

Abstract
We report the identification of three new transcription regulatory elements that are associated with muscle gene expression in the nematode Caenorhabditis elegans. Starting from a subset of well-characterized nematode muscle genes, we identified conserved DNA motifs in the promoter regions using computational DNA pattern-recognition algorithms. These were considered to be putative muscle transcription regulatory motifs. Using the green-fluorescent protein (GFP) as a reporter, experiments were done to determine the biological activity of these motifs in driving muscle gene expression. Prediction accuracy of muscle expression based on the presence of these three motifs was encouraging; nine of 10 previously uncharacterized genes that were predicted to have muscle expression were shown to be expressed either specifically or selectively in the muscle tissues, whereas only one of the nine that scored low for these motifs expressed in muscle. Knockouts of putative regulatory elements in the promoter of the mlc-2 and unc-89 genes show that they significantly contribute to muscle expression and act in a synergistic manner. We find that these DNA motifs are also present in the muscle promoters of C. briggsae, indicating that they are functionally conserved in the nematodes.
Understanding the regulatory mechanisms that drive expression of genes during development or in specific tissues is one of the central problems in biology. Annotating noncoding genomic sequences is an equally challenging issue in computational genomics. The temporal and spatial expression pattern of genes is encoded in the genome in the form of organized arrays of cis-acting DNA elements that act as target sites for transcription factors (TFs). These DNA elements are recognized and bound by the cognate TFs that are responsible for the control of transcription of the genes. We have been interested in studying transcription control mechanisms that guide the expression of the muscle-specific genes in the nematode Caenorhabditis elegans. Some of the transcription factors, which are critical for muscle specification and function, e.g., the MyoD class of bHLH factors, and the NK-2 class of homeodomains (Chen et al. 1994; Okkema and Fire 1994; Okkema et al. 1997; Harfe and Fire 1998; Harfe et al. 1998), are conserved across distant phyla, suggesting that the knowledge gained from model organisms like the nematodes can be extrapolated to understanding the functional biology in higher eukaryotes.
A substantial amount of work has been done previously to elucidate some of the transcription factors and regulatory elements that are responsible for modulating gene expression in C. elegans muscle. Screening for TFs that bind to known regulatory elements, or searches for some of the known TFs that are responsible in myogenesis and muscle function in vertebrates and insects have resulted in the identification of some TFs that are critical for C. elegans muscle function (Chen et al. 1994; Okkema and Fire 1994; Harfe and Fire 1998; Harfe et al. 1998; Zhang et al. 1999). The identified factors include proteins of the bHLH class of transcription factors (hlh-1, Ce-Twist), which bind to the socalled `E-boxes' (consensus CAnnTG) (Chen et al. 1994; Harfe et al. 1998); ceh-22, a homeodomain belonging to the NK-2 class (Okkema and Fire 1994) that binds to the `NdE-box' motif (CATATG), which is related to, but distinct from, the standard E-box motif (Okkema and Fire 1994; Okkema et al. 1997). It is thought that several of the muscle transcription factors act in combination with other, more ubiquitous, TFs (Okkema and Fire 1994; Zhang et al. 1999). In some cases, these muscle-specific TFs may act in combination with organ-specific TFs to activate muscle gene expression in certain tissues, as is seen in the case of ceh-22, which shows a strong synergistic pattern of pharyngeal-muscle gene expression with the pharynx-specific transcription factor pha-1 (Okkema et al. 1997).
The regulatory elements of a few of the muscle-specific genes have been studied in detail using sequence deletions or mutations. For example, several DNA regulatory elements of the myosin heavy-chain isoforms in pharyngeal muscle (myo-1 and myo-2), and body-wall muscle (myo-3, unc-54, hlh-1) have been identified by deletion studies (Okkema et al. 1993). Two types of elements were discovered for the myosins. The first set contained general signals that allow high levels of expression without any obvious contribution to tissue specificity. The second set consists of promoter and enhancer elements, which appear to generate the observed tissue specificity. Tissue-specific enhancers were found to be present in the upstream regions and introns for both the body-wall and pharyngeal muscle genes (Jantsch-Plunger and Fire 1994). Apart from the myosin genes, the regulatory elements of hlh-1 (body-wall muscle) and ceh-24 (pharyngeal muscle) have also been studied (Krause et al. 1994; Harfe and Fire 1998). The study of the hlh-1 gene is particularly interesting; it exhibits distinctly different sequence requirements in the upstream region for embryonic and adult body-wall muscle expression (Krause et al. 1994), showing the complexity of TF–DNA interactions during different stages of development of the body-wall muscle. These studies not only identify the muscle regulatory sequences of some individual genes, but also delineate some of the complexities in organization of these elements and the sequence regions in which they may be located (upstream sequences and some introns).
Though detailed studies on a few individual genes have been done, a study investigating the global transcription regulatory elements that may guide the expression of most genes in the muscle tissues has not been performed. Identification of the regulatory elements in a few individual muscle genes has been important in understanding the regulatory sequences and TF target sites that guide gene expression in different muscle tissues, but it does not mean that all or most of the muscle genes will contain those motifs. It is possible that many of the muscle genes may contain a set of common regulatory elements that are yet unknown. These global muscle elements may then interact with organ-specific TFs to regulate expression of different genes in different muscle tissue types. In order to better understand the transcription regulation and development in the muscle, a more systematic approach involving the identification of common regulatory elements in muscle genes would be of benefit.
Over the past several years, DNA pattern-recognition methods (Brazma et al. 1998; Stormo 2000) have been successfully applied to the detection of regulatory elements (e.g., Hughes et al. 2000; McCue et al. 2001; GuhaThakurta et al. 2002b). We have used two such DNA pattern-recognition methods to identify putative DNA regulatory elements in the upstream regions of a collection of well-characterized C. elegans muscle genes. We identified three novel motifs using the DNA pattern-recognition programs. The significance of the identified motifs was evaluated using several independent test sets that contained both muscle and nonmuscle genes from C. elegans. These elements were also found to be significantly over-represented in the promoters of muscle genes of the related nematode, Caenorhabditis briggsae. The functional role of the putative DNA motifs in transcription regulation in muscle genes was examined by experiments using the GFP (green fluorescent protein) reporter technology (Chalfie et al. 1994). Based on the presence of these regulatory motifs, nine of the 10 genes that were enriched in muscle-related motifs were found to express in the muscle, whereas only one of the nine genes that were poor in such elements, expressed in the muscle. Site knockouts were created in the promoter of two well-known muscle genes to determine the contribution of each of these motifs in muscle expression. These knockouts resulted in their significantly reduced muscle expression. In addition, from double-site knockouts we observed that the contributions of these sites to muscle expression were nonindependent, suggesting cooperative action of the elements.
Results
DNA regulatory elements identified by computational DNA pattern-recognition algorithms
For a training set, we chose 19 well-characterized genes known to be expressed in muscle from previous studies (Table 1, details given in Methods section). We used the -2000 to -1 region of the training set genes to identify potential muscle-DNA regulatory motifs (GuhaThakurta et al. 2002a). We chose to focus on the upstream regions, since this region is almost always important for regulation of expression. Some of the muscle-specific enhancer sequences may be present in introns (Jantsch-Plunger and Fire 1994). But, including the intron regions could potentially add more sequence data without a concomitant increase in the signal for the regulatory elements, making computational identification of DNA motifs more difficult. We hope to examine the intronic regions for regulatory elements in future studies. Given the relatively closely spaced gene distribution in C. elegans, the selected upstream regions are likely to contain most of the relevant promoter elements, and most of the known regulatory elements are within these regions. However, it is possible that the selected regions may exclude relevant motifs in genes with large promoters, long 5′ UTRs, or membership in operons. Because transcriptional start sites have not been determined for many C. elegans genes, we have used the translation start site (the 1 position) to select the candidate promoter regions, because it is nearly unambiguous.
Table 1.
C. elegans muscle genes used as training and test sets
| Serial | Gene symbol | Elegans gene ID | Briggsae gene ID | Operon | Rank | Motif1 | Mofit2 | Motif3 |
|---|---|---|---|---|---|---|---|---|
| 1 | mlc-3 | F09F7.2 | CBG24046 | N | 1 | + | + | + |
| 2 | unc-22 | ZK617.1 | ND | N | 6 | + | + | + |
| 3 | unc-87 | F08B6.4 | CBG12778 | N | 15 | + | + | + |
| 4 | gpd-2 | K10B3.8 | ND | Y | 16 | + | + | + |
| 5 | unc-54 | F11C3.3 | CBG19730 | N | 29 | + | + | + |
| 6 | unc-120 | D1081.2 | CBG12542 | N | 38 | + | + | + |
| 7 | myo-3 | K12F2.1 | CBG23416 | N | 42 | + | + | + |
| 8 | mup-2 | T22E5.5 | CBG05057 | N | 46 | + | + | + |
| 9 | lev-11 | Y105E8B.1 | CBG19793 | N | 85 | + | + | + |
| 10 | deb-1 | ZC477.9 | CBG05763 | N | 91 | + | + | + |
| 11 | tni-1 | F42E11.4 | CBG17351 | N | 166 | + | + | + |
| 12 | unc-89 | C09D1.1 | CBG12078 | N | 227 | + | + | + |
| 13 | mlc-1 | C36E6.3 | ND | N | 329 | + | + | + |
| 14 | unc-112 | C47E8.7 | CBG04558 | N | 405 | + | + | + |
| 15 | act-4 | M03F4.2 | ND | N | 573 | + | + | - |
| 16 | unc-97 | F14D12.2 | CBG14705 | N | 964 | + | + | + |
| 17 | let-2 | F01G12.5 | CBG16372 | N | 974 | + | + | + |
| 18 | unc-105 | C41C4.5 | CBG00750 | N | 1514 | + | + | + |
| 19 | myo-1 | R06C7.10 | CBG21911 | N | 1631 | + | + | + |
| 20 | unc-15 | F07A5.7 | CBG11932 | N | 1955 | + | + | - |
| 21 | pat-3 | ZK1058.2 | CBG03601 | N | 2117 | + | + | + |
| 22 | unc-45 | F30H5.1 | CBG15283 | N | 2238 | + | + | - |
| 23 | mef-2 | W10D5.1 | CBG12442 | N | 2320 | + | + | - |
| 24 | pat-4 | C29F9.7 | CBG15792 | N | 2449 | + | + | - |
| 25 | unc-60 | C38C3.5 | CBG06572 | N | 2491 | + | + | - |
| 26 | pat-10 | F54C1.7 | CBG10771 | N | 2523 | + | + | - |
| 27 | act-2 | T04C12.5 | ND | N | 2672 | + | + | - |
| 28 | sup-10 | R09G11.1 | CBG01870 | N | 2806 | + | + | - |
| 29 | act-1 | T04C12.4 | ND | N | 3161 | + | + | - |
| 30 | atn-1 | W04D2.1 | CBG23504 | N | 3399 | + | + | - |
| 31 | lam-1 | W03F8.5 | CBG20003 | N | 3463 | - | + | + |
| 32 | unc-52 | ZC101.2 | CBG11064 | N | 3546 | + | + | - |
| 33 | act-3 | T04C12.6 | ND | N | 3913 | + | + | - |
| 34 | unc-68 | K11C4.5 | CBG19042 | N | 4139 | + | - | + |
| 35 | myo-2 | T18D3.4 | CBG00120 | N | 4154 | + | + | - |
| 36 | hlh-1 | B0304.1 | CBG13470 | N | 8517 | - | + | + |
| 37 | epi-1 | K08C7.3 | CBG04423 | N | 11,340 | - | + | - |
| 38 | gpd-3 | K10B3.7 | ND | Y | 11,342 | - | + | - |
| 39 | emb-9 | K04H4.1 | CBG10116 | Y | 11,503 | - | + | - |
| 40 | mec-8 | F46A9.6 | CBG03748 | N | 12,040 | - | + | - |
| 41 | egl-19 | C48A7.1 | CBG05858 | N | 12,377 | - | + | - |
Genes that are shaded were included in the training set for motif discovery, and remaining genes were used as a test set. Putative C. briggsae orthologs of C. elegans muscle genes are given. Presence (+) or absence (-) of site predictions in the upstream 2000 bp of the genes are given, along with the rank of the gene when ordered according to the combined score of the three motifs in their upstream regions (equation 4). C. elegans genes that were inside operons according to Blumenthal et al. (2002) are indicated with a Y (for yes) or N (for no) in the Operon column.
There are several computational DNA pattern-recognition methods currently available (Brazma et al. 1998; Stormo 2000). We have decided to use weight matrix-based methods rather than DNA sequence patterns with IUPAC alphabets, since they are likely to capture more information about the variability of the DNA-binding sites. We have used two methods, one based on a greedy algorithm (CONSENSUS, Hertz and Stormo 1999) and another based on Gibbs sampling procedure (ANN-SPEC, Workman and Stormo 2000). Using these computational DNA pattern-recognition methods on the training set, three different motifs were found to be significant in the training set (initially reported in GuhaThakurta et al. 2002a; Fig. 1). Motif 1 (CCCGCGGGAGCCCG) and motif 3 (AAGAAGAAGC) were identified by CONSENSUS, while motif 2 (TCTCTCTAACCC) was identified by ANN-SPEC. The underlined part of motif 1, which is the most conserved part of that motif, was also identified by ANN-SPEC. These motifs did not correspond to any known transcription factor binding sites from a search of known motifs from the TRANSFAC database (Matys et al. 2003), or the regulatory sequences identified previously using sequence-deletion studies. The motifs identified were considered novel muscle gene-enriched regulatory elements.
Figure 1.

Logos (Schneider and Stephens 1990) for the three putative DNA muscle regulatory motifs identified by computational methods and their information contents in bits.
Overrepresentation of the identified DNA motifs in training and test sets
In order to assess the significance of the binding probability of the TFs to the upstream regions of muscle genes, we determined the TF–DNA-binding probabilities for the sites corresponding to the putative DNA regulatory elements in a number of sequence sets in C. elegans (Table 2), and compared them with those obtained from a random set (randomly selected 2000 genes). These probabilities (which are given by the probability proportionality values [PPVs], see equation 2 for details) take into account both the site `strength' (i.e., the match of the model weight matrix to a site) and frequencies. Multiple sites (even though weak), or one particular site with high score, can both result in high-binding probability of a TF to a sequence.
Table 2.
TF-DNA binding PPVs (probability–proportionality values) as an indicator TF–DNA binding (refer to equation 3)
| Sites | CE training | CE test | CB muscle |
|---|---|---|---|
| motif 1 | 5094.98 | 111.67 | 22.27 |
| motif 2 | 24.89 | 12.26 | 6.72 |
| motif 3 | 9.49 | 5.95 | 34.79 |
| ces-2 | 0.58 | 0.75 | 0.48 |
| skn-1 | 0.52 | 0.96 | 1.18 |
| gata | 0.50 | 0.71 | 1.27 |
Motifs 1 through 3 are putative C. elegans muscle regulatory elements. Motifs ces-2, skn-1, and gata are C. elegans regulatory motifs unrelated to muscle expression and used as controls. Different gene-sets tested are given. CE training, C. elegans muscle training set (refer to Table 1); CE test, C. elegans muscle test set; CB Muscle, putative orthologs in C. briggsae of known C. elegans muscle genes (Table 1). The mean of the ratios of binding probabilities are given for the test or training gene set versus 500 random sets. Values greater than 5 are in bold.
Table 2 gives the average ratio of putative TF–DNA-binding probabilities (PPVs) of muscle gene upstream sequences to the upstream sequences from 500 random sets of genes in C. elegans or C. briggsae. The ratios were determined 500 different times with the training and test sets and different random sets, each random set containing 2000 genes selected randomly from the genome. Table 2 shows that the binding probabilities of the identified DNA motifs to their cognate TFs are higher in the two C. elegans muscle gene sets as compared with random sequences. In the C. elegans muscle upstream sequences, motif 1 has significantly higher scores (more than three orders of magnitude in the training set, and two orders of magnitude in the test set binding probability compared with random sequences). Motifs 2 and 3 also contribute to TF-binding probability that is at least one order of magnitude higher in the training set muscle genes, and sixfold or more in the test set genes. It is not surprising that the trainingset genes show higher scores compared with the test sets, since the motif discovery was done in the training set. However, the big difference in motif 1 scores between the training and test sets in C. elegans perhaps cannot be entirely accounted for by the above fact. One plausible explanation could be differences in temporal expression patterns of some of the training and test set genes in the muscle as described in the Discussion.
For the purpose of comparison, the ratios of binding probabilities are also shown for three other C. elegans DNA regulatory motifs, which are not related to muscle regulation, viz., the GATA (consensus, ACTGATAA), a potential intestine-specific regulatory motif (Egan et al. 1995) and two other DNA motifs, skn-1 and ces-2, taken from the TRANSFAC database. skn-1 represents the DNA-binding site (consensus, TAATGTCATCCA) for the C. elegans skn-1 protein, which is a TF required for the correct specification of certain blastomere fates in early C. elegans embryos (Blackwell et al. 1994), and ces-2 represents the DNA-binding site (consensus, ATTACGTAAT) for ces-2, a TF that controls the cell-death fate of individual cell types in programmed cell death (Metzstein et al. 1996). None of the three unrelated motifs show higher binding probability for the muscle genes compared with random genes in C. elegans.
To determine whether the DNA regulatory motifs may be functionally conserved in the phylogenetically related nematode, C. briggsae, we determined the average ratio of binding probabilities for the C. briggsae muscle orthologous upstream sequences as compared with random sets of C. briggsae upstream sequences. For C. briggsae, the ratios vary in magnitude from ∼7 to 35, whereas for the three unrelated DNA motifs (GATA, skn-1, and ces-2), the ratios are not more than ∼1.3. This shows that the putative DNA regulatory motifs are also overrepresented in the muscle promoters of C. briggsae.
Since transcription factors almost always work in concert, we tested the statistical significance of the combined scores from the three motifs in the muscle promoters. We observed statistically significant scores in the nematode muscle genes using the Mann-Whitney test (Zar 1974), a simple nonparametric procedure frequently used for testing whether differences exist between two sampled populations. We looked at the upstream regions (-2000 to -1) of all genes from the genome, determined the DNA sites for a motif, m, above the cutoff using the PATSER program, and calculated the PPV for each of the sequences (equation 2). A combined PPV for the three motifs was also calculated for the upstream sequence of each gene in the genome (equation 4). All of the upstream sequences were sorted according to the decreasing log of the combined PPV, ln(Pseq_M) (equation 4). We calculated the Mann-Whitney statistic (Mann and Whitney 1947; Zar 1974) for testing the hypothesis, HA: genes in a given test set have significantly higher binding probability values (PPVs) when compared with a randomly selected set of genes. Based on the calculation of the Mann-Whitney statistic and z-scores (described in more detail in Zar 1974; GuhaThakurta et al. 2002b), the p-value for the null-hypothesis (H0: test genes and random genes do not differ in their PPVs) can be determined. Using combined scores from motifs 1, 2, and 3, the p-values for accepting the null hypothesis in several independent data-sets were as follows: (1) 4.8 × 10-7 for the C. elegans muscle test set, (2) 4.8 × 10-5 for the set of around 1200 genes that have been shown to be overexpressed in the C. elegans muscle using RNA tagging and c-DNA microarray experiments (Roy et al. 2002), and (3) 1 × 10-9 for the C. briggsae muscle genes (Table 1). As a control, we tried a combination of two other C. elegans regulatory motifs that are not muscle related (skn-1 and ces-2); the p-value was observed to be ∼0.5 for the C. elegans muscle-test set, and 0.16 with the 1200 muscle-expressed genes in Roy et al. (2002). In all cases, the z-scores and p-values were computed with 500 different background sets (each background set consisting of 2000 randomly selected genes form the genome) and the average of 500 z-scores were taken to report the p-values. These highly significant p-values with several independent test sets strongly suggested that the identified motifs are over-represented in the promoters of nematode muscle genes, and are therefore likely to be functional elements.
Regulatory site clustering and investigation of muscle regulatory module
We have investigated the distribution of sites, looking for evidence of site clustering and the possibility of a regulatory module in the promoters of the C. elegans muscle genes. We find that compared with a random set of genes, the predicted muscle regulatory sites are more frequent in the immediate upstream region of C. elegans muscle genes (Fig. 2). As expected, for the random genes, roughly 20% of the sites are present in each 200-nt window, but in muscle genes, the percent of sites are higher near to the gene start sites (TSS).
Figure 2.

Fraction of total sites observed in the upstream 1000 nts mapped as a function of distance from the start site of the 41 known muscle (▪) and 2000 random genes (□) in C. elegans. Averages over the gene sets are shown. Motifs 1 (i), motif 2 (ii), motif 3 (iii), and combined frequency of motifs 1, 2, and 3 (iv).
In human skeletal muscle genes, the known regulatory sites tend to cluster within a distance of roughly 200 nts, forming a muscle regulatory module (Wasserman and Fickett 1998). We investigated the presence of a similar optimal window size for a muscle regulatory module in C. elegans using predicted sites from the three motifs we identified here. Several computational methods now exist for identification of putative DNA regulatory modules in input sequences given a set of transcription-factor binding-site profiles. Most can be grouped in two classes, viz., a sliding window approach, and hidden Markov model approach (Bailey and Noble 2003). We have used two of those methods, one from each class, viz., MSCAN (Johansson et al. 2003) and COMET (Frith et al. 2002). MSCAN computes the statistical significance of observing regulatory modules in different sequence windows based on the distribution of hits for the individual DNA profiles in a random sequence. COMET uses an HMM process, where it assumes that cis-elements occur in a Poisson process embedded in random DNA. Given that the motifs are all clustered near the AUG end of the promoter region, we did not observe any significant combinations of motif clusters. It is possible that we have not yet discovered all of the muscle regulatory motifs (see below), and this issue of whether there are particular combinations that define important modules should be revisited at a later time.
Muscle regulatory sites in cross-species conserved regions
In human muscle genes, 98% of experimentally defined sequence-specific binding sites of skeletal-muscle transcription factors are confined to the 19% of human sequences that are most conserved in the orthologous rodent sequence using a Bayesian alignment method (Wasserman et al. 2000). Since only a few functional binding sites in C. elegans are known, it is impossible to deduce such numbers for the nematodes. Another issue is that C. elegans and C. briggsae are further apart in terms of evolutionary distance compared with human–rodent, with significantly larger nucleotide substitution rates (Hardison 2004; Stein et al. 2003; Waterston et al. 2002). This could make it difficult to find extended alignments in the promoter regions of these two nematodes like the ones that are observed between human and mouse. However, we wanted to see whether the predicted binding sites are enriched in conserved upstream regions in C. elegans and C. briggsae muscle sequences. We aligned the orthologous promoters from the two nematodes using a local (BLASTZ, Schwartz et al. 2003) and a global alignment tool (GLASS, Batzoglou et al. 2000). The noncoding upstream regions of orthologous genes were repeat-masked and then aligned using BLASTZ and GLASS. The results of alignments were post-processed with a sliding window of 50 bp with 70% identity (65% for BLASTZ). Only those alignments that met these criteria for alignment length and percent identity were retained as blocks of sequence conservation. The average fraction of the muscle upstream sequences that fall in these conserved regions is ∼9% using GLASS, and ∼6% using BLASTZ (which is significantly below what has been observed for human–mouse alignments as described in the Discussion). The percentage of predicted muscle-regulatory sites in the upstream sequences, corresponding to the three muscle motifs, varied from 6% to 10% within BLASTZ conservation (average ∼7%), and 19%–55% within blocks of GLASS conservation (average ∼32%). Thus, using the global alignment tool, we get an enrichment of the predicted sites in the regions of conservation, which is not observed using the local alignment tool. However, this enrichment still misses the majority of the predicted sites.
Expression patterns of genes predicted to express in C. elegans muscle
We calculated the combined PPV using motifs 1, 2, and 3 for all gene upstream regions in C. elegans (equation 4); all of the genes were then sorted according to their combined PPVs. We decided to check the expression of 10 high-ranking genes (Table 3). We used GFP technology (Chalfie et al. 1994) to evaluate the expression by fusing the promoter region of the genes to GFP, creating promoter::GFP constructs. Nine of the 10 promoter::GFP constructs are expressed in the muscle. Most are expressed only in muscle, but a few are also expressed in a handful of other tissues, mainly neuronal cells (Fig. 3; detailed expression patterns of several genes are also given at http://ural.wustl.edu/~dg/Nematode_Muscle_Regulation.html). It is worth noting here that many of the known muscle genes are frequently observed to express also in neuronal tissues. As a caveat, it should also be mentioned that experimentally proving that a gene is expressed in only one tissue is difficult. This is because the observance of expression in one tissue does not rule out low or transient expression of that gene in other tissues, and the detection of expression in some of the tissues can be inherently problematic. We also tested nine genes that ranked lowly for the presence of three motifs (between ranks 4800 and 17,000). We did not simply pick the worst ranked genes by our ranking criteria, because we thought that even if those were all negative, it would not be as convincing as picking a more random sampling from a wider range of rankings that we expected to be negatives. Among these nine constructs, only one (F09C8.2) showed expression in the muscle.
Table 3.
The list of genes whose expression has been characterized using promoter::GFP constructs
| Serial | Rank | Gene ID | Gene name | Muscle |
|---|---|---|---|---|
| 1 | 4 | C49A1.10 | Y | |
| 2 | 9 | F55C7.2 | Y | |
| 3 | 10 | Y44A6D.3 | Y | |
| 4 | 13 | R08B4.2 | Neuronal | |
| 5 | 14 | B0513.1 | gei-1 | Y |
| 6 | 17 | T22C1.7 | Y | |
| 7 | 21 | W06F12.1a | Y | |
| 8 | 50 | F41E7.6 | Y | |
| 9 | 52 | F02E9.2b | lin-28 | Y |
| 10 | 60 | C05D11.4 | let-756 | Y |
Muscle expression is indicated by Y. Rank of the genes, based on the three muscle motifs, are given. For figures of GFP expression patterns, see http://ural.wustl.edu/~dg/Nematode_Muscle_Regulation.html. One gene is expressed in neurons but not in muscle. lin-28 did not express in any tissue in our experiments, but we found it to have muscle expression in Moss et al. (1997).
Figure 3.

GFP-expression patterns of genes B0513.1 (A), C05D11.4 (B), C49A1.10 (C), T22C1.7 (D), Y44A6D.3 (E), W06F12.1a (F), F41E7.6 (G), F55C7.2 (H), and R08B4.2 (I). (A–C) GFP expression in the cells and nuclei of body-wall muscle cells. (D,E) GFP expression in the nuclei of body-wall muscle cells. (F) GFP expression in the cells and nuclei of pharyngeal muscle cells. (G) GFP expression in the anal depressor muscle cell. (H) GFP expression in the intestinal muscle cell. (I) GFP in the nuclei and neuronal process of a neuronal cell.
Expression of promoter::GFP constructs with site knockouts
We assessed the contribution of motifs 1, 2, and 3 to muscle expression of the C. elegans myosin light chain protein, mlc-2, which shows localized expression only in the muscle (Rushforth et al. 1998). It is worth noting that mlc-2, which was not in our initial training or test sets, shares its upstream region with mlc-1, the two genes being divergent in the genomic sequence separated by ∼2500 nts. The immediate promoter regions (first 400 bp) of mlc-2 and mlc-1 are not identical. The ranks of the promoters for these genes among all C. elegans genes, when scored for the three motifs, are thus different. mlc-2 is ranked at 399, while mlc-1 is at position 329 (Table 1). Sites corresponding to motifs 1, 2, and 3 in the mlc-2 promoter (Fig. 4A) were replaced with mutated sequences within the upstream 400 nts. Knocking out the individual sites had varied effects on the expression of this gene (Fig. 4B,C,D). Individually, motifs 1, 2, or 3 knockouts reduced expression of mlc-2 to about 35%, 60%, and 31% of the wild type, respectively (Fig. 4B,C). The promoter with all three motifs mutated still retained about 5% of the wild-type expression, and it remained muscle specific. The double knockouts all reduced expression of the gene more than what would be expected from a simple multiplication of independent effects. For example, expected expression of the double knockout construct eliminating sites for motifs 1 and 2 is ∼21% based on independent effects of individual motifs, where the observed expression level was down to ∼5% of the wild type, indistinguishable from knocking out of all three motifs. The same is true for the other two double-site knockouts. Thus, multiple double-site knockouts suggest synergistic effect of these motifs in the gene expression regulation. To verify that the motifs are functional in other muscle genes, we did a knockout of motif 3 in the promoter region of unc-89, another well-known muscle gene (Benian et al. 1996). This single-site knockout reduces the expression of GFP in body-wall muscle to about 7% of the wild type (Fig. 4B,C,D). Interestingly, the motif 3 knockout did not reduce the expression in pharyngeal muscle cells, suggesting a different mode of regulation in that tissue.
Figure 4.

(A) Location of predicted sites corresponding to motifs 1, 2, and 3 in the immediate upstream region of genes mlc-2 and unc-89. Sites on the reverse strand are shown below the sequence line. The numbers above or below each site indicate the site score given by the PATSER program upon alignment of the matrix to a sequence. (B–D, left) mlc-2 Expression; (B–D, right) unc-89 expressions. (B) GFP intensity vs. pixel measurements for wild-type and different site knockouts in the promoters of mlc-2 and unc-89 genes. Data from one line and one photograph is shown for each of the constructs. (C) Total intensity measurements for wild-type and different site knockouts for the mlc-2 and unc-89 gene promoters. The GFP intensity data for the wild-type and different knockouts were obtained from a minimum of 11 animals in case of mlc-2 and 30 animals for unc-89. Three different lines were generated for mlc-2 wild type, mlc-2 motif 2 knockout, mlc-2 motif knockouts 1+2+3, unc-89 wild type, and unc-89 motif 3 knockout. In all other cases, one line was used. (D) GFP expression photographs with wild-type mlc-2 promoter::GFP construct and with the triple knockout (motifs 1, 2, and 3 eliminated), and GFP expression with wild-type unc-89 promoter::GFP construct and with the knockout of motif 3 site. The left two panels of each figure are the photographs of wild-type proteins, while the right panels give the site knock-outs.
Discussion
We have reported here the identification of three new muscle DNA-regulatory elements in C. elegans. Candidates for regulatory elements were first determined using computational DNA pattern-recognition methods that were then experimentally validated. None of the identified motifs corresponded to known regulatory elements in the TRANSFAC database, or DNA elements previously found to be driving muscle expression in some individual genes based on sequence deletion studies, suggesting that many of the global regulators of muscle genes, which are found in the promoters of most muscle genes, could be distinct from the gene-specific elements that have been found by sequence-deletion analyses of a specific gene before. We observe the following facts with the identified motifs: (1) genes enriched in these motifs usually express specifically or selectively in the muscle, and (2) the motifs contribute significantly to the expression of muscle-specific genes. This, however, does not necessarily mean that the motifs give muscle-specific expression; we have not demonstrated that the motifs are not present in any other groups of genes nor experimentally verified that they do not contribute to the expression of genes in any other tissue.
Motif scores and muscle expression
We observe significantly higher scores for the motifs in muscle training and test sets when compared with background sets (Table 2). While evaluating the significance of individual motifs, we observed a significant difference in the scores of motif 1 in the training and test sets (Table 2). While a higher score for the training set is not surprising, as the motifs were discovered using that set, upon further investigation, we find another plausible explanation that can contribute to this difference. The training set was enriched in genes that predominantly encode proteins that were part of the thick and thin filaments (15 of 19) while, in comparison, the test set was enriched in genes that predominantly encode proteins that were part of the basement membrane (10 of 22) or part of the dense body and M-line (5 of 22). While these structures are all in muscle cells, the basement membrane develops early in the muscle cell, followed later by the dense body and M-line to which, still later, the thin filament and thick filament, respectively, attach (Waterston 1988). This suggests that the difference between the training set and the test set with respect to motif 1 scores could have a temporal explanation, although we have not verified this experimentally. It also points to the fact that understanding the full complexity of the gene regulation in a tissue will require studies of both the spatial and temporal components.
In a ranked list of C. elegans genes, ordered according to the combined scores of the three motifs, many of the known muscle genes appear near the top, and only a few are below 10,000, the rank that would be expected of a randomly chosen gene (given the C. elegans genome has nearly 20,000 genes). However, since not all muscle genes rank near the top, we think there are additional regulatory motifs that contribute to muscle expression that escaped discovery in our study, or a more appropriate computational model than the simple PPV statistic that we have used here is needed with the currently identified motifs that can better explain the expression of the muscle genes. Other complicating factors include the organization of C. elegans genes into operons. Two of the genes (gpd-3 and emb-9) that score below rank 10,000 are inside operons (see Table 1 and Blumenthal et al. 2002), where they are not the first gene in the operon, so it is not surprising their immediate upstream regions do not contain the regulatory sites. In the case of gene emb-9, the promoter of the first gene (K04H4.2) in the operon scores high with respect to the muscle motifs, ranking 374 in the ordered list of genes. Genes gpd-2 and gpd-3 are the second and third genes of an operon, but upon examination of the operon and the genes within it, the start of the transcription and the regulatory region appears to be included in the 2 kb upstream of the gpd-2 gene, which explains its high ranking (rank 16, Table 1).
Based on experimental evidence for muscle expression, we find the top scoring genes on our list (sorted by the combined scores of the three motifs) to be highly enriched in genes expressed specifically or selectively in muscle. Among the genes that scored highly for these motifs, nine of the 10 previously uncharacterized genes that we tested expressed in the muscle, whereas in a sample of genes that scored poorly, only one of the nine tested showed muscle expression. These numbers do not represent false-positive and false-negative rates of our prediction, since we have not determined a cutoff for classification and we have only tested a few predictions, but they are encouraging results. Using logistic regression analysis, with the five, well-characterized TF–DNA binding sites, which constitute the human-muscle regulatory module, the false-positive and false-negative rates were reported as 52% and 40%, respectively (Wasserman and Fickett 1998). Regulatory modules in higher eukaryotes like vertebrates are likely to be much more complex, however, with more TF–DNA interactions per gene.
Experimental validation of individual motifs
The decrease in muscle expression by the site mutations in mlc-2 and unc-89 indicate that the identified motifs contribute toward muscle expression. The double-site mutations have a nonindependent effect on expression. Most transcription factors work in a combinatorial fashion with others, so mutating individual regulatory elements is not only going to affect the binding of the cognate TF to that site, but disturb the binding of other factors that bind to nearby sites, thereby affecting the transcription regulatory complex in that region.
Enrichment of motifs in the promoters of C. briggsae muscle genes
Based on the highly significant p-values from Mann-Whitney statistics, it appears that the identified regulatory motifs are conserved in promoters of the muscle genes in the nematode, C. briggsae. Upon consideration of the ranks of the muscle genes in the two organisms, we observe that the median rank of these genes in C. elegans is 2100 and in C. briggsae is 3700, which are in the top ∼10% and 18% of the total number of genes in the genome (expected median rank is 10,000 if we consider 20,000 genes in the genome). Though the C. briggsae gene ranks are higher, which is not surprising considering that the motif discovery was done in C. elegans, the ranks are still significantly lower than expected by chance.
In spite of the fact that the identified motifs are overrepresented in the muscle genes of both the nematodes, there are some differences between them. If we compare the ranks of individual genes between the two organisms, we find that they are quite variable. This is not too surprising given that only about 32% of the predicted sites occur with the regions aligned by GLASS, and indicates that the number of sites, and their specific sequences can vary between the species. Nonetheless, only two genes of the 33 orthologs rank poorly (rank >4000) in both organisms and one of those (emb-9) is inside an operon, so there appears to be one true outlier (epi-1). Most of the genes have low ranks in both species, but there are several cases where the rank is much poorer in C. briggsae than in C. elegans (and a couple of the opposite). An extreme example is the mlc-3 gene, which has a rank of 1 in C. elegans but 12,583 in C. briggsae. Assuming mlc-3 is also muscle specific in C. briggsae (which we do not know for sure, but the assumption seems reasonable), there are several possible explanations for this. (1) Scores for some of the functional sites in C. briggsae fall below the threshold we have used here and were not detected while scanning with the motifs; (2) the same regulatory sites still control its expression, but they are now located outside of the region we included in the scoring (in the first two introns of mlc-3 gene there are 16 sites corresponding to the three motifs in C. briggsae as opposed to six sites in C. elegans); (3) there are additional motifs beside the three we have obtained thus far, and in the C. briggsae mlc-3 gene, those have replaced the motifs used in C. elegans. The observation that there are a few genes with high ranks in C. elegans, and even a few more in C. briggsae, provides additional evidence that there are likely to be more motifs involved in the specification of muscle expression than just the three we have identified and studied so far. In fact, preliminary searches with another motif discovery tool that used orthologous promoter sequences from both nematodes (Wang and Stormo 2003) have identified a few other potential motifs in addition to the three described here, but they have not been experimentally verified yet (T. Wang and G.D. Stormo, unpubl.).
While the overrepresentation of the motifs in the C. briggsae muscle genes indicates a functional conservation, the evidence for conservation of individual binding sites is weak, and the overall alignments of the promoter regions are much less informative than for human and mouse comparisons. Whereas in one study about 19% of the upstream 10 kb was alignable between human and mouse, we only obtain 6%–9%, depending on the alignment algorithm used. And, while 98% of the known regulatory sites in the human–mouse comparison were within the alignable segments, we only find an average of 32% of our predicted sites in the aligned 9% using GLASS, thereby giving an enrichment of approximately threefold. While that represents a significant enrichment, it means that the majority of probable regulatory sites are not to be found in the regions aligned by programs such as BLASTZ and GLASS that are the standard methods in the field today. This observation could be due to the following: (1) individual sites are not conserved in C. briggsae and C. elegans, i.e, a specific site in C. elegans could disappear over evolutionary time and reappear at a different position; (2) there are limitations to the currently available traditional phylogenetic footprinting methods and their reliance on “the” alignment of the promoter regions. Whether one uses global or local alignment methods, approaches that return a single optimal alignment, though enriched in regulatory sites, can still miss some fraction of them. In phylogenetic footprinting studies with bacteria, which are more highly diverged than the species used here, alignments of the promoter regions are not relied on to identify the motifs, because only the motifs themselves are significantly conserved (McCue et al. 2001).
When, instead of returning a single alignment for each promoter region, several (optimal and suboptimal) ungapped, local alignments were kept (T. Wang, L.A. Schriefer, and G.D. Stormo, unpubl.), all of the three motifs reported here are obtained along with a few others, indicating both the value of these suboptimal alignments as well as increased motif identification sensitivity from having sequences from two species. Sequences from additional nematodes that are evolutionarily between C. elegans and C. briggsae should help in identification of functional regulatory and cross-species analyses.
One cannot rule out the possibility that the majority of sites in aligned regions are biologically functional, whereas those that are outside of these regions are not. Based on our preliminary site knockout analysis in mlc-2 and unc-89 genes, this appears unlikely, since none of the sites that we determined to reduce expression on mutation are in the aligned regions. A more complete analysis with many more known functional elements is needed before conclusions can be reached on this issue.
Future directions and conclusions
Identification of functional DNA regulatory motifs remains a challenging problem. The DNA-binding sites for transcription factors tend to be degenerate and often function only in the context of other sites. Thus, not all biologically functional motifs are likely to be statistically significant enough by themselves to be detected by computational methods. Despite the challenges, we have shown that DNA-pattern recognition methods and simple statistical tests give biologically meaningful results in the nematodes. We expect that more functional motifs are yet to be discovered; in fact, some previously characterized motifs were not identified in our analysis. One such element could be the E-box motif (CAnnTG), a short and variable motif that is bound by the bHLH factors and frequently observed in multiple copies in the promoters of muscle genes (Yutzey and Konieczny 1992). It was not picked up by computational methods nor did it appear to be overrepresented in the nematode muscle genes. In an initial analysis with an expanded set of muscle-specific genes and incorporating information from C. briggsae, the motifs we report here were confirmed and a few others have been tentatively identified (T. Wang, L.A. Schriefer, and G.D. Stormo, unpubl.), that remain to be experimentally tested. Using a larger set of motifs, we also expect to find significant combinations that function as regulatory modules for controlling gene expression. But, even at this point, using only C. elegans and a moderately sized set of training examples, we have shown that computational motif discovery algorithms can identify sites that are critical to the proper expression of muscle-specific genes, and can also be utilized as a search tool to identify additional, previously unknown, muscle-specific genes. Our current studies are focused on validating the larger set of putative motifs, analyzing them for significant clusters, and applying experimental approaches to identify the transcription factor that binds to each motif.
Methods
Identification of C. elegans muscle genes
Of the thousands of genes that are expressed in the different muscle tissues, the most useful genes for the purpose of this study are the ones that are preferentially expressed in the muscle. Preferential expression can be either specific (expression only in the muscle tissue) or selective (expression in muscle and a few other tissues like neurons). Both kinds of genes are likely to contain regulatory elements that are muscle specific.
We identified a total of 41 muscle-specific or selective genes from the literature (Table 1) and from our previous work, 19 of which were put in our training set from which we did motif discovery, and 22 were put in the test set as described below. The motif discovery effort was done when we could collect a substantially large number of genes for the motif-finding programs. This set consisted of 19 experimentally characterized genes (`training set') with well-defined intron–exon boundaries, which was important for accurate identification of the promoter regions. We gradually added 22 more genes to this initial list, which we used as an independent `test set' for evaluation of the significance of the motifs. For evaluation of the statistical significance of the motifs through a Mann-Whitney test, we used 500 background sets, each set consisting of 2000 randomly selected genes from the C. elegans genome. The test set for C. briggsae consisted of 33 genes that were orthologous to the 41 C. elegans genes (Table 1). The 500 background sets for C. briggsae were prepared in the same way as they were for C. elegans.
C. briggsae orthologs of C. elegans muscle genes
The C. briggsae genome sequence and annotation has recently been completed (Stein et al. 2003; ftp://ftp.wormbase.org/pub/wormbase/briggsae/). Using syntenic markers and reciprocal BLAST runs, the Washington University Genome Sequencing Center determined the putative orthologs of more than 11,000 C. elegans genes in C. briggsae. From this list, orthologs for 33 of the 41 C. elegans muscle genes were obtained (Table 1).
Obtaining upstream sequences
The C. elegans chromosomal sequence and the gene structures were downloaded from the WormBase ftp-site (ftp://ftp.wormbase.org/pub/wormbase/). These were then used to obtain -2000 to -1 upstream region of the genes.
Identification of putative regulatory elements using computational DNA pattern recognition methods
We used two DNA pattern recognition programs, viz., CONSENSUS and ANN-SPEC. CONSENSUS and ANN-SPEC are local multiple-sequence alignment programs that run on a given set of sequences (training set) to identify conserved motifs commonly present in those sequences. Both of these programs use position-weight matrix-based models (Stormo 2000) to represent ungapped DNA sequence motifs. The programs were run on upstream regions (-2000 to -1) of the C. elegans training set muscle genes.
CONSENSUS
The CONSENSUS program (Hertz and Stormo 1999) uses a greedy algorithm and searches for a matrix with a low probability of occurring by chance or, equivalently, having a high information content. Version 6.c of CONSENSUS was used and the top scoring result was reported. Different pattern lengths were tested, and both strands of the DNA were searched for motifs, because TFs can bind in either orientation. The patterns with high information content and the lowest expected frequency were considered.
ANN-SPEC
ANN-SPEC (Workman and Stormo 2000) uses a simple artificial neural network and Gibbs sampling method to define DNA binding-site patterns. The program searches for the parameters of a simple perception network (weight matrix) that maximize the specificity for protein (TF) binding to a positive sequence set (or training set) compared with a background sequence set. The use of background sequences allows the method to find patterns with greater discriminatory capability and specificity when compared with the original version of the Gibbs sampling method (Workman and Stormo 2000; GuhaThakurta and Stormo 2001). ANN-SPEC Version 1.0 was used. A background sequence set of upstream regions from 3000 randomly picked genes was used for the runs. Different motif lengths were tried and both strands of the DNA were searched for motifs. Because of the nondeterministic nature of the algorithm, multiple training runs were performed (100), with each run iterating 2000 times. The results were sorted by their best-attained objective function values. Weight matrices corresponding to the 10 highest scoring runs were compared, and if more than five of these top scoring 10 runs gave a motif with one consistent consensus pattern, that pattern was considered significant.
Calculation of site scores, cutoffs, and searching for sites in sequences
A position-weight matrix (PWM) has been found to be a good model for describing protein-binding sites in DNA (Stormo 2000). An l-long DNA binding-site pattern is described by a 4 × l weight matrix, with four weights (for four DNA nucleotides) per pattern position. The score for any particular site is the sum of matrix values corresponding to the sequence of the site. Under the simplifying assumption that the positions contribute independently to the binding affinity and the matrix elements are log-odds scores, then the score for an individual site should be proportional to its binding energy (Berg and von Hippel 1987; Stormo and Fields 1998; Benos et al. 2002).
The PATSER program (G.Z. Hertz and G.D. Stormo, unpubl.) takes as input a weight matrix and a set of sequences. For each sequence, the score of every subsequence (i.e., for every possible binding site) is determined, and those that exceed the user-defined cutoff score are identified in the output. We used the default cutoff determined by PATSER based on the information content of the weight matrix. The information content of the matrix is related to the probability of observing a site by chance (Schneider et al. 1986; Stormo 2000), and given the weight matrix, it is possible to calculate the probability of observing a sequence with a particular score or greater (Staden 1989; Hertz and Stormo 1999). The default cutoff from PATSER is the score with a probability set by the information content.
Determination of promoter binding probabilities from `site' scores
A “site” corresponding to a particular motif is simply a high-scoring subsequence that is obtained by the PATSER program using the appropriate motif weight matrix as an input. Weight matrices for the motifs were determined using the CONSENSUS and ANN-SPEC programs or were obtained from the TRANSFAC database (Matys et al. 2003). From a consideration of the thermodynamics of protein–DNA interactions and the statistics of the scores (Stormo and Fields 1998), we expect that the score should be proportional to the free energy of binding. Therefore, at equilibrium, the probability of the protein binding to a site with a score, s, is given by:
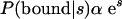 |
1 |
The exact proportionality factor depends on a number of things, including the availability of binding sites within the genome and the concentration of the TF in the nucleus, but because we only use it to rank different potential binding sites, we can ignore it. We also know that there are commonly multiple-binding sites in the promoter region for a regulatory TF, so we calculate the probability that it will bind at any of those sites (probability–proportionality value, or PPV), as
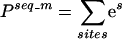 |
2 |
where, m denotes the DNA-binding motif for the TF. This treatment is likely oversimplified, given the known cooperative binding of TFs to promoter elements. Nevertheless, more complicated models have not proven more effective for the analysis presented here, and this simplified approach has produced meaningful results.
We want to consider the probability that all sequences in a given set are regulated or bound by a TF. Then, for that given set of N sequences, the average PPV should be given by the geometric mean of the PPVs of the sequences:
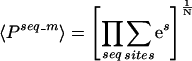 |
3 |
When no sites are observed above threshold for a motif, m, we simply set s = 0, so that es = 1 and the product is not equal to zero. A combined PPV for a multiple motif set model, M, can also be calculated for the upstream sequence of each gene in the C. elegans genome. For lack of more specific information regarding the mode of TF binding and interaction with the putative DNA regulatory sites, we assumed that for up-regulation of genes in the muscle (1) relevant TFs (corresponding to the motifs being considered) need to bind to the upstream sequence, and (2) if there are multiple sites scoring above the cutoff for a particular motif, any one of those binding sites may be occupied by the corresponding TF. For a particular upstream sequence, the combined PPV for multiple motifs (M) is calculated by taking a product of individual PPVs (from equation 2 above) for the motifs:
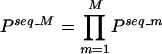 |
4 |
Sorting genes by PPVs (probability–proportionality values) and nonparametric analysis
Nonparametric, or distribution-free tests may be applied in any situation in which actual measurements are not used, but instead, the ranks of the measurements are used. The data may be ranked either from highest to lowest or from lowest to highest values. In our case, we have the Pseq_(m/M) (probability–proportionality values) for all C. elegans genes, based on the DNA binding-site motifs, arranged in decreasing order. We use the nonparametric analog of the two-sampled t test, commonly known as the Mann-Whitney test (Mann and Whitney 1947; Zar 1974).
We take the sorted list of Pseq_M calculated using combinations of motifs. We then consider two sets of ranks, that of the muscle (training or test set) genes and that of the random genes. Based on the rank positions of the genes in the list, a z-score can be calculated (details in Zar 1974 and GuhaThakurta et al. 2002b). From the z-score, the significance of the hypothesis, HA, muscle genes have higher PPVs compared with randomly selected genes, can be assessed.
Alignment of C. elegans and C. briggsae upstream regions
For all ∼11,000 C. elegans/C. briggsae orthologous gene pairs, we obtained the noncoding upstream regions, repeat masked the sequences for common C. elegans repeats using the RepeatMasker program (http://ftp.genome.washington.edu/RM/webrepeatmaskerhelp.html), and then aligned each upstream pair using the two programs BLASTZ (Schwartz et.al. 2003) and GLASS (Batzoglou et. al. 2000). The BLASTZ program is a local alignment tool, whereas GLASS gives a global alignment. The chaining option was used for BLASTZ. Default parameters were used for GLASS. The results of alignments were post-processed with a sliding window of 50 bp with 70% identity (65% for BLASTZ). Only those alignments that met these criteria for alignment length and percent identity were retained as blocks of sequence conservation.
Putative muscle regulatory module detection
MSCAN (Johansson et al. 2003) was run through its Web-interface http://tfscan.cgb.ki.se/cgi-bin/MSCAN, whereas the COMET (Frith et al. 2002) software code was downloaded, compiled on Linux, and run from the command line. The programs were run with the input-weight matrices that were determined from the C. elegans muscle genes. For both programs, we used a p-value threshold of 0.01, but different window sizes were tried. We required a minimum number of two sites to be in a window for MSCAN. For COMET, we tried several different average distances between sites.
Promoter::GFP constructs and gene expression characterization
To determine the in vivo expression pattern of muscle expressing candidate genes and the negative control gene set, we constructed promoter::GFP expression fusions (Yuan et al. 2000). Genes to be tested were fused with the promoterless GFP vector pLS43. pLS43 is an adaptation of pPD95.67 (A. Fire, S. Xu, J. Ahnn, and G. Seydoux, pers. comm.) with additional nuclear localization signals. Gene-specific primers were used to amplify the promoter cassettes from genomic DNA using a high-fidelity thermostable polymerase. Two PCR products were amplified for each gene. The first product was ∼500 bases long and had HindIII and BamHI sites introduced by the amplifying oligonucleotides and overlapped the second exon of the gene to be analyzed with the GFP gene remaining in-frame. The 500-base amplified product was digested with HindIII and BamHI, then cloned into pLS43 digested with HindIII and BamHI. The second amplified product was longer, containing ∼6 Kb (-6000 to -1) 5′ of the start Met and extending 3′ to overlap the first product by a minimum of 250 bases. Transformed lines carrying extrachromosomal arrays were generated as described (Mello et al. 1991) using the collagen gene rol-6 as a coinjection marker. Transformation was done using a 20:1:1 ratio of rol-6(pRF4)/experimental construct plasmid/large PCR fragment, respectively, at 200 ng/μL in 10 mM Tris, 1 mM EDTA (pH 8). These constructs were injected into N2 where recombination occurred, resulting in an in-vivo promoter::GFP. Rolling GFP-expressing progeny were isolated, then studied for in-vivo GFP expression.
Knockout of putative DNA regulatory elements
Gene-specific primers with HindIII or BamHI added were used to amplify the promoter cassettes from genomic DNA using a high-fidelity thermostable polymerase.
Mlc-2 (HindIII) 5′-GACACAAGCTTGGGACACATTATCTCT GCTGG-3′
Mlc-2 (BamHI) 5′-TCCAACATGTCCAAGGCCGCGGATCC GGGG-3′
Unc-89 (HindIII) 5′-GACACAAGCTTCGCCTAAAACACCG CAGCTG-3′
Unc-89 (BamHI) 5′-CCTTACCATCATGGCTAGTCGGGATC CGGGG-3′
The PCR-amplified fragments were digested with HindIII and BamHI and cloned into the HindIII and BamHI sites of the promoterless GFP vector pPD95.67 to create pLS45.005 and pLS45.018 for mlc-2 and unc-89, respectfully.
All knockout constructs were made by altering the above constructs using the QuickChange kit (Strategene) as described by the manufacturer. Underlined bases in the following oligonucleotides do not match wild-type sequence.
Mlc-2 (Motif 1) 5′-CACTCTATCTCAAACGGCAGTGATGGAATCTGCCACCCTCCACC-3′
Mlc-2 (Motif 2) 5′-CTACTAACTTTGCCCGCCGTGAGCTCGGCACCTCCTCTCGGTCTC-3′
Mlc-2 (Motif 3) 5′-GATCGGGACTTGGAAAAGGCTATGAGTTCATACTTTTCATGGGTG-3′
Unc-89 (Motif 3) 5′-CTCATAGTGGGGTGAGAACACTCATCGCGCAGACGCTAACAGAG-3′
All constructs were confirmed by sequencing using BigDye Terminator v3.0 (Applied Biosystems) on an ABI Prizm 3100 Genetic Analyzer (Applied Biosystems).
Transformed lines carrying extrachromosomal arrays were generated as described above, using a 20:1 ratio of rol-6(pRF4)/experimental construct at 200 ng/μL in 10 mM Tris, 1 mM EDTA (pH 8). These constructs were injected into N2 and the rolling, GFP-expressing animals were isolated, then studied for in-vivo GFP expression.
Photographic analysis of GFP expression
Animals that displayed the rolling phenotype of rol-6 were photographed; mlc-2 lines were staged at the larval L3 stage, unc-89 lines were staged at the larval L2 stage. Animals were anesthetized and mounted as described (McCarter et al. 1999), then photographed on a Bmax-BX60 (Olympus, Inc.) microscope with a Quantix (Photmetrics, Ltd.) cooled CCD camera using OpenLab version 3.0.9 software (Improvision, Ltd.) for microscope and photographic control. All images for the same gene were photographed under identical conditions for accurate comparison. InSpeck fluorescent bead standards (Molecular Probes, Inc.) were used to confirm that identical exposures were maintained.
The TIF images were analyzed on an iMac (Apple, Inc.) using public domain NIH Image program version 1.60 (developed at the U.S. National Institutes of Health and available on the internet at http://rsb.info.nih.gov/nih-image/). The histogram analysis of NIH Image generated the frequency of occurrences of each pixel value (0 = black, 255 = white) of each photograph. To help visualize the difference between the wild-type and knockout GFP-expression lines, pixel values that represented the black background and darkest gray of the GFP expression (pixel values <19) were excluded from the analysis, as were pixel values that exceeded those seen in most of the knockout lines (pixel values >118).
A graphic comparison between the wild-type and knockout expression lines was done by taking the nonexcluded pixel values (19–118) and resetting their values as 1–100 on the x-axis. The y-axis is calculated by multiplying the pixel value by the frequency of occurrence of that pixel value to yield the total intensity for that pixel value. The cumulative total intensity is the area under the curve or the sum of all of the total intensity for pixel values from 1 to 100.
Acknowledgments
We thank LaDeana Hillier and Todd Harris for providing the C. briggsae sequence data, including the comprehensive list of elegans-briggsae orthologs prior to publication. This work was supported by the National Institutes of Health (NIH) grants GM28755 and HG00249 to G.D.S.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.2961104.
References
- Bailey, T.L. and Noble, W.S. 2003. Computational detection of cis-regulatory modules. Bioinformatics 19: II5-II14. [DOI] [PubMed] [Google Scholar]
- Batzoglou, S., Patcher, L., Mesirov, J., Berger, B., and Lander, E.S. 2000. Human and mouse gene structure: Comparative analysis and application to exon prediction. Genome Res. 10: 950-958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benian, G.M., Tinley, T.L., Tang, X., and Borodovsky, M. 1996. The Caenorhabditis elegans gene unc-89, required for muscle M-line assembly, encodes a giant modular protein composed of Ig and signal transduction domains. J. Cell. Biol. 132: 835-848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benos, P.V., Bulyk, M.L., and Stormo, G.D. 2002. Additivity in protein-DNA interactions: How good an approximation is it? Nucleic Acids Res. 15: 4442-4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg, O.G. and von Hippel, P.H. 1987. Selection of DNA binding sites by regulatory proteins: Statistical-mechanical theory and application to operators and promoters. J. Mol. Biol. 193: 723-750. [DOI] [PubMed] [Google Scholar]
- Blackwell, T.K., Bowerman, B., Priess, J.R., and Weintraub, H. 1994. Formation of a monomeric DNA binding domain by Skn-1 bZIP and homeodomain elements. Science 266: 621-628. [DOI] [PubMed] [Google Scholar]
- Blumenthal, T.D., Evans, D., Link, C.D., Guffanti, A., Lawson, D., Thierry-Mieg, J., Thierry-Mieg, D., Chiu, W.L., Duke, K., Kiraly, M., et al. 2002. A global analysis of Caenorhabditis elegans operons. Nature 417: 851-854. [DOI] [PubMed] [Google Scholar]
- Brazma, A., Jonassen, I., Eidhammer, I., and Gilbert, D. 1998. Approaches to the automatic discovery of patterns in biosequences. J. Comput. Biol. 5: 279-305. [DOI] [PubMed] [Google Scholar]
- Chalfie, M., Tu, Y., Euskirchen, G., Ward, W.W., and Prasher, D.C. 1994. Green fluorescent protein as a marker for gene expression. Science 263: 802-805. [DOI] [PubMed] [Google Scholar]
- Chen, L., Krause, M., Sepanski, M., and Fire, A. 1994. The Caenorhabditis elegans MYOD homologue HLH-1 is essential for proper muscle function and complete morphogenesis. Development 120: 1631-1641. [DOI] [PubMed] [Google Scholar]
- Egan, C.R., Chung, M.A., Allen, F.L., Heschl, M.F., Van Buskirk, C.L., and McGhee, J.D. 1995. A gut-to-pharynx/tail switch in embryonic expression of the Caenorhabditis elegans ges-1 gene centers on two GATA sequences. Dev. Biol. 170: 397-419. [DOI] [PubMed] [Google Scholar]
- Frith, M.C., Spouge, J.T., Hanse, U., and Weng, Z. 2002. Statistical significance of clusters of motifs represented by position specific scoring matrices in nucleotide sequences. Nucleic Acids Res. 30: 3214-3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GuhaThakurta, D. and Stormo, G.D. 2001. Identifying target sites for cooperatively binding factors. Bioinformatics 17: 608-621. [DOI] [PubMed] [Google Scholar]
- GuhaThakurta, D., Schriefer, L.A., Hresko, M.C., Waterston, R.H., and Stormo, G.D. 2002a. Identifying muscle regulatory elements and genes in the nematode Caenorhabditis elegans. Proc. Pacific Symp. Biocomput. 7: 425-436. [DOI] [PubMed] [Google Scholar]
- GuhaThakurta, D., Palomar, L., Stormo, G.D., Tedesco, P., Johnson, T.E., Walker, D.W., Lithgow, G., Kim, S., and Link, C.D. 2002b. Identification of a novel cis-regulatory element involved in the heat shock response in Caenorhabditis elegans using microarray gene expression and computational methods. Genome Res. 12: 701-712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardison, R.C. 2004. Comparative genomics. PLoS Biol. 1: 156-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harfe, B.D. and Fire, A. 1998. Muscle and nerve-specific regulation of a novel NK-2 class homeodomain factor in Caenorhabditis elegans. Development 125: 421-429. [DOI] [PubMed] [Google Scholar]
- Harfe, B.D., Gomes, A.V., Kenyon, C., Liu, J., Krause, M., and Fire, A. 1998. Analysis of a Caenorhabditis elegans Twist homolog identifies conserved and divergent aspects of mesodermal patterning. Genes & Dev. 12: 2623-2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz, G.Z. and Stormo, G.D. 1999. Identifying DNA and protein patterns with statistically significant alignments of multiple sequences. Bioinformatics 15: 563-577. [DOI] [PubMed] [Google Scholar]
- Hughes, J.D., Estep, P.W., Tavazoie, S., and Church, G.M. 2000. Computational identification of cis-regulatory elements associated with groups of functionally related genes in Saccharomyces cerevisiae. J. Mol. Biol. 296: 1205-1214. [DOI] [PubMed] [Google Scholar]
- Jantsch-Plunger, V. and Fire, A. 1994. Combinatorial structure of a body muscle-specific transcriptional activator in Caenorhabditis elegans. J. Biol. Chem. 269: 27021-27028. [PubMed] [Google Scholar]
- Johansson, O., Alkema, W., Wasserman, W.W., and Lagergren, J. 2003. Identification of functional cluster of transcription factor binding sites in genome sequences: The MSCAN algorithm. Bioinformatics 19: i169-i176. [DOI] [PubMed] [Google Scholar]
- Krause, M., Harrison, S.A., Xu, S-Q., Chen, L., and Fire, A. 1994. Elements regulating cell- and stage-specific expression of C. elegans MyoD family homolog hlh-1. Dev. Biol. 166: 133-148. [DOI] [PubMed] [Google Scholar]
- Mann, H.B. and Whitney, D.R. 1947. On a test of whether one of two random variables is stochastically larger than the other. Ann. Math. Stat. 18: 50-60. [Google Scholar]
- Matys, V., Fricke, E., Geffers, R., Gößling, E., Haubrock, M., Hehl, R., Hornischer, K., Karas, D., Kel, A.E., Kel-Margoulis, O.V., et al. 2003. TRANSFAC: Transcriptional regulation, from patterns to profiles. Nucleic Acids Res. 31: 374-378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarter, J., Bartlett, B., Dang, T., and Schedl, T. 1999. On the control of oocyte meiotic maturation and ovulation in Caenorhabditis elegans. Dev. Biol. 205: 111-128. [DOI] [PubMed] [Google Scholar]
- McCue, L., Thompson, W., Carmack, C., Ryan, M.P., Liu, J.S., Derbyshire, V., and Lawrence, C.E. 2001. Phylogenetic footprinting of transcription factor binding sites in proteobacterial genomes. Nucleic Acids Res. 29: 774-782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mello, C.J., Kramer, J.M., Stinchcomb, D., and Ambros, V. 1991. Efficient gene transfer in C. elegans: Extrachromosomal maintenance and integration of transforming sequences. EMBO J. 10: 3959-3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzstein, M.M., Hengartner, M.O., Tsung, N., Ellis, R.E., and Horvitz, H.R. 1996. Transcriptional regulator of programmed cell death encoded by Caenorhabditis elegans genes ces-2. Nature 382: 545-547. [DOI] [PubMed] [Google Scholar]
- Moss, E.G., Lee, R.C., and Ambros, V. 1997. The cold shock domain protein LIN-28 controls developmental timing in C. elegans and is regulated by the lin-4 RNA. Cell 88: 637-646. [DOI] [PubMed] [Google Scholar]
- Okkema, P.G. and Fire, A. 1994. The Caenorhabditis elegans NK-2 class homeoprotein CEH-22 is involved in combinatorial activation of gene expression in pharyngeal muscle. Development 120: 2175-2186. [DOI] [PubMed] [Google Scholar]
- Okkema, P., Harrison, S.A., Plunger, V., Aryana, A., and Fire, A. 1993. Requirements for myosin gene expression and regulation in Caenorhabditis elegans. Genetics 135: 385-404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okkema, P., Ha, E., Haun, C., Chen, W., and Fire, A. 1997. The Caenorhabditis elegans NK-2 homeobox gene ceh-22 activates pharyngeal muscle gene expression in combination with pha-1 and is required for normal pharyngeal development. Development 124: 3965-3973. [DOI] [PubMed] [Google Scholar]
- Roy, P.J., Stuart, J.M., Lund, J., and Kim, S.K. 2002. Chromosomal clustering of muscle-expressed genes in Caenorhabditis elegans. Nature 418: 975-979. [DOI] [PubMed] [Google Scholar]
- Rushforth, A.M., White, C.C., and Anderson, P. 1998. Functions of the Caenorhabditis elegans regulatory myosin light chain genes mlc-1 and mlc-2. Genetics 150: 1067-1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider, T.D. and Stephens, R.M. 1990. Sequence logos: A new way to display CONSENSUS sequences. Nucleic Acids Res. 18: 6097-6100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider, T.D., Stormo, G.D., Gold, L., and Ehrenfeucht, A. 1986. Information content of binding sites of nucleotide sequences. J. Mol. Biol. 188: 415-431. [DOI] [PubMed] [Google Scholar]
- Schwartz, S., Kent, W.J., Smit, A., Zhang, Z., Baertsch, R., Hardison, R.C., Haussler, D., and Miller, W. 2003. Human–mouse alignments with BLASTZ. Genome Res. 13: 103-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staden, R. 1989. Methods for calculating the probabilities of finding patterns in sequences. Comput. Appl. Biosci. 5: 89-96. [DOI] [PubMed] [Google Scholar]
- Stein, L.D., Bao, Z., Blasiar, D., Blumenthal, T., Brent, M.R., Chen, N., Chinwalla, A., Clarke, L., Clee, C., Coghlan, A., et al., 2003. The genome sequence of Caenorhabditis briggsae: A platform for comparative genomics. PLoS Biol. 1: 166-192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stormo, G.D. 2000. DNA binding sites: Representation and discovery. Bioinformatics 16: 16-23. [DOI] [PubMed] [Google Scholar]
- Stormo, G.D. and Fields, D.S. 1998. Specificity, free energy and information content in protein-DNA interactions. Trends Biochem. Sci. 23: 109-113. [DOI] [PubMed] [Google Scholar]
- Wang, T. and Stormo, G.D. 2003. Combining phylogenetic data with co-regulated genes to identify regulatory motifs. Bioinformatics 19: 2369-2380. [DOI] [PubMed] [Google Scholar]
- Wasserman. W.W. and Fickett, J.W. 1998. Identification of regulatory regions which confer muscle-specific gene expression. J. Mol. Biol. 278: 167-181. [DOI] [PubMed] [Google Scholar]
- Wasserman, W.W., Palumbo, M., Thomson, W., Fickett, J.W., and Lawrence, C.E. 2000. Human–mouse genome comparisons to locate regulatory sites. Nat. Genet. 26: 225-228. [DOI] [PubMed] [Google Scholar]
- Waterston, R.H. 1988. Muscle. In The nematode Caenorhabditis elegans (ed. W.B. Wood), pp. 281-335. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY.
- Waterston, R.H., Lindblad-Toh, K., Birney, E., Rogers, J., Abril, J.F., Agarwal, R., Ainscough, R., Alexandersson, M., An, P., Antonarakis, S.E., et al. 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 240: 520-562. [DOI] [PubMed] [Google Scholar]
- Workman, C.T. and Stormo, G.D. 2000. ANN-SPEC: A method for discovering transcription factor binding sites with improved specificity. Pacific Symp. Biocomput. 5: 467-478. [DOI] [PubMed] [Google Scholar]
- Yuan, A., Dourado, M., Butler, A., Walton, N., Wei, A., and Salkoff, L. 2000. SLO-2, a K+ channel with an unusual Cl- dependence. Nature Neurosci. 3: 771-779. [DOI] [PubMed] [Google Scholar]
- Yutzey, K. and Konieczny, S. 1992. Different E-box regulatory sequences are functionally distinct when placed within the context of the troponin I enhancer. Nucleic Acids Res. 20: 5105-5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zar, J.H. 1974. Biostatistical analysis, pp. 108-113. Prentice-Hall, Inc., Englewood Cliffs, NJ.
- Zhang, J-M., Chen, L., Krause, M., Fire, A., and Paterson, B.M. 1999. Evolutionary conservation of MyoD function and differential utilization of E proteins. Dev. Biol. 206: 465-472. [DOI] [PubMed] [Google Scholar]
Web site references
- http://ural.wustl.edu/~dg/Nematode_Muscle_Regulation.html; Characterizing muscle expression of genes from Table 3.
- http://ftp.genome.washington.edu/RM/webrepeatmaskerhelp.html; RepeatMasker Web site.
- http://tfscan.cgb.ki.se/cgi-bin/MSCAN; MSCAN Web site.
- http://rsb.info.nih.gov/nih-image/; Fluorescence image analysis software.