across sequence position and homogeneous across branches, with branch lengths proportional to those in a particular tree (Eizirik et al. 2001), scaled to fit rates estimated from a particular region (the CFTR region) (Thomas et al. 2003). If the substitution rates were grossly underestimated, or there were very strong clustering of mutations or “hotspots,” i.e., regions whose mutation rate was, say, double the average nonfunctional parts of the CFTR locus, there would be more genome positions where key information was irrevocably lost in the early branches, and the accuracy of the reconstruction would be reduced. (2) Different modes of selection are not modeled, including specific types of purifying selection in codons and other functional regions, and positive selection for new functions. The former is likely to help reconstruction, but the latter may inhibit the ability to accurately reconstruct the ancestor in certain critical sites. (3) Some nucleotide-level mutational processes like DNA polymerase slippage effects (Nishizawa and Nishizawa 2002) or gene conversion are not included in the simulation. These may change patterns of molecular evolution in some areas and reduce our ability to infer ancestral states. Nonallelic gene conversion in particular could, in principle, make it difficult to apply the reconstruction method we use to find ancestral versions of repetitive regions in some cases. However, we saw no evidence that this is a serious problem in our analysis of the alignment of ancestral repeats, such as the MER20 shown in Figure 4. (4) Large-scale mutational processes like tandem and segmental duplication, inversion, and translocation are not included in the simulations. The alignment of the one multimegabase mammalian genome region where we have data from many species, the CFTR region, shows a dearth of such changes. However, it is estimated that perhaps 10% of the euchromatic human genome has been subject to recent duplications (Samonte and Eichler 2002) and/or an excess of rearrangements (Kent et al. 2003; Pevzner and Tesler 2003), suggesting that at least a similar proportion of the ancestral euchromatic genome would be difficult to reconstruct without additional data and better techniques.
Despite these shortcomings, our validation of the reconstruction by both simulation and ancestral repeat and codon analysis on actual data suggests that for regions like CFTR, which are likely to be typical, the above issues are not severe enough to prevent a reasonably accurate reconstruction.
More significant technical challenges remain if we wish to conduct in vivo functional tests of reconstructed ancestral genomic regions, either in cell lines or in mouse models. Multikilobase sequences of transgenic DNA can be inserted into mouse embryonic stem cells via homologous recombination (“knockin”) methods (Prosser and Rastan 2003; Robertson et al. 2003) and BAC transgenics (Yang and Seed 2003). “Humanized” mice, which have specific individual genes replaced by their human versions, have been produced by these methods. Multimegabase transgenic sequences have been introduced in mammalian artificial chromosomes, e.g., for the human CFTR locus (Auriche et al. 2002). However, these methods of introducing foreign DNA are expensive even when using available genomic sequences, and new methods for synthesizing large segments of DNA de novo would be needed to apply them to ancestral genomic reconstruction, e.g., to produce what might be called “retrovolved” mice that harbor the ancestral versions of specific gene loci. Furthermore, multiple loci would have to be changed to explore co-evolving sets of genes. However, if these obstacles can be overcome, it would be quite interesting to attempt in vivo tests of reconstructed ancestral genomic regions in a mouse model, especially in cases where phenotypic differences between mice and the placental ancestor are hypothesized.
Extant eutherian species are variations on a common “mammalian theme.” Accurate reconstruction of large genomic regions of an eutherian ancestor may help us identify and understand the common functional elements of that theme, as well as the lineage-specific evolutionary innovations that led to the modern variations on it. Because distances are reduced and direction of change can be resolved, much can be learned by comparing mammalian genomes to their common ancestor rather than pair-wise among themselves. Because the number of substitutions per site leading from the placental ancestral genome to the human genome is only one third of that from the ancestor to mouse (Cooper et al. 2003; Thomas et al. 2003; RGSPC 2004), the ancestral genome is much closer to our own genome than is the mouse model. While the present work is only a small feasibility study, in the long run, we expect that an accurate ancestral reconstruction of the euchromatic genomic regions of our placental ancestor will prove extremely valuable for studying the evolutionary processes and specific evolutionary events that shaped our own genome, as well as the genomes of other modern mammalian species.
Methods
Simulation procedure
We built a simulation procedure, based on the Rose program (Stoye et al. 1997), that mimics the evolution of mammalian sequences under no selective pressure. The simulations are based on the phylogenetic tree inferred by Eizirik et al. (2001) on a set of genes for a large set of mammals. The branch lengths are uniformly scaled by a factor of K = 2.1, chosen to fit as closely as possible the substitution rates observed in neutral DNA of a 1.87-Mb region of human chromosome 7 with orthologous sequences in eight other mammals (Siepel and Haussler 2003; Thomas et al. 2003). Given this phylogenetic tree, we simulate sequence evolution by performing random substitutions, deletions, and insertions along each branch, in proportion to its length. Substitutions follow a context-independent HKY model (Hasegawa et al. 1985) with Ts/Tv = 2, p(a) = p(t) = 0.3, and p(c) = p(g) = 0.2, except that substitution rates of CpG pairs are 10 times higher than other rates (Siepel and Haussler 2003). Deletions are initiated at a rate about 0.056 times the substitution rate, their length is chosen according to a previously reported empirical distribution (Kent et al. 2003) that ranges between one and 5000 nucleotides, and their starting point is uniformly distributed. Insertions occur randomly according to a mixture model. Small insertions (of size between 1 and 20 nt) occur at half the rate of deletions, their size distribution is empirically determined (Kent et al. 2003) and their content is a random sequence where each nucleotide is chosen independently from the background distribution. We also simulate the insertion of retrotransposons. For this, we used a library of 15 different types of transposable elements chosen to cover the large majority of repetitive elements observed in well-studied mammals (Jurka 2000). Each insertion was accomplished by randomly selecting a part of the consensus sequence for a given type of element and inserting it at a random location in the sequence, i.e., we did not model preferences for particular insertion locations (IHGSC 2001). The rate of insertion of each repeat varies from branch to branch, so that certain retrotransposons (like ALUs, SINEs B2, BOV) are lineage specific, while others (L1, LTR, DNA) are both present in the sequence at the root of the tree (with a range of decaying level) and can be inserted along any branch. Care was taken to ensure that the rate of repeat insertion yields a set of sequences whose repetitive content and repeat age distribution resembles closely those previously reported for human (IHGSC 2001) and mouse (IMGSC 2002), and resembles those observed in the greater CFTR region for other mammals. In cases where no lineage-specific repeat information was available, we used repetitive element consensi of species not used in this study (monotremes and marsupials) and used an insertion rate equal to the insertion rate of ALUs in the primate lineage.
We use the above methods to simulate evolution from an ancestral mammalian sequence forward to modern versions of that sequence, simulating speciation events at the branch points of the tree, and substitutions, insertions, and deletions along each branch. To initiate such a simulation, we first need to generate a hypothetical ancestral mammalian sequence to go at the root of the tree. This is the sequence that we will later try to reconstruct from the sequences at the leaves of the tree. This hypothetical ancestral mammalian sequence is generated by another simulation, i.e., starting with a repeat-free 40% GC-rich random sequence, we simulate its evolution for a time and at a rate similar to those between human and mouse, using the same set of mutational operations as previously described, but inserting transposons that are believed to predate the mammalian radiation. This simulated ancestral sequence thus has a repeat content and age distribution that should approximate that of the actual ancestral mammalian genome.
Alignment and reconstruction
After generating a set of simulated sequences, the sequences are first soft-repeat-masked using RepeatMasker (Smit and Green 1999) and then aligned using the Threaded Block Aligner (TBA) multiple-alignment program (Schwartz et al. 2003; Blanchette et al. 2004). The TBA alignment of the CFTR region can be interactively explored on the human genome browser (Kent et al. 2002) at http://genome.ucsc.edu (under the ENCODE tracks), and is updated as new species become available. An archival version of the alignment used in this study is available at http://genome.ucsc.edu/ancestors. The ancestral sequence is predicted based on this multiple alignment. To determine which positions in the multiple alignment correspond to bases that were in the common ancestor and which represent lineage-specific insertions we start by using RepeatMasker (Smit and Green 1999) to soft-mask repetitive regions. Lineage-specific like ALUs are excised as their insertion came after the Boreoeutherian ancestor. The repeat-masked multiple alignment is then fed to a greedy algorithm that attempts to explain the remaining indels with the plausible scenario. For the most part, the algorithm assumes that the alignment is phylogenetically correct, i.e., that two bases are aligned if and only if they derive from a common ancestral base. See below for a departure from this assumption. Originally, all of the gaps in the alignment are marked as unexplained. The algorithm iteratively selects the insertion or deletion, performed along a specific edge of the tree and spanning one or more columns of the alignment, that yields the largest number of alignment gaps explained per unit of cost. The number of gaps explained by a deletion is the number of unexplained gaps in the subtree above which the deletion occurs. The number of gaps explained by an insertion is the number of unexplained gaps in the complement of the subtree above which the insertion occurs. The costs are defined heuristically. The cost of a deletion is given by 1 + 0.01 log(L) -0.01 b, where L is the length of the deletion and b is the length of the branch along which the event takes place. The cost of an insertion is given by 1 + 0.01 log(L) -0.01 b -r, where L and b are defined as above and r is a term that takes value 0.5 if the repetitive content of the segment inserted is >90%. Once the best insertion or deletion has been identified, its gaps are marked as “explained.” This does not preclude them from being part of other indels, but they will not count in their evaluation. Finally, heuristics are used to reduce errors due to incorrect alignment, in particular to reduce the problems caused by two repetitive regions from two distantly related species mistakenly aligned to each other, with other species having gaps in that region. More precisely, a subtree containing at least six leaves, <20% of which have bases at a given position, will never be predicted to have an ancestral base at that position. See Fredslund et al. (2003) for an alternative approach to the same problem. After having established which positions of the multiple alignment correspond to bases in the ancestor, we predict which nucleotide was present at each position in the ancestor using the standard posterior probability approach (Yang et al. 1995) based on a simple dinucleotide substitution model where substitutions at the two positions are independent except for CpG, whose substitution rate to TpG is 10 times higher than those of other transitions (Siepel and Haussler 2003). The branch lengths are inferred from the data using PHYML (Guindon and Gascuel 2003). The equilibrium frequencies are estimated from the data. The only parameter given to the reconstruction algorithm is the transition/transversion ratio of the HKY model, which is set at 2.
In experiments using actual sequence data from present day mammals, the simulation steps are omitted, and the same alignment and reconstruction procedure is followed.
Base-by-base confidence estimates
The probability that a given ancestral base is incorrect due to a mismatch or added base can be approximated empirically based on two indicators of reconstruction errors. The first indicator is the theoretical substitution-based reconstruction error probability pe calculated as the sum of the posterior probabilities of the three least likely ancestral nucleotides at that position (Yang et al. 1995). The second indicator nid is the number of insertion and deletion events that span the site, as estimated by our reconstruction method. Each reconstruction error observed during the simulation was recorded, together with pe (rounded to the closest percentage point) and nid for the corresponding site. As seen in Supplemental Figure S3, (a), pe turns out to be an excellent predictor of mismatch errors, but a poor predictor of added bases. On the other hand, Figure S3, (b) shows that nid is good at predicting added bases, but quite inefficient at predicting mismatch errors. The probability of error of each type can in fact be estimated jointly for each pair (pe, ni)), which provides a reasonable confidence estimate for both types of errors at any reconstructed base, making it possible to identify high-confidence or low-confidence bases in the reconstructed sequence.
Acknowledgments
We thank Jim Kent, Arian Smit, Adam Siepel, Gill Bejerano, Elliot Margulies, Brian Lucena, Leonid Chindelevitch, and Ron Davis for helpful discussions and suggestions. W.M. was supported by grant HG-02238 from the National Human Genome Research Institute, E.G. was supported by NHGRI, D.H. and M.B. were supported by NHGRI Grant 1P41HG02371 and the Howard Hughes Medical Institute. Finally, we thank the NISC Comparative Sequencing Program for providing multispecies comparative sequence data.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.2800104.
Footnotes
[Supplemental material is available online at www.genome.org and http://genome.ucsc.edu/ancestors.]
References
- Adey, N.B., Tollefsbol, T.O., Sparks, A.B., Edgell, M.H., and Hutchison III, C.A. 1994. Molecular resurrection of an extinct ancestral promoter for mouse L1. Proc. Natl. Acad. Sci. 91: 1569-1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auriche, C., Carpani, D., Conese, M., Caci, E., Zegarra-Moran, O., Donini, P., and Ascenzioni, F. 2002. Functional human CFTR produced by a stable minichromosome. EMBO Rep. 3: 862-868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin, J.J., Ross, A.J., Smith, A.B., Fortey, R.A., and Thomas, R.H. 1997. Problems of reproducibility—Does geologically ancient DNA survive in amber-preserved insects. Proc. R. Soc. Lond. B Biol. Sci. 264: 467-474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejerano, G., Pheasant, M., Makunin, I., Stephen, S., Kent, W.J., Mattick, J.S., and Haussler, D. 2004. Ultraconserved elements in the human genome. Science 304: 1321-1325. [DOI] [PubMed] [Google Scholar]
- Bienvenu, T., Petitpretz, P., Beldjord, C., and Kaplan, J.C. 1994. A missense mutation (F87L) in exon 3 of the cystic fibrosis transmembrane conductance regulator gene. Hum. Mutat. 3: 395-396. [DOI] [PubMed] [Google Scholar]
- Birnbaum, D., Coulier, F., Pebusque, M.J., and Pontarotti, P. 2000. Paleogenomics: Looking in the past to the future. J. Exp. Zool. 288: 21-22. [DOI] [PubMed] [Google Scholar]
- Blanchette, M., Kunisawa, T., and Sankoff, D. 1999. Gene order breakpoint evidence in animal mitochondrial phylogeny. J. Mol. Evol. 49: 193-203. [DOI] [PubMed] [Google Scholar]
- Blanchette, M., Kent, W.J., Riemer, C., Elnitski, L., Smit, A.F., Roskin, K.M., Baertsch, R., Rosenbloom, K., Clawson, H., Green, E.D., et al. 2004. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res. 14: 708-715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boffelli, D., McAuliffe, J., Ovcharenko, D., Lewis, K.D., Ovcharenko, I., Pachter, L., and Rubin, E.M. 2003. Phylogenetic shadowing of primate sequences to find functional regions of the human genome. Science 299: 1391-1394. [DOI] [PubMed] [Google Scholar]
- Bourque, G., Pevzner, P.A., and Tesler, G. 2004. Reconstructing the genomic architecture of ancestral mammals: Lessons from human, mouse, and rat genomes. Genome Res. 14: 507-516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Challem, J.J. 1997. Did the loss of endogenous ascorbate propel the evolution of Anthropoidea and Homo sapiens? Med. Hypotheses 48: 387-392. [DOI] [PubMed] [Google Scholar]
- Chang, B.S. and Donoghue, M.J. 2000. Recreating ancestral proteins. Trends Ecol. Evol. 15: 109-114. [DOI] [PubMed] [Google Scholar]
- Chang, B.S., Jonsson, K., Kazmi, M.A., Donoghue, M.J., and Sakmar, T.P. 2002. Recreating a functional ancestral archosaur visual pigment. Mol. Biol. Evol. 19: 1483-1489. [DOI] [PubMed] [Google Scholar]
- Cooper, G.M., Brudno, M., Green, E.D., Batzoglou, S., and Sidow, A. 2003. Quantitative estimates of sequence divergence for comparative analyses of mammalian genomes. Genome Res. 13: 813-820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham, C.W., Omland, K.E., and Oakley, T.H. 1998. Reconstructing ancestral states, a critical reappraisal. Trends Ecol. Evol. 13: 361-368. [DOI] [PubMed] [Google Scholar]
- Eizirik, E., Murphy, W.J., and O'Brien, S.J. 2001. Molecular dating and biogeography of the early placental mammal radiation. J. Hered. 92: 212-219. [DOI] [PubMed] [Google Scholar]
- El-Mabrouk, N. and Sankoff, D. 1999. On the reconstruction of ancient doubled circular genomes using minimum reversals. Genome Inform. Ser. Workshop, Genome Inform. 10: 83-93. [PubMed] [Google Scholar]
- Enard, W., Przeworski, M., Fisher, S.E., Lai, C.S., Wiebe, V., Kitano, T., Monaco, A.P., and Paabo, S. 2002. Molecular evolution of FOXP2, a gene involved in speech and language. Nature 418: 869-872. [DOI] [PubMed] [Google Scholar]
- Evans, W., Kenyon, C., Peres, Y., and Schulman, L. 2000. Broadcasting on trees and the Ising model. Ann. Appl. Probab. 10: 410-433. [Google Scholar]
- Fredslund, J., Hein, J., and Scharling, T. 2003. A large version of the small parsimony problem. Lecture Notes in Bioinformatics, Proc. WABI'03. 2812: 417-432. [Google Scholar]
- Gaucher, E.A., Thomson, J.M., Burgan, M.F., and Benner, S.A. 2003. Inferring the palaeoenvironment of ancient bacteria on the basis of resurrected proteins. Nature 425: 285-288. [DOI] [PubMed] [Google Scholar]
- Goodman, M., Barnabas, J., Matsuda, G., and Moore, G.W. 1971. Molecular evolution in the descent of man. Nature 233: 604-613. [DOI] [PubMed] [Google Scholar]
- Graphodatsky, A.S., Yang, F., Perelman, P.L., O'Brien, P.C., Serdukova, N.A., Milne, B.S., Biltueva, L.S., Fu, B., Vorobieva, N.V., Kawada, S.I., et al. 2002. Comparative molecular cytogenetic studies in the order Carnivora: Mapping chromosomal rearrangements onto the phylogenetic tree. Cytogenet Genome Res. 96: 137-145. [DOI] [PubMed] [Google Scholar]
- Guindon, S. and Gascuel, O. 2003. PHYML—A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. System. Biol. 52: 696-704. [DOI] [PubMed] [Google Scholar]
- Hardison, R.C. 2000. Conserved noncoding sequences are reliable guides to regulatory elements. Trends Genet. 16: 369-372. [DOI] [PubMed] [Google Scholar]
- Hardison, R.C., Roskin, K.M., Yang, S., Diekhans, M., Kent, W.J., Weber, R., Elnitski, L., Li, J., O'Connor, M., Kolbe, D., et al. 2003. Covariation in frequencies of substitution, deletion, transposition, and recombination during eutherian evolution. Genome Res. 13: 13-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa, M., Kishino, H., and Yano, T. 1985. Dating of the human–ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 22: 160-174. [DOI] [PubMed] [Google Scholar]
- Hein, J. 1989. A new method that simultaneously aligns and reconstructs ancestral sequences for any number of homologous sequences, when the phylogeny is given. Mol. Biol. Evol. 6: 649-668. [DOI] [PubMed] [Google Scholar]
- Hein, J., Wiuf, C., Knudsen, B., Moller, M.B., and Wibling, G. 2000. Statistical alignment: Computational properties, homology testing and goodness-of-fit. J. Mol. Biol. 302: 265-279. [DOI] [PubMed] [Google Scholar]
- Hillis, D.M., Huelsenbeck, J.P., and Cunningham, C.W. 1994. Application and accuracy of molecular phylogenies. Science 264: 671-677. [DOI] [PubMed] [Google Scholar]
- Hoeffding, W. 1963. Probability inequalities for sums of bounded random variables. J. Amer Statist. Assoc. 58: 13-27. [Google Scholar]
- Huelsenbeck, J.P. and Bollback, J. 2001. Empirical and hierarchical Bayesian estimation of ancestral states. Syst. Biol. 50: 351-366. [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium (IHGSC). 2001. Initial sequencing and analysis of the human genome. Nature 409: 860-921. [DOI] [PubMed] [Google Scholar]
- International Mouse Genome Sequencing Consortium (IMGSC). 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520-562. [DOI] [PubMed] [Google Scholar]
- Ivics, Z., Hackett, P.B., Plasterk, R.H., and Izsvak, Z. 1997. Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell 91: 501-510. [DOI] [PubMed] [Google Scholar]
- Jermann, T.M., Opitz, J.G., Stackhouse, J., and Benner, S.A. 1995. Reconstructing the evolutionary history of the artiodactyl ribonuclease superfamily. Nature 374: 57-59. [DOI] [PubMed] [Google Scholar]
- Jurka, J. 2000. Repbase update: A database and an electronic journal of repetitive elements. Trends Genet. 16: 418-420. [DOI] [PubMed] [Google Scholar]
- Kent, W.J., Sugnet, C.W., Furey, T.S., Roskin, K.M., Pringle, T.H., Zahler, A.M., and Haussler, D. 2002. The human genome browser at UCSC. Genome Res. 12: 996-1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent, W.J., Baertsch, R., Hinrichs, A., Miller, W., and Haussler, D. 2003. Evolution's cauldron: Duplication, deletion, and rearrangement in the mouse and human genomes, Proc. Natl. Acad. Sci. 100: 11484-11489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura, M. 1980. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences, J. Mol. Evol. 16: 111-120. [DOI] [PubMed] [Google Scholar]
- Koshi, J. and Goldstein, R. 1996. Probabilistic reconstruction of ancestral protein sequences. J. Mol. Evol. 42: 313-320. [DOI] [PubMed] [Google Scholar]
- Le Cam, L. 1986. Asymptotic methods in statistical decision theory. Springer-Verlag, New York.
- Lunter, G.A., Miklos, I., Song, Y.S., and Hein, J. 2003. An efficient algorithm for statistical multiple alignment on arbitrary phylogenetic trees. J. Comput. Biol. 10: 869-889. [DOI] [PubMed] [Google Scholar]
- Margulies, E.H., Blanchette, M., NISC Comparative Sequencing Program, Haussler, D., and Green, E. 2003. Identification and characterization of multi-species conserved sequences. Genome Res. 13: 2507-2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marota, I. and Rollo, F. 2002. Molecular paleontology. Cell. Mol. Life Sci. 59: 97-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossel, E. 2003. On the impossibility of reconstructing ancestral data and phylogenies. J. Comput. Biol. 10: 669-676. [DOI] [PubMed] [Google Scholar]
- Ng, P.C. and Henikoff, S. 2002. Accounting for human polymorphisms predicted to affect protein function. Genome Res. 12: 436-446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishizawa, M. and Nishizawa, K. 2002. A DNA sequence evolution analysis generalized by simulation and the Markov chain Monte Carlo method implicates strand slippage in a majority of insertions and deletions. J. Mol. Evol. 55: 706-717. [DOI] [PubMed] [Google Scholar]
- Onay, T., Topaloglu, O., Zielenski, J., Gokgoz, N., Kayserili, H., Camcioglu, Y., Cokugras, H., Akcakaya, N., Apak, M., Tsui, L.C., et al. 1998. Analysis of the CFTR gene in Turkish cystic fibrosis patients: Identification of three novel mutations (3172delAC, P1013L and M1028I). Hum. Genet. 102: 224-230. [DOI] [PubMed] [Google Scholar]
- Pevzner, P. and Tesler, G. 2003. Genome rearrangements in mammalian evolution: Lessons from human and mouse genomes. Genome Res. 13: 37-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosser, H. and Rastan, S. 2003. Manipulation of the mouse genome: A multiple impact resource for drug discovery and development. Trends Biotechnol. 21: 224-232. [DOI] [PubMed] [Google Scholar]
- Pupko, T., Pe'er, I., Shamir, R., and Graur, D. 2000. A fast algorithm for joint reconstruction of ancestral amino acid sequences. Mol. Biol. Evol. 17: 890-896. [DOI] [PubMed] [Google Scholar]
- Pupko, T., Pe'er, I., Hasegawa, M., Graur, D., and Friedman, N. 2002. A branch-and-bound algorithm for the inference of ancestral amino-acid sequences when the replacement rate varies among sites: Application to the evolution of five gene families. Bioinformatics 18: 1116-1123. [DOI] [PubMed] [Google Scholar]
- Rat Genome Sequencing Project Consortium (RGSPC). 2004. Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature 428: 493-521. [DOI] [PubMed] [Google Scholar]
- Robertson, G.R., Field, J., Goodwin, B., Bierach, S., Tran, M., Lehnert, A., and Liddle, C. 2003. Transgenic mouse models of human CYP3A4 gene regulation, Mol. Pharmacol. 64: 42-50. [DOI] [PubMed] [Google Scholar]
- Samonte, R.V. and Eichler, E.E. 2002. Segmental duplications and the evolution of the primate genome. Nat. Rev. Genet. 3: 65-72. [DOI] [PubMed] [Google Scholar]
- Schaner, P., Richards, N., Wadhwa, A., Aksentijevich, I., Kastner, D., Tucker, P., and Gumucio, D. 2001. Episodic evolution of pyrin in primates: Human mutations recapitulate ancestral amino acid states. Nat. Genet. 27: 318-321. [DOI] [PubMed] [Google Scholar]
- Schultz, T.R and Churchill, G.A 1999. The role of subjectivity in reconstructing ancestral character states: A Bayesian approach to unknown rates, states, and transformation asymmetries. Syst. Biol. 48: 651-664. [Google Scholar]
- Schultz, T.R., Crocroft, R.B., and Churchill, G.A. 1996. The reconstruction of ancestral character states. Evolution 50: 504-511. [DOI] [PubMed] [Google Scholar]
- Schwartz, S., Kent, W.J., Smit, A., Zhang, Z., Baertsch, R., Hardison, R.C., Haussler, D., and Miller, W. 2003. Human–mouse alignments with BLASTZ. Genome Res. 13: 103-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shedlock, A.M., Milinkovitch, M.C., and Okada, N. 2000. SINE evolution, missing data, and the origin of whales. Syst. Biol. 49: 808-817. [DOI] [PubMed] [Google Scholar]
- Shi, Y. and Yokoyama, S. 2003. Molecular analysis of the evolutionary significance of ultraviolet vision in vertebrates. Proc. Natl. Acad. Sci. 100: 8308-8313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel, A. and Haussler, D. 2003. Combining phylogenetic and hidden markov models in biosequence analysis. In Proceedings of the 7th Annual International Conference on Research in Computational Molecular Biology. 277-286. [DOI] [PubMed]
- Smit, A. and Green P. 1999. ReapeatMasker, http://ftp.genome.washington.edu/RM/RepeatMasker.html
- Smit, A. and Riggs, A.D. 1996. Tiggers and other DNA transposon fossils in the human genome. Proc. Natl. Acad. Sci. 93: 1443-1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, H.O., Hutchison III, C.A., Pfannkoch, C., and Venter, J.C. 2003. Generating a synthetic genome by whole genome assembly: PhiX174 bacteriophage from synthetic oligonucleotides. Proc. Natl. Acad. Sci. 100: 15440-15445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer, M.S., Murphy, W.J., Eizirik, E., and O'Brien, S.J. 2003. Placental mammal diversification and the Cretaceous-Tertiary boundary. Proc. Natl. Acad. Sci. 100: 1056-1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoye, J., Evers, D., and Meyer, F. 1997. Generating benchmarks for multiple sequence alignments and phylogenetic reconstructions. Proc. Int. Conf. Intell. Syst. Mol. Biol. 5: 303-306. [PubMed] [Google Scholar]
- Sunyaev, S., Ramensky, V., Koch, I., Lathe III, W., Kondrashov, A.S., and Bork, P. 2001. Prediction of deleterious human alleles. Hum. Mol. Genet. 10: 591-597. [DOI] [PubMed] [Google Scholar]
- Thomas, J.W., Touchman, J.W., Blakesley, R.W., Boufford, G.G., Beckstrom-Sternberg, S.M., Margulies, E.H., Blanchette, M., Siepel, A.C., Thomas, P.J., McDowell, J.C., et al. 2003. Comparative analyses of multi-species sequences from targeted genomic regions. Nature 424: 788-793. [DOI] [PubMed] [Google Scholar]
- Thornton, J.W. 2004. Resurrecting ancient genes: Experimental analysis of extinct molecules. Nat. Rev. Genet. 5: 366-375. [DOI] [PubMed] [Google Scholar]
- Thornton, J.W., Need, E., and Crews, D. 2003. Resurrecting the ancestral steroid receptor: Ancient origin of estrogen signaling. Science 301: 1714-1717. [DOI] [PubMed] [Google Scholar]
- Yang, F., Alkalaeva, E.Z., Perelman, P.L., Pardini, A.T., Harrison, W.R., O'Brien, P.C., Fu, B., Graphodatsky, A.S., Ferguson-Smith, M.A., and Robinson, T.J. 2003. Reciprocal chromosome painting among human, aardvark, and elephant (superorder Afrotheria) reveals the likely eutherian ancestral karyotype. Proc. Natl. Acad. Sci. 100: 1062-1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Y. and Seed, B. 2003. Site-specific gene targeting in mouse embryonic stem cells with intact bacterial artificial chromosomes. Nat. Biotechnol. 21: 447-451. [DOI] [PubMed] [Google Scholar]
- Yang, Z., Kumar, S., and Nei, M. 1995. A new method of inference of ancestral nucleotide and amino acid sequences. Genetics 141: 1641-1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, J. and Nei, M. 1997. Accuracies of ancestral amino acid sequences inferred by parsimony, likelihood and distance methods. J. Mol. Evol. 44: S139-S146. [DOI] [PubMed] [Google Scholar]
- Zhang, J. and Rosenberg, H.F. 2002. Complementary advantageous substitutions in the evolution of an antiviral RNase of higher primates. Proc. Natl. Acad. Sci. 99: 5486-5491. [DOI] [PMC free article] [PubMed] [Google Scholar]
Web site references
- www.nisc.nih.gov; NISC Comparative Sequencing Program.
- http://genome.ucsc.edu/ancestors; Author's Supplemental information site.
- http://genome.ucsc.edu; Interactive browser for alignments.
- http://www.genet.sickkids.on.ca/cftr/; Cystic Fibrosis Mutation Database.