

Abstract
Aim
To evaluate the pharmacokinetics, pharmacodynamics, nasal tolerance and effects on sedation of a highly concentrated aqueous intranasal midazolam formulation (Nazolam) and to compare these to intravenous midazolam.
Methods
In this four‐way crossover, double‐blind, double‐dummy, randomized, placebo‐controlled study, 16 subjects received 2.5 mg Nazolam, 5.0 mg Nazolam, 2.5 mg intravenous midazolam or placebo on different occasions. Pharmacokinetics of midazolam and α‐hydroxy‐midazolam were characterized and related to outcome variables for sedation (saccadic peak velocity, the Bond and Lader visual analogue scale for sedation, the simple reaction time task and the observer's assessment of alertness/sedation). Nasal tolerance was evaluated through subject reporting, and ear, nose and throat examination.
Results
Nazolam bioavailability was 75%. Maximal plasma concentrations of 31 ng ml−1 (CV, 42.3%) were reached after 11 min (2.5 mg Nazolam), and of 66 ng ml−1 (coefficient of variability, 31.5%) after 14 min (5.0 mg Nazolam). Nazolam displayed a significant effect on OAA/S scores. Sedation onset (based on SPV change) occurred 1 ± 0.7 min after administration of 2.5 mg intravenous midazolam, 7 ± 4.4 min after 2.5 mg Nazolam, and 4 ± 1.8 min after 5 mg Nazolam. Sedation duration was 118 ± 95.6 min for 2.5 mg intravenous midazolam, 76 ± 80.4 min for 2.5 mg Nazolam, and 145 ± 104.9 min for 5.0 mg Nazolam. Nazolam did not lead to nasal mucosa damage.
Conclusions
This study demonstrates the nasal tolerance, safety and efficacy of Nazolam. When considering the preparation time needed for obtaining venous access, conscious sedation can be achieved in the same time span as needed for intravenous midazolam. Nazolam may offer important advantages in conscious sedation.
Keywords: conscious sedation, intranasal, midazolam, pharmacodynamics, pharmacokinetics
What is Already Known about this Subject
The nasal route appears to be a very convenient route of administration for conscious sedation and for the lay treatment of epileptic seizures, which may save time until intravenous access can be established. Most previous studies were performed with intranasal formulations that led to nasal run‐off or nasal mucosal damage, or failed due to flaws in the experimental design. A study with this particular formulation had not been done previously and is essential for the establishment of intranasal midazolam in conscious sedation and as a treatment for epilepsy.
What this Study Adds
This study reports on the pharmacokinetic and pharmacodynamic properties of an aqueous nasal formulation of midazolam. This new formulation is well tolerated and capable of delivering small enough administration volumes to be fully absorbed by the nasal mucosa, yet allowing for clinically relevant systemic midazolam concentrations in healthy adult subjects. Application of this novel formulation may offer important advantages in conscious sedation and epilepsy.
Tables of Links
| TARGETS | |
|---|---|
| Ligand‐gated ion channels 2 | G protein‐coupled receptors 3 |
| GABAA | α1D‐adrenoceptor |
| α1A‐adrenoceptor | |
| TRH2 receptor |
| LIGANDS | |
|---|---|
| Midazolam |
These Tables lists key protein targets and ligands in this article that are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 1, and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 2, 3.
Introduction
Midazolam is a short‐acting benzodiazepine with anxiolytic, sedative, anticonvulsant and skeletal muscle relaxant properties 4. Due to its fast onset and recovery profile, it is the preferred medication for obtaining conscious sedation and management of epileptic seizures 5, 6. Midazolam is used in a wide range of indications for conscious sedation, including sedation for the majority of outpatient diagnostic, therapeutic and endoscopic procedures, and sedation for the preparation of general anaesthesia in hospitalized patients 5. Administration of midazolam is generally intravenous as other administration routes such as oral, rectal, subcutaneous and buccal lead to a delayed onset of efficacy and to a large interindividual variability in efficacy onset 7, 8, 9, 10. The nasal route therefore appears to be a very convenient route of administration for conscious sedation 11 and for the lay treatment of acute epileptic seizures 12. However, until now, no nasal formulation is available that: (1) has a volume that is small enough to be effectively received and retained by the nasal mucosa; and (2) does not lead to nasal toxicity 13, 14, 15, 16, 17, 18, 19, 20, 21, 22.
As conscious sedation is usually applied in procedural settings, its induction time is critical for optimizing the efficient use of hospital personnel and utilities. For this reason, intravenous administration of midazolam currently is preferred over nonintravenous administration routes 5. Despite its fast and predictable onset of action, intravenous administration has important disadvantages, including the requirement of intravenous access and the intermittent dosing protocol, which is necessary to avoid high initial peak midazolam plasma concentrations 4.
Nasal administration of midazolam is a simple, useful and reliable alternative to the parenteral route 11. It offers several practical advantages, as it allows for direct, easy and needle free administration, and can be safely administered without the need for professional assistance 11, 12. Compared to intravenous administration, nasal administration is particularly advantageous in children, patients with needle phobia, patients with varicose (difficult accessible) veins, and in emergency room settings, in dentistry and other nonhospital settings. In addition, it provides potential for rapid systemic drug absorption and quick onset of action without initial high peak concentrations, that in the presence of comedication such as opioids may lead to adverse events 4.
Unfortunately, nasal delivery of midazolam has not been very successful until now 13, 14, 15, 16, 17, 18, 19, 20, 21. This is due to the absence of solvents that are able to dissolve midazolam at efficacious dosages without leading to nasal mucosal damage 13, 14, 15, 16, 17, 18, 19, 20, 21. The maximal volume of nasal application is ideally restricted to approximately 100 μL 22, requiring the efficacious dose of midazolam to be dissolved within this volume. Higher volumes lead to nasal drop‐off or swallowing, which in turn may lead to gastrointestinal absorption followed by first‐pass metabolism resulting in lower and unpredictable concentrations, a high variability in midazolam bioavailability, and a relatively long onset of action. This in turn can cause overdosing if a second dose is applied because the first one did not act fast enough. The most concentrated form of the midazolam injection fluid only contains 5 mg ml−1. As a result, for conscious sedation, at least 1 ml (= 5 mg midazolam) needs to be administered intranasally, a volume that is 10 times larger than the nasal mucosa can receive and retain. This therefore leads to leakage and/or swallowing of the fluid, which in turn leads to inaccurate and inadequate dosing. The intranasal administration of midazolam requires highly concentrated solutions with a high bioavailability, in order to achieve clinically relevant plasma concentrations through the administration of a single nasal spray 13, 14, 15, 16, 17, 18, 19, 20, 21. Attempts to overcome this limitation by formulating midazolam in organic solvents or absorption enhancers that allow for dosing volumes as low as 100 μl have largely failed due to the fact that these solvents are typically irritating to the highly sensitive and easily disrupted nasal mucosa 13, 14, 15, 16, 17, 18, 19, 20, 21, and/or require administration volumes that are much larger than 100 μl 14, 15, 16.
Recently, a highly concentrated, aqueous midazolam formulation (Nazolam) that allows dosing of 100 μl or below has become available. Because of the aqueous nature of the midazolam formulation, nasal tolerance was expected to be good. If proven successful, this aqueous midazolam administration will be the first to address all limitations of currently intravenous and intranasal applications of midazolam. In this study, the pharmacokinetics (PK), efficacy and tolerability of this nasal midazolam formulation were evaluated and compared to intravenous midazolam.
Methods
Study design
This was a randomized, double‐blind, double‐dummy placebo‐controlled, four‐way crossover study in 16 healthy volunteers. The study was conducted in adherence to the guidelines of the International Conference on Harmonisation Guideline for Good Clinical Practice and in accordance with the principles of the Declaration of Helsinki. The study was performed at the Centre for Human Drug Research in Leiden, the Netherlands, and approved by the local ethics committee of Leiden University Medical Center (Leiden; ref: P10.215) and registered with EudraCT (ref: 2010–023 425‐38). The subjects consented in writing to the study after full explanation of what was involved.
Subjects
Inclusion criteria for this study were for healthy male or female volunteers aged 18–55 years, with a body mass index of 18–33 kg m−2. All subjects had to be willing and able to comply with study procedures. Exclusion criteria included: history of central nervous system or psychiatric disease; history of drug, substance and/or alcohol abuse; and abnormal findings on screening medical history, physical examination, electrocardiography (ECG), vital signs, and/or blood and urine laboratory profile. Subjects with anatomical anomalies causing obstruction of the nares, recent (<4 weeks) nose bleeds, or with a history of chronic nasal obstruction or clinically significant nasal surgery that could affect absorption of or tolerance to midazolam were excluded. Subjects with clinically significant upper respiratory infection, common cold or flu‐like symptoms and/or rhinitis at screening were also excluded. Subjects were not allowed to use any medication which could affect the metabolism of midazolam or the performance of central nervous system (CNS) measurements from 2 weeks prior to the start of the study days. Subjects were not allowed to consume more than 8 units of xanthine‐containing products per day. Subjects had to refrain from consumption of xanthine‐ or alcohol‐containing products and smoking from 1 day prior to admission until the end of the study day. On study days, intake of medication, alcohol or drugs was questioned, and a urine drug screen, pregnancy test and an alcohol breath test were performed before any study‐related procedures were started.
Study treatments
Study treatments were administered as a unit‐dose nasal spray containing 2.5 or 5.0 mg midazolam (Nazolam) or the same formulation without midazolam, and intravenous solution containing 2.5 mg midazolam or saline. On each study day, subjects received both an intranasal and intravenous study drug administration. The subjects on each study occasion received one of four treatments: (1) 2.5 mg intranasal midazolam and intravenous placebo; (2) 5 mg intranasal midazolam and intravenous placebo; (3) intranasal placebo and 2.5 mg intravenous midazolam; and (4) intranasal placebo and intravenous placebo. During administration subjects were asked to be seated and not to move nor to inhale. Delivery of the unit dose spray was assessed by the physician administering the nasal doses. The sequence of treatments was randomized, defined by a Williams square design, and study days were separated by washout periods of at least 6 days. Doses were administered in the nonfasted state.
The recommended starting dose for conscious sedation is 2–2.5 mg 4. The lowest dose administered intranasally was therefore 2.5 mg, based on the assumption of a high bioavailability. In addition, a 5‐mg dose was tested because this dose is relevant in the treatment of epilepsy. Moreover, by studying both dosages, an indication of dose proportionality and dose–response relationships of the intranasal formulation could be obtained. The concentration of the aqueous midazolam spray was 55.6 mg ml−1 midazolam HCl (50 mg midazolam base per ml) and the volume was 50 μl for the 2.5‐mg dose and 100 μl for the 5‐mg dose. The spray was administered in the same nostril throughout the study by the supervising physician.
The dosing regimen for the administration of 2.5 mg intravenous midazolam was 1 mg/30 seconds, which is in accordance with the approved label of midazolam for the indication of conscious sedation 4.
PK methods
Venous blood samples for PK analyses were obtained via an indwelling catheter before administration and at 1 min and 15 s, and at every 3 min (until 30 min), every 10 min (until 60 min), every 30 min (until 2 h), and at 3, 4, 6, 8 and 12 h after drug administration. The Vacutainer tubes with lithium heparin containing the blood samples were gently mixed by inversion (~8–10 times) and kept on wet ice thereafter. The samples were processed by centrifugation (10 min at 2–8°C at 2000 × g) within 30 min after the sample was drawn and the plasma was stored −80°C until analysis. Bioanalytical analysis was performed by ABL (Analytisch Biochemisch Laboratorium BV, Assen, The Netherlands) using a validated method on a validated API 4000 LC–MS/MS system. Plasma concentrations of midazolam and its active metabolite α‐hydroxy‐midazolam were determined using liquid chromatography coupled with tandem mass spectrometry. Quality control (QC) concentrations included QC‐Low (target midazolam or α‐hydroxy‐midazolam concentrations in human heparin plasma of 0.300 ng ml−1), QC‐Medium (target concentration of 3.00 ng ml−1) and QC‐High (target concentration of 75.0 ng ml−1). Assay specifics included acceptable precision (total coefficients of variability [CVs] of 15% for all QC target concentrations), good accuracy (mean absolute biases values for all QC target concentrations of 15%) and adequate incurred sample reproducibility (difference of ≤20% for all reanalyzed midazolam samples and 98% of reanalyzed α‐hydroxy‐midazolam samples). The analytical range of the assay for both the parent and the metabolite was 0.100–100 ng ml−1. The analyses were conducted in accordance with good laboratory practice principles.
PD methods
The ‘Neurocart’ is a battery of sensitive tests for a wide range of CNS domains that was developed to examine different kinds of CNS‐active drugs including benzodiazepines 23, 24, 25, 26. All tests were performed twice at baseline, and repeated in the following order at the same time points as the PK blood sampling. Measurements were performed in a quiet room with ambient illumination with only one subject per session in the same room.
Saccadic peak velocity
Saccadic peak velocity (SPV) is one of the most sensitive parameters for sedation 27, 28, 29 and was therefore used to evaluate the onset and duration of pharmacological effect of intranasal midazolam. The use of a computerized measurement of saccadic eye movements has been described elsewhere 29, 30, 31. The definition of the onset and duration of pharmacological effect (sedation) was based on the subject's individual variability in SPV. Onset of sedation was defined as the (linearly interpolated) time point at which the SPV reached minus 2 standard deviations (SDs) of the prevalue (baseline) SPV level for the occasion. Duration was defined as the total amount of time that the response was below the minus 2 SDs threshold; this total time could be made up of a number of episodes if the threshold was crossed repeatedly before complete termination of the effect.
Visual analogue scales
Visual analogue scales (VASs) as originally described by others 32, have been previously used to quantify subjective effects of benzodiazepines 29 and to evaluate sedative effect of both intranasal and intravenous midazolam 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45. In this study, by using the Bond and Lader VAS, the ‘directions’ of different scales on a form were alternated, to avoid habitual scoring by subjects 46.
Simple reaction time task
The simple reaction time task (SRTT) measures the attention and speed of information processing of the subject. In this task, participants view a black computer screen. At random intervals (0.5–1.5 s), a white circle appears in the centre of the computer screen. Subjects were instructed to press the space bar with the index finger of their dominant hand each time the circle appears. They were instructed to respond as quickly as possible after appearance of the circle. A total of 40 circles were presented, and the duration of the task was approximately 1 min. The outcome of the task is the time between stimulus display and response. It has been shown to respond to several classes of sedative drugs 47. Several previous studies also showed the positive application of SRTT in the investigation of midazolam's sedative effect 47, 48, 49. The SRTT can be regarded as a clinically relevant measure that represents the level of dysfunctioning that may be caused by sedation and was included in this trial to evaluate whether recovery from sedation was similar for intravenous and intranasal dosing.
Observer's assessment of alertness/sedation
The observer's assessment of alertness/sedation scale (OAA/S) was previously developed to objectively measure the level of alertness in subjects who are sedated. The OAA/S Scale has been shown to be reliable and valid and to be sensitive to the level of midazolam administered 51. The OAA/S has been used extensively in studies with intravenous midazolam 34, 35, 39, 43, 44, 45, 52.
Adverse event profile
Adverse events (AE) monitoring, 12‐lead ECGs, and laboratory safety tests were conducted at intervals throughout the study. Transcutaneous oxygen saturation levels were monitored during the first 6 h after administration and blood pressure and heart rate until discharge (12 h after administration).
Nasal tolerance assessments
The nasal adverse event profile was monitored by inspection of the nasal mucosa by a physician trained by an ear, nose and throat medical specialist and by subject self‐assessment (subjective monitoring of congestion, irritation, pain, runniness and loss of smell). Nasal symptoms were assessed prior to and after nasal application and, if present, specified as: (1) congestion or stuffiness; (2) irritation or itchiness; (3) runniness; (4) pain or discomfort; and/or (5) loss of or abnormal smell. In case of a nasal symptom, the severity was graded as: (1) mild symptoms (minor awareness of symptoms, lasting up to 1 h); (2) moderate symptoms (moderate awareness of symptoms, lasting up to 12 h), or (3) severe symptoms (strong awareness of symptoms, lasting more than 12 h). In addition, local nasal tolerance was assessed by means of a regular nasal examination using the following scoring system: (1) no visible abnormality; (2) mild abnormality (<1 cm of erythema and/or swelling and/or other visible abnormality); (3) moderate abnormality (1–2 cm of erythema and/or swelling and/or other visible abnormality); or (4) severe abnormality (>2 cm of erythema and/or swelling and/or other visible abnormality). All observed or reported AEs were recorded for all subjects and classified as mild, moderate or severe, their relationship to study drug was assessed by the investigator.
PK analysis
PK parameters for midazolam and α‐hydroxy‐midazolam were estimated using noncompartmental modelling (WinNonlin 5.2; Pharsight, Mountain View, CA, USA). The distributions of the dose‐normalized PK parameters were compared using anova. For each subject and each treatment, the following PK parameters were determined: maximum plasma concentration (C max), the time at which the maximum concentration was reached, relative to dosing (T max), the terminal halflife (t1/2) and the area under the plasma concentration‐time curve up to the last quantifiable concentration (AUClast) and extrapolated to infinity (AUCinf). Descriptive statistics were reported for each PK parameter.
Statistical methods
Sample size was determined based on a presumed onset of sedative effect of midazolam as defined based on a decrease in saccadic SPV of >2SD from baseline. In a previous study performed by our research group, the intersubject CV of the time of onset defined on the basis of a decline in SPV was 62.5% 53. In this previous study the intrasubject CV could not be calculated; however, assuming the intrasubject CV to be smaller than the intersubject CV, an intrasubject CV of 50% was used for sample size calculations. Sample size calculations were performed in nQuery (version 7.0). It was determined that a sample size of 16 would have 80% power to detect a difference in mean time of onset of sedative effect of 3.283 min, assuming a standard deviation of differences of 4.380, using a paired t test with a 0.05 two‐sided significance level.
SRTT and OAA/S data were log‐transformed prior to analysis to correct for the expected log‐normal distribution of the data and analysis was performed on log‐transformed data. Repeatedly measured PD data (SPV, VAS, SRTT and OAA/S) were compared with a mixed model analysis of variance (using SAS PROC MIXED) with fixed factors treatment, period, time and treatment by time, random factors subject, subject by treatment and subject by time, and the baseline value (average over all measurements at or before time = 0) as covariate. The contrast between the midazolam treatments and placebo were calculated within the statistical model. For onset of sedation based on SPV, the SD of the SPV during the whole placebo period was calculated for each subject, and the threshold of sedation was determined as the baseline value per period minus 2 SDs. Onset and duration of sedative effect were compared between treatment groups, assuming that the effect sizes of the different treatment groups were comparable. As OAA/S is a categorical variable, it was not assumed to be normally distributed. Therefore, an additional analysis was performed using the GLIMMIX procedure. In this procedure, all OAA/S data (including placebo) after dosing (time = 0) followed a multinomial distribution and were compared with a mixed model analysis of variance with fixed factors treatment and period and random factors subject and subject by time. All calculations were performed using SAS for windows V9.1.3 (SAS Institute, Inc., Cary, NC, USA).
Results
Subjects
Sixteen healthy subjects (eight male, eight female) were enrolled in this study. They were on average aged 26 years (range 19–53 years), and had an average body mass index of 23.2 kg m−2 (range 19.6–28.1 kg m−2). All subjects had negative predose urine tests for drugs of abuse, including benzodiazepines. Concomitant medication used during the study period included paracetamol (up to 1.0 g per day), noscapine, acetylsalicylic acid and xylometazoline (one subject, stopped more than 1 day before study drug administration). All subjects completed the study.
PK results
The PK parameters and mean concentration–time profiles of midazolam and α‐hydroxy‐midazolam are shown in Table 1 and Figure 1. The intravenous data of two subjects had to be excluded due to sampling failures on two occasions (both 2.5 mg intravenous midazolam). Inclusion of the data gathered during these occasions did not significantly change the PK parameter estimates. Doubling of the intranasal midazolam dose resulted in dose‐proportional increases in AUC and maximum concentration (AUC: mean increase, 2.0‐fold; Cmax: mean increase, 2.2‐fold). Dose‐normalized Cmax and AUC(0‐t) and AUC(0‐∞) were higher for the intravenous formulation than for the intranasal formulations. The overall concentration–time profiles of the intranasal formulations showed no second peak and the formation of metabolite was low and the relative amounts formed compared to the parent compound were comparable with the formation of metabolite after intravenous administration. Mean ratio of α‐hydroxy‐midazolam AUC to midazolam AUC after intranasal midazolam administration was 0.2 for all formulations and dosages.
Table 1.
Pharmacokinetic parameters of midazolam and α‐hydroxy‐midazolam in healthy subjects after administration of a single dose of 2.5 mg midazolam intravenous (IV) or 2.5 or 5 mg midazolam intranasal (IN)
| Treatment | AUC 0–∞ (ng h ml −1 ) | C max ( ng ml −1 ) | t 1/2 (h) | T max (min) | F | |
|---|---|---|---|---|---|---|
| Midazolam | Midazolam 2.5 mg IV | 93.9 (33.8) | 219.2 (68.1) | 3.6 (29.4) | 2.0 (1.2–3.0) | 1 |
| Midazolam 2.5 mg IN | 65.6 (49.0)** | 30.6 (42.3) | 6.3 (123.4)* | 10.9 (6.0–24.0) | 0.74 (0.28–1.85) | |
| Midazolam 5 mg IN | 131.9 (26.0) | 66.2 (31.5) | 4.3 (31.0) | 13.8 (9.0–24.0) | 0.76 (0.45–1.20) | |
| α‐hydroxy‐midazolam | Midazolam 2.5 mg IV | 15.83 (36.9) | 6.1 (37.2) | 4.6 (45.5) | 14.4 (9.0–21.0) | |
| Midazolam 2.5 mg IN | 10.9 (54.1) | 2.4 (55.5) | 5.3 (40.0) | 45.4 (24.0–240.0) | ||
| Midazolam 5 mg IN | 24.0 (37.5) | 5.3 (34.5) | 6.3 (44.2) | 50.6 (21.0–121.2) |
For one (*) or two (**) subjects, adjusted r‐squared was <0.800 and/or %AUC extrapolated to infinity was >20%. When parameters were excluded, AUC0‐infinity was 56.5 (37.9) ng h ml−1 and t1/2 was 4.4 (24) hours.
AUC, C max and half‐lives are expressed as geometric mean (CV%); T max and F are expressed as geometric mean (range); AUC, area under the curve; C max, peak plasma concentration; CV, coefficient of variation; F, bioavailability; IV, intravenous; IN, intranasal; t1/2, elimination half‐life; Tmax, time to reach Cmax
Figure 1.
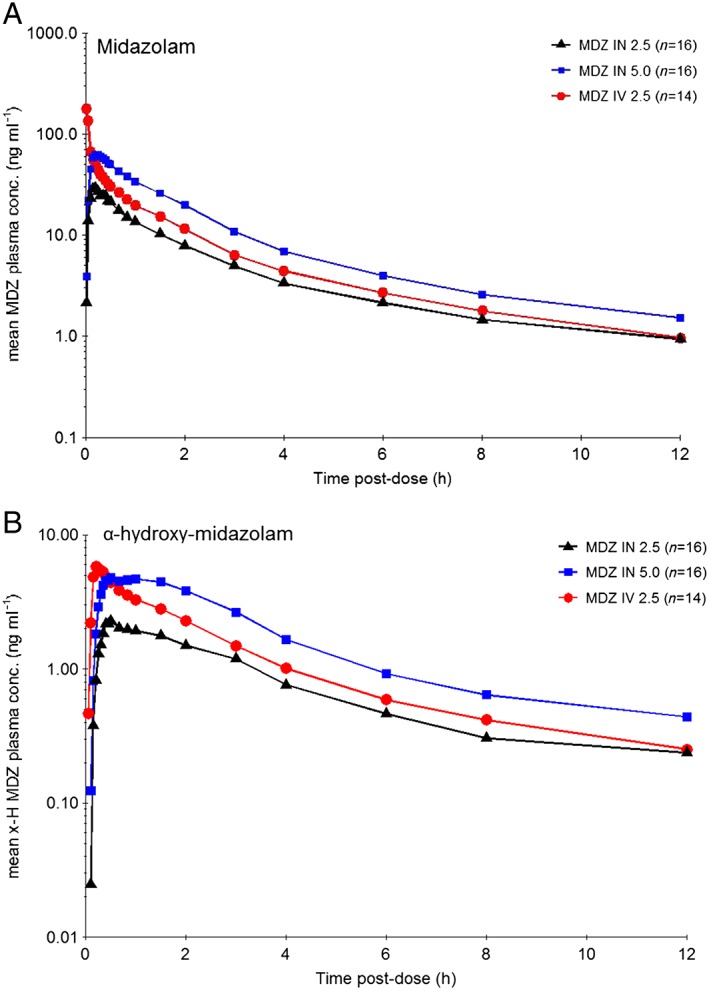
Geometric mean concentration‐time profiles (log‐linear) of (A) midazolam (MDZ) and (B) α‐hydroxy‐midazolam after intravenous (IV) (2.5 mg) and intranasal (IN) (2.5 and 5.0 mg) midazolam administration in healthy subjects
The midazolam intravenous administration displayed a nine‐fold ratio between the highest and lowest observed C max value, whereas the ratio between the highest and lowest C max for the intranasal 2.5 mg administration was seven‐fold, and for the 5.0 mg intranasal administration three‐fold, which led to the CV in C max included in Table 1. The bioavailability of intranasal midazolam with 74% for the 2.5 mg dose and 76% for the 5.0 mg dose indicates a constant bioavailability over the investigated dose range.
PD results
SPV
A marked and time‐dependent decrease in SPV was seen after midazolam administration until 3 h after administration (Figure 2). There was a statistically significant difference in SPV between midazolam 2.5 mg intranasal and 5.0 mg intranasal (P < 0.001; 35.3, 95% CI = 20.6, 50.0).
Figure 2.
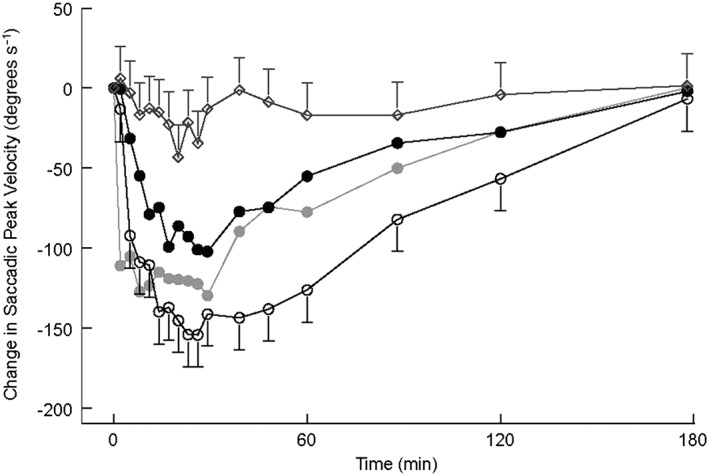
Saccadic peak velocity Least Squared Means (LSMs) change from baseline profile with 95% confidence interval as error bars (first 3 h after administration). Open rhombus represents placebo; grey closed circle represents midazolam 2.5 mg intravenous (IV); black closed circle represents midazolam 2.5 mg intranasal (IN); open circle represents midazolam 5.0 mg IN
Onset of action of midazolam (as defined by a decrease in SPV of more than 2SD below baseline) occurred 7 ± 4.4 min after administration of midazolam 2.5 mg intranasal, 4 ± 1.8 min after midazolam 5.0 mg intranasal. Onset of action after administration of midazolam 2.5 intravenous occurred on average 1 ± 0.7 min after administration. There was a statistically significant difference between midazolam 2.5 mg intranasal and 2.5 mg intravenous (P < 0.001; 6.2, 95% CI = 4.2, 6,2), midazolam 5.0 mg intranasal and 2.5 mg intravenous (P = 0.007; 2.7, 95% CI = 0.8, 4.7) and midazolam 2.5 mg intranasal and 5.0 mg intranasal (P = 0.001; 3.5, 95% CI = 1.5, 5.4).
Duration of action as defined by a 2SD decrease in SPV was on average 76 ± 80.4 min after administration of midazolam 2.5 mg intranasal and 145 ± 104.9 min of midazolam 5.0 mg intranasal. Duration of action was on average 118 ± 95.6 min after 2.5 mg intravenous midazolam. There was a statistically significant difference between 2.5 mg intranasal and intravenous midazolam (P = 0.03; −38.5, 95% CI = –60.6, −3.9) and between the two intranasal dose levels (P = 0.001; −53.4, 95% CI = –69.9, −28.0), but not between 2.5 mg intravenous and 5.0 mg intranasal.
VASs
Subjective alertness decreased after administration in a time‐dependent manner in all midazolam groups. There was a statistically significant difference between the intranasal dose levels (P = 0.0009; 1.9, 95% CI = 0.8, 2.9; see Figure 3). There were no effects on VAS Calmness or VAS Mood.
Figure 3.
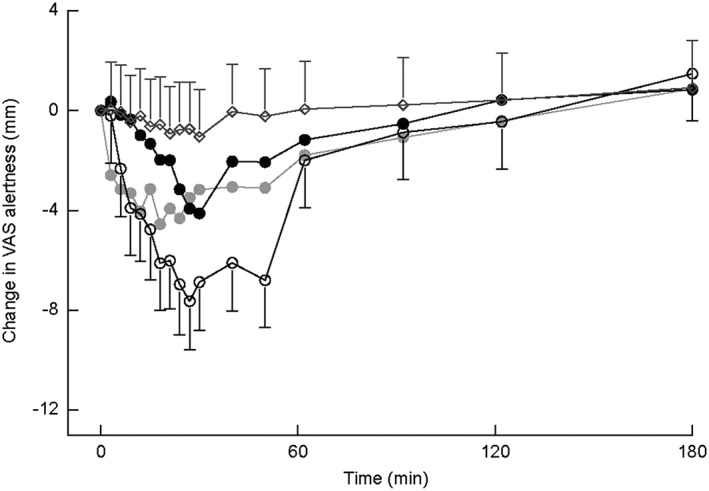
Visual analogue scale Alertness LSMs change from baseline profile with 95% confidence interval as error bars (first 3 h after administration). Open rhombus represents placebo; grey closed circle represents midazolam 2.5 mg intravenous (IV); black closed circle represents midazolam 2.5 mg intranasal (IN); open circle represents midazolam 5.0 mg IN
Simple reaction time task
Midazolam had a marked effect on the reaction time with a statistically significant difference between the intranasal dose levels (P = 0.0005; −10.8, 95% CI = –16.2, −5.2; see Figure 4).
Figure 4.
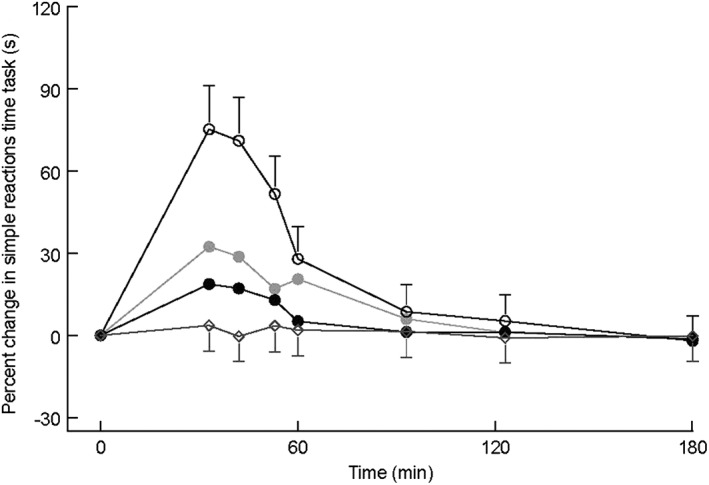
Simple reactions time task LSMs change from baseline profile with 95% confidence interval as error bars (first 3 h after administration). Open rhombus represents placebo; grey closed circle represents midazolam 2.5 mg intravenous (IV); black closed circle represents midazolam 2.5 mg intranasal (IN); open circle represents midazolam 5.0 mg IN
Observer's assessment of alertness/sedation
Intranasal midazolam displayed a significant effect on sedation as measured using OAA/S. Levels of sedation after midazolam intranasal 2.5 mg and midazolam intravenous 2.5 mg administration were comparable, whereas midazolam intranasal 5.0 mg led to higher sedation levels (see Figure 5). The odds ratio (defined as the chance [odds] that a subject scored an OAA/S of 1 [awake/orientated, indicating no sedation] during one treatment vs. the other) was 2.3 (P < 0.0001; 95% CI = 1.63, 3.18) for the contrast midazolam 2.5 mg intranasal vs. midazolam 2.5 mg intravenous, 0.8 (P = 0.30; 95% CI = 0.62, 1.16) for the contrast midazolam 5.0 mg intranasal vs. midazolam 2.5 mg intravenous, and 2.7 (P < 0.0001; 95% CI = 1.92, 3.75) for the contrast midazolam 2.5 mg intranasal vs. midazolam 5.0 mg intranasal.
Figure 5.
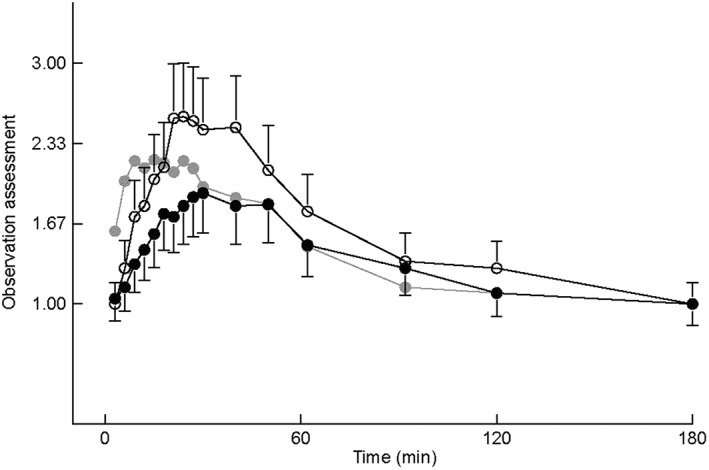
Observation assessment LSMs profile with 95% confidence interval as error bars (first 3 h after administration). Grey closed circle represents midazolam 2.5 mg intravenous (IV); black closed circle represents midazolam 2.5 mg intranasal (IN); open circle represents midazolam 5.0 mg IN. Score 1 = awake/oriented; score 2 = drowsy/normal speech; score 3 = slow reaction to verbal; score 4 = inability 2 saccades
Adverse event profile
General
There were few AEs. They were mild, transient and equally distributed over the 2.5 mg and 5.0 mg intranasal and intravenous groups. The most common AEs were somnolence and headache, which were reported 46 and 13 times in total and in 31–94% (somnolence) and 6–25% (headache) of subjects (including placebo) and reported in all treatment groups. Administration of a single dose of midazolam did not result in clinically significant changes in physical findings or ECG recordings.
In general, types of adverse events for the intranasal and intravenous formulations of 2.5 mg midazolam were similar. There were more cases of diplopia in the intranasal treatment groups (one case in 2.5 mg group and six cases in 5 mg group), which could be explained by midazolam's characteristic (dose‐related) benzodiazepine effects on GABAA‐receptors in the central nervous system. Since GABAA‐receptors do not occur peripherally, it is unlikely that this is due to local effects of the intranasal formulation. The larger number of cases of sleep‐related symptoms in the 5.0 mg intranasal midazolam group probably results from the higher AUC in this treatment group. There were more cases of attention disturbances in the 2.5 mg intranasal midazolam treatment group compared to the 5.0 mg intranasal and 2.5 mg intravenous midazolam groups. Attention disturbance represents the lower end of the spectrum of GABAA effects, and subjects in the low‐dose intranasal treatment group may have not been sedated to such a level that somnolence occurred, but enough to experience attention problems.
Nasal AEs
No significant abnormalities were found during nasal examination in any of the subjects. Mild and transient visible abnormalities (<1 cm) were observed for one subject almost 12 h after administration of 2.5 mg intranasal midazolam and for one subject 2 h after administration of 5.0 mg intranasal midazolam, which all resolved spontaneously. Mild nasal symptoms were observed in two subjects 1 h after administration of 5.0 mg intranasal midazolam, which resolved within 1–2 h. In one subject, these symptoms may be related to mild runniness already observed before dosing. One subject reported rhinorrhoea, and one reported sneezing after 2.5 mg intranasal midazolam. After 5.0 mg intranasal midazolam, one subject reported cough, one reported irritation, one reported observing some blood in a tissue (after blowing his nose) on the day after administration (during the study day no nasal symptoms or visible abnormalities were observed) and two reported sneezing.
Oxygen saturation
No clinically relevant decreases in transcutaneous oxygen saturation or blood pressure were observed.
Discussion
This study is the first to report on the PK and effects on sedation of Nazolam. Nazolam is a new aqueous nasal formulation of midazolam that does not lead to nasal tissue damage and delivers small enough volumes to be fully absorbed by the nasal mucosa, yet containing sufficiently high concentrations of midazolam to establish clinically relevant systemic midazolam concentrations. For all midazolam treatment groups, effects were seen on PD outcome variables of sedation and clinically relevant levels of sedation as measured using OAA/S (≥ score 2, or drowsy/normal speech) were achieved within minutes after administration. It is therefore clear that use of Nazolam is an effective, convenient and safe method of inducing conscious sedation for a wide range of applications.
Most previous studies used formulations that led to nasal run‐off or nasal mucosal damage, or were hampered by flaws in the experimental design. Single administration of Nazolam, however, was well tolerated and safe in healthy adult subjects. Nasal symptoms were mild and transient and only observed in two patients. Furthermore, in the slightly acidic, mainly aqueous Nazolam formulation, midazolam is partly present in an open‐ring structure formed by the acid‐catalyzed ring opening of the 4,5‐double bond of the diazepine ring. Once administered to the physiological pH buffered nasal mucosa, the pH of the formulation rapidly increases. This causes midazolam to revert to its pharmacologically active closed ring structure that, due to the low solubility of the closed‐ring structure, causes a pH driven transport of midazolam to the nasal mucosa that leads to efficacy onset in a similar time window as observed after intravenous administration 4. Mean absolute bioavailability of Nazolam was high (approximately 75%) and clinically effective concentrations were reached within minutes after nasal administration. Observed maximal systemic midazolam concentrations were comparable to those observed after oral midazolam administration 54, 55. Lower (and thus more favourable) and less variable peak concentrations were seen after intranasal compared to intravenous administration of midazolam. Nazolam showed dose proportional PK in the investigated dose range (2.5–5.0 mg). Several PK studies have been published using intranasal formulations 13, 14, 15, 16, 17, 18, 19, 20, 21. Although some showed comparable PK results, different nasal formulations were used, mainly using very large volumes, or high concentrations of organic solvents or absorption enhancers 13, 14, 15, 16, 17, 18, 19, 20, 21. Few nasal adverse events occured after administration of the Nazolam formulation. This finding is of clinical relevance as all previously studied midazolam formulations lead to high levels of nasal toxicity. For example, the administration of midazolam in formulations containing cyclodextrines has been reported to lead to mild to moderate nasal irritation 14, 15 and to lead to throat issues in 83% of subjects 16. Formulations containing cyclodextrin in combination with the absorption enhancer chitosan cause nasal irritation in 92% of subjects 20, and lead to tearing eyes in 65% of subjects 19. Nasal administration of midazolam in organic solvents also leads to substantial nasal toxicity. The recently developed organic solvent‐based midazolam formulation USL‐261, for example, displays nasal discomfort in 84% of subjects, throat irritation in 84%, increased lacrimation in 76% and dysgeusia in 72% of subjects 21.
In the present work, there were no signs of important contribution of ingestion‐related intestinal absorption, as the overall concentration–time profiles of intranasal formulations did not show a second peak and the formation of metabolite was low and comparable with intravenous levels. The absence of clinically relevant decreases in transcutaneous oxygen saturation parameters and blood pressure in this study indicate that the AE profile of nasal midazolam is comparable to that observed after oral midazolam administration.
SPV is generally considered as a sensitive and reproducible biomarker for the sedative effects of benzodiazepines 56 and was therefore used as a biomarker of pharmacological effect of midazolam in this study. SPV has already been used as an outcome variable in several previous studies with intravenous midazolam 57, 58, 59, 60, 61, 62, 63, 64 and changes in saccadic eye movements allow the accurate recognition of the wake–sleep transition 28, 65. In this study, an attempt was made to compare the onset and duration of pharmacological effect of the different midazolam formulations as accurately and realistically as possible. In a recent review on biomarkers for the effects of benzodiazepines in healthy subjects, a relationship between SPV reduction and clinical efficacy was described, as all reviewed benzodiazepines caused an impairment of saccadic peak velocity, which was closely related to the therapeutic dose 56. Therefore, SPV was used in this study to evaluate the onset and duration of pharmacological effect of intranasal midazolam. The definition of the onset and duration of pharmacological effect (sedation) was based on the subject's individual variability in SPV under placebo. As expected, intranasal midazolam led to a marked decrease in saccadic peak velocity at both investigated doses. Nazolam 2.5 mg led to conscious sedation in all individuals as reflected in the clear SPV decline observed for all subjects, showing that SPV is a sensitive biomarker and a good choice for a proof‐of‐pharmacology study such as the current one.
The effects of intranasal midazolam on SPV and subjective VAS alertness increased in a dose proportional fashion. The time effect curves of SPV and VAS alertness were comparable, which supports the appropriateness of the use of SPV as a surrogate marker for the sedative effect of midazolam. However, SPV was clearly more sensitive to midazolam effects than VAS alertness, as the observed effects of midazolam on SPV started earlier and returned to baseline later than those on VAS alertness. The use of SPV to define onset of pharmacological effect is therefore supported by the current data. Onset and duration of sedation were compared between treatment groups. The duration of sedation was slightly shorter for intranasal than for the intravenous formulation, but this was not statistically significant.
No effects were seen on subjective mood and calmness (as assessed by VAS), but this was not unexpected as it is in accordance with our experience with studies in healthy nonanxious subjects and probably related to a floor effect in the assessment of subjective calmness.
To assess the impact of ongoing sedation due to midazolam on normal functioning, the effect on the SRTT was assessed. Reaction time was increased by midazolam for about as long as the other PD effects (slowing of SPV and decrease in VAS alertness). The effects on SSRT of both the intranasal and the intravenous formulation returned to baseline in almost 2 h for all formulations.
In conclusion, this study demonstrates that clinically effective concentrations can be reached within minutes after nasal application of a highly concentrated midazolam formulation with sedation profiles comparable to those observed after intravenous midazolam administration. When considering the preparation time needed for obtaining venous access, conscious sedation onset and duration can be achieved in the same time span for nasal as for intravenous administration of midazolam. Potential applications of this new formulation are not limited to settings where midazolam is currently being used intravenously, but could also include settings in which intravenous access is not feasible such as in children and patients with needle phobia, and in urgent/emergency room situations. With the demonstrated absence of initial high peak plasma concentrations, nasal delivery also allows for safe and efficacious conscious sedation outside hospital settings such as the general practitioner office and dentistry settings. Finally, as the absorption capacity of the nasal mucosa is limited to 100 μl per nostril, nasal administration is relatively safe to overdosing. The immediate and noninvasive characteristics of this new formulation offer important advantages for clinical use in conscious sedation and in epilepsy.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare: This study was funded by Medir B.V. and executed by the Centre for Human Drug Research (CHDR). CHDR is a Clinical Research Organisation and as such receives payment for execution of phase I studies, including the study that led to the current manuscript. CHDR personnel (including L.S., R.Z., E.K., Z.G., J.v.G. and G.J.G.) receive regular salaries. L.S., R.Z., E.K., Z.G., J.v.G. and G.J.G. had no other relationships or activities that could appear to have influenced the submitted work. R.G. was a paid contractor of MEDIR B.V. (the company developing this intranasal midazolam formulation) for this study and a wide range of development activities. B.T. is a paid consultant to MEDIR B.V. for the study reported in this manuscript and outside the submitted work. F.M. has an issued patent (licensed to MEDIR B.V.) relevant to the work reported in this manuscript with no other relationships or activities that could appear to have influenced the submitted work.
This study was funded by MEDIR B.V.
Contributors
L.S. conducted the clinical study, reviewed the data, and drafted the study report and manuscript. R.Z. managed the regulatory submission and responses, conducted the clinical study, and reviewed the data, study report and manuscript. F.W.H.M.M. contributed to study design and reviewed the data, study report and manuscript. E.S.K. performed the analysis of the pharmacodynamic data and reviewed the study report and manuscript. Z.G. contributed to conduction of the clinical study and reviewed the manuscript. B.T. reviewed the data and manuscript. J.M.A.G. contributed to study design, reviewed the data, study report and manuscript. R.G. contributed to study design, and reviewed the data, study report and manuscript. G.J.G. contributed to study design, managed the regulatory submission and responses, reviewed the data, and contributed to the study report and manuscript.
Schrier, L. , Zuiker, R. , Merkus, F. W. H. M. , Klaassen, E. S. , Guan, Z. , Tuk, B. , van Gerven, J. M. A. , van der Geest, R. , and Groeneveld, G. J. (2017) Pharmacokinetics and pharmacodynamics of a new highly concentrated intranasal midazolam formulation for conscious sedation. Br J Clin Pharmacol, 83: 721–731. doi: 10.1111/bcp.13163.
References
- 1. Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP, et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 2016; 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alexander SPH, Peters JA, Kelly E, Marrion N, Benson HB, et al. The Concise Guide to PHARMACOLOGY 2015/16: Ligand‐gated ion channels. Br J Pharmacol 2015; 172: 5870–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, et al. The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 2015; 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Midazolam FDA label. Available at http://www.fda.gov/ohrms/dockets/dailys/01/Mar01/032101/cp00001_exhibit_02.pdf (last accessed 30 May 2016).
- 5. Pitetti R, Davis PJ, Redlinger R, White J, Wiener E, Calhoun KH. Effect on hospital‐wide sedation practices after implementation of the 2001 JCAHO procedural sedation and analgesia guidelines. Arch Pediatr Adolesc Med 2006; 160: 211–216. [DOI] [PubMed] [Google Scholar]
- 6. Glauser T, Shinnar S, Gloss D, Alldredge B, Arya R, Bainbridge J, et al. Evidence‐based guideline: treatment of convulsive status epilepticus in children and adults: report of the guideline committee of the American Epilepsy Society. Epilepsy Currents 2016; 16: 48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Link B, Haschke M, Grignaschi N, Bodmer M, Aschmann YZ, Wenk M, et al. Pharmacokinetics of intravenous and oral midazolam in plasma and saliva in humans: usefulness of saliva as matrix for CYP3A phenotyping. Br J Clin Pharmacol 2008; 66: 473–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Clausen TG, Wolff J, Hansen PB, Larsen F, Rasmussen SN, Dixon JS, et al. Pharmacokinetics of midazolam and alpha‐hydroxy‐midazolam following rectal and intravenous administration. Br J Clin Pharmacol 1988; 25: 457–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pecking M, Montestruc F, Marquet P, Wodey E, Homery M‐C, Dostert P. Absolute bioavailability of midazolam after subcutaneous administration to healthy volunteers. Br J Clin Pharmacol 2002; 54: 357–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schwagmeier R, Alincic S, Striebel HW. Midazolam pharmacokinetics following intravenous and buccal administration. Br J Clin Pharmacol 1998; 46: 203–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wilton NCT, Leigh J, Rosen DR, Pandit UA. Preanesthetic sedation of preschool children using intranasal midazolam. Anesthesiology 1988; 69: 972–975. [DOI] [PubMed] [Google Scholar]
- 12. Wilson MT, Macleod S, O'Regan ME. Nasal/buccal midazolam use in the community. Arch Dis Child 2004; 89: 50–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Burstein AH, Modica R, Hatton M, Forrest A, Gengo FM. Pharmacokinetics and pharmacodynamics of midazolam after intranasal administration. J Clin Pharmacol 1997; 37: 711–718. [DOI] [PubMed] [Google Scholar]
- 14. Loftsson T, Gudmundsdottir H, Sigurjonsdottir JF, Sigurdsson HH, Sigfusson SD, Masson M, et al. Cyclodextrin solubilization of benzodiazepines: formulation of midazolam nasal spray. Int J Pharm 2001; 212: 29–40. [DOI] [PubMed] [Google Scholar]
- 15. Gudmundsdottir H, Sigurjonsdottir JF, Masson M, Fjalldal O, Stefansson E, Loftsson T. Intranasal administration of midazolam in a cyclodextrin based formulation: bioavailability and clinical evaluation in humans. Pharmazie 2001; 56: 963–966. [PubMed] [Google Scholar]
- 16. Dale O, Nilsen T, Loftsson T, Hjorth TH, Klepstad P, Kaasa S, et al. Intranasal midazolam: a comparison of two delivery devices in human volunteers. J Pharm Pharmacol 2006; 58: 1311–1318. [DOI] [PubMed] [Google Scholar]
- 17. Knoester PD, Jonker DM, Van Der Hoeven RT, Vermeij TA, Edelbroek PM, Brekelmans GJ, et al. Pharmacokinetics and pharmacodynamics of midazolam administered as a concentrated intranasal spray. A study in healthy volunteers. Br J Clin Pharmacol 2002; 53: 501–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wermeling DP, Record KA, Kelly TH, Archer SM, Clinch T, Rudy AC. Pharmacokinetics and pharmacodynamics of a new intranasal midazolam formulation in healthy volunteers. Anesth Analg 2006; 103: 344–349. [DOI] [PubMed] [Google Scholar]
- 19. Haschke M, Suter K, Hofmann S, Witschi R, Frohlich J, Imanidis G, et al. Pharmacokinetics and pharmacodynamics of nasally delivered midazolam. Br J Clin Pharmacol 2010; 69: 607–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hardmeier M, Zimmermann R, Ruegg S, Pfluger M, Deuster S, Suter K, et al. Intranasal midazolam: pharmacokinetics and pharmacodynamics assessed by quantitative EEG in healthy volunteers. Clin Pharmacol Ther 2012; 91: 856–862. [DOI] [PubMed] [Google Scholar]
- 21. Bancke LL, Dworak HA, Rodvold KA, Halvorsen MB, Gidal BE. Pharmacokinetics, pharmacodynamics, and safety of USL261, a midazolam formulation optimized for intranasal delivery, in a randomized study with healthy volunteers. Epilepsia 2015; 56: 1723–1731. [DOI] [PubMed] [Google Scholar]
- 22. Marx D, Birkhoff M. Multi‐dose container for nasal and ophthalmic drugs: a preservative free future? In: Drug Development – A Case Study Based Insight into Modern Strategies, ed Rundfeldt C. Rijeka, Croatia: InTech, 2011; 509–524. [Google Scholar]
- 23. de Haas SL, de Visser SJ, van der Post JP, de Smet M, Schoemaker RC, Rijnbeek B, et al. Pharmacodynamic and pharmacokinetic effects of TPA023, a GABA(A) alpha(2,3) subtype‐selective agonist, compared to lorazepam and placebo in healthy volunteers. J Psychopharmacol 2007; 21: 374–383. [DOI] [PubMed] [Google Scholar]
- 24. van Steveninck AL, Gieschke R, Schoemaker HC, Pieters MS, Kroon JM, Breimer DD, et al. Pharmacodynamic interactions of diazepam and intravenous alcohol at pseudo steady state. Psychopharmacology (Berl) 1993; 110: 471–478. [DOI] [PubMed] [Google Scholar]
- 25. van Steveninck AL, Gieschke R, Schoemaker RC, Roncari G, Tuk B, Pieters MS, et al. Pharmacokinetic and pharmacodynamic interactions of bretazenil and diazepam with alcohol. Br J Clin Pharmacol 1996; 41: 565–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zoethout RW, Schoemaker RC, Zuurman L, van Pelt H, Dahan A, Cohen AF, et al. Central nervous system effects of alcohol at a pseudo‐steady‐state concentration using alcohol clamping in healthy volunteers. Br J Clin Pharmacol 2009; 68: 524–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Van Steveninck AL, Schoemaker HC, Pieters MS, Kroon R, Breimer DD, Cohen AF. A comparison of the sensitivities of adaptive tracking, eye movement analysis and visual analog lines to the effects of incremental doses of temazepam in healthy volunteers. Clin Pharmacol Ther 1991; 50: 172–180. [DOI] [PubMed] [Google Scholar]
- 28. Van Steveninck AL, van Berckel BN, Schoemaker RC, Breimer DD, van Gerven JM, Cohen AF. The sensitivity of pharmacodynamic tests for the central nervous system effects of drugs on the effects of sleep deprivation. J Psychopharmacol 1999; 13: 10–17. [DOI] [PubMed] [Google Scholar]
- 29. Van Steveninck AL. Methods of assessment of central nervous system effects of drugs in man. Thesis State University Leiden; 1994.
- 30. Baloh RW, Sills AW, Kumley WE, Honrubia V. Quantitative measurement of saccade amplitude, duration, and velocity. Neurology 1975; 25: 1065–1070. [DOI] [PubMed] [Google Scholar]
- 31. Van Steveninck AL, Cohen AF, Ward T. A microcomputer based system for recording and analysis of smooth pursuit and saccadic eye movements. Br J Clin Pharmacol 1989; 27: 712–713. [Google Scholar]
- 32. Norris H. The action of sedatives on brain stem oculomotor systems in man. Neuropharmacology 1971; 10: 181–191. [DOI] [PubMed] [Google Scholar]
- 33. Tschirch FT, Gopfert K, Frohlich JM, Brunner G, Weishaupt D. Low‐dose intranasal versus oral midazolam for routine body MRI of claustrophobic patients. Eur Radiol 2007; 17: 1403–1410. [DOI] [PubMed] [Google Scholar]
- 34. Shannon M, Albers G, Burkhart K, Liebelt E, Kelley M, McCubbin MM, et al. Safety and efficacy of flumazenil in the reversal of benzodiazepine‐induced conscious sedation. The Flumazenil Pediatric Study Group. J Pediatr 1997; 131: 582–586. [DOI] [PubMed] [Google Scholar]
- 35. Overly FL, Wright RO, Connor FA, Jay GD, Linakis JG. Bispectral analysis during deep sedation of pediatric oral surgery patients. J Oral Maxillofac Surg 2005; 63: 215–219. [DOI] [PubMed] [Google Scholar]
- 36. Marx CM, Stein J, Tyler MK, Nieder ML, Shurin SB, Blumer JL. Ketamine‐midazolam versus meperidine‐midazolam for painful procedures in pediatric oncology patients. J Clin Oncol 1997; 15: 94–102. [DOI] [PubMed] [Google Scholar]
- 37. Louon A, Reddy VG. Nasal midazolam and ketamine for paediatric sedation during computerised tomography. Acta Anaesthesiol Scand 1994; 38: 259–261. [DOI] [PubMed] [Google Scholar]
- 38. Ljungman G, Kreuger A, Andreasson S, Gordh T, Sorensen S. Midazolam nasal spray reduces procedural anxiety in children. Pediatrics 2000; 105: 73–78. [DOI] [PubMed] [Google Scholar]
- 39. Liu J, Singh H, White PF. Electroencephalogram bispectral analysis predicts the depth of midazolam‐induced sedation. Anesthesiology 1996; 84: 64–69. [DOI] [PubMed] [Google Scholar]
- 40. Lejus C, Renaudin M, Testa S, Malinovsky JM, Vigier T, Souron R. Midazolam for premedication in children: nasal vs. rectal administration. Eur J Anaesthesiol 1997; 14: 244–249. [DOI] [PubMed] [Google Scholar]
- 41. Hollenhorst J, Munte S, Friedrich L, Heine J, Leuwer M, Becker H, et al. Using intranasal midazolam spray to prevent claustrophobia induced by MR imaging. AJR Am J Roentgenol 2001; 176: 865–868. [DOI] [PubMed] [Google Scholar]
- 42. Connors K, Terndrup TE. Nasal versus oral midazolam for sedation of anxious children undergoing laceration repair. Ann Emerg Med 1994; 24: 1074–1079. [DOI] [PubMed] [Google Scholar]
- 43. Calderon E, Pernia A, Roman MD, Perez AC, Torres LM. Analgesia and sedation in the subarachnoid anesthesia technique: comparative study between remifentanil and fentanyl/midazolam. Rev Esp Anestesiol Reanim 2003; 50: 121–125. [PubMed] [Google Scholar]
- 44. Bergese SD, Patrick BS, McSweeney TD, Fernandez S, Dzwonczyk R, Sage K. A comparative study of dexmedetomidine with midazolam and midazolam alone for sedation during elective awake fiberoptic intubation. J Clin Anesth 2010; 22: 35–40. [DOI] [PubMed] [Google Scholar]
- 45. Avramov MN, Smith I, White PF. Interactions between midazolam and remifentanil during monitored anesthesia care. Anesthesiology 1996; 85: 1283–1289. [DOI] [PubMed] [Google Scholar]
- 46. Bond A, Lader M. The use of analogue scales in rating subjective feelings. Br J Med Psychol 1974; 47: 211–218. [Google Scholar]
- 47. Wezenberg E, Sabbe BG, Hulstijn W, Ruigt GS, Verkes RJ. The role of sedation tests in identifying sedative drug effects in healthy volunteers and their power to dissociate sedative‐related impairments from memory dysfunctions. J Psychopharmacol 2007; 21: 579–587. [DOI] [PubMed] [Google Scholar]
- 48. Motsch J, Epple J, Fresenius M, Neff S, Schmidt W, Martin E. Desflurane versus isoflurane in geriatric patients. A comparison of psychomotor and postoperative well‐being following abdominal surgical procedures. Anasthesiol Intensivmed Notfallmed Schmerzther 1998; 33: 313–320. [DOI] [PubMed] [Google Scholar]
- 49. Polster MR, Gray PA, O'Sullivan G, McCarthy RA, Park GR. Comparison of the sedative and amnesic effects of midazolam and propofol. Br J Anaesth 1993; 70: 612–616. [DOI] [PubMed] [Google Scholar]
- 50. Dudchenko P, Paul B, Sarter M. Dissociation between the effects of benzodiazepine receptor agonists on behavioral vigilance and responsitivity. Psychopharmacology (Berl) 1992; 109: 203–211. [DOI] [PubMed] [Google Scholar]
- 51. Chernik DA, Gillings D, Laine H, Hendler J, Silver JM, Davidson AB, et al. Validity and reliability of the observer's assessment of alertness/sedation scale: study with intravenous midazolam. J Clin Psychopharmacol 1990; 10: 244–251. [PubMed] [Google Scholar]
- 52. Deng XM, Xiao WJ, Luo MP, Tang GZ, Xu KL. The use of midazolam and small‐dose ketamine for sedation and analgesia during local anesthesia. Anesth Analg 2001; 93: 1174–1177. [DOI] [PubMed] [Google Scholar]
- 53. Dingemanse J, van Gerven JM, Schoemaker RC, Roncari G, Oberyé JJ, van Oostenbruggen MF, et al. Integrated pharmacokinetics and pharmacodynamics of Ro 48-6791, a new benzodiazepine, in comparison with midazolam during first administration to healthy male subjects. Br J Clin Pharmacol 1997; 44: 477–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Smith MT, Eadie MJ, Brophy TO . The pharmacokinetics of midazolam in man. Eur J Clin Pharmacol 1981; 19: 271–278. [DOI] [PubMed] [Google Scholar]
- 55. Allonen H, Ziegler G, Klotz U. Midazolam kinetics. Clin Pharmacol Ther 1981; 30: 653–661. [DOI] [PubMed] [Google Scholar]
- 56. de Visser SJ, van der Post JP, de Waal PP, Cornet F, Cohen AF, van Gerven JM. Biomarkers for the effects of benzodiazepines in healthy volunteers. Br J Clin Pharmacol 2003; 55: 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Aho M, Erkola O, Kallio A, Scheinin H, Korttila K. Comparison of dexmedetomidine and midazolam sedation and antagonism of dexmedetomidine with atipamezole. J Clin Anesth 1993; 5: 194–203. [DOI] [PubMed] [Google Scholar]
- 58. Ayuse T, Hoshino Y, Kurata S, Ayuse T, Schneider H, Kirkness JP, et al. The effect of gender on compensatory neuromuscular response to upper airway obstruction in normal subjects under midazolam general anesthesia. Anesth Analg 2009; 109: 1209–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ball DM, Glue P, Wilson S, Nutt DJ. Pharmacology of saccadic eye movements in man. 1. Effects of the benzodiazepine receptor ligands midazolam and flumazenil. Psychopharmacology (Berl) 1991; 105: 361–367. [DOI] [PubMed] [Google Scholar]
- 60. Bevan JC, Veall GR, Macnab AJ, Ries CR, Marsland C. Midazolam premedication delays recovery after propofol without modifying involuntary movements. Anesth Analg 1997; 85: 50–54. [DOI] [PubMed] [Google Scholar]
- 61. Salmon JF, Mets B, James MF, Murray AD. Intravenous sedation for ocular surgery under local anaesthesia. Br J Ophthalmol 1992; 76: 598–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Song YS, Song ES, Lee KH, Park YH, Shin WC, Ku JH. Sleep‐related nocturnal erections and erections during midazolam‐induced sedation in healthy young men. Int J Impot Res 2006; 18: 522–526. [DOI] [PubMed] [Google Scholar]
- 63. Sundstrom I, Nyberg S, Backstrom T. Patients with premenstrual syndrome have reduced sensitivity to midazolam compared to control subjects. Neuropsychopharmacology 1997; 17: 370–381. [DOI] [PubMed] [Google Scholar]
- 64. van Gerven JM, Roncari G, Schoemaker RC, Massarella J, Keesmaat P, Kooyman H, et al. Integrated pharmacokinetics and pharmacodynamics of Ro 48‐8684, a new benzodiazepine, in comparison with midazolam during first administration to healthy male subjects. Br J Clin Pharmacol 1997; 44: 487–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Griffiths AN, Marshall RW, Richens A. Saccadic eye movement analysis as a measure of drug effects on human psychomotor performance. Br J Clin Pharmacol 1984; 18 (Suppl. 1): 73S–82S. [DOI] [PMC free article] [PubMed] [Google Scholar]