

Abstract
Aims
To assess pharmacokinetics (PK) and safety of GP2015, a proposed etanercept biosimilar, in two studies: comparison with etanercept originator (ETN, bioequivalence study) and comparison of GP2015 administered via an autoinjector (AI) or prefilled syringes (PFS, delivery study).
Methods
Both studies were randomized, two‐sequence, two‐period, crossover studies conducted in healthy male subjects. In the bioequivalence study, subjects were randomized to receive a single 50 mg subcutaneous (s.c.) injection of GP2015 or ETN. In the delivery study, subjects were randomized to receive a single 50 mg s.c. injection of GP2015 via AI or PFS. Following a wash‐out period of 35 days, subjects in the bioequivalence study received single 50 mg s.c. injection of GP2015 or ETN, and subjects in the delivery study received single 50 mg s.c. injection of GP2015 via AI or PFS.
Results
The geometric mean ratios (90% confidence interval) of GP2015/ETN for Cmax (1.11 [1.05–1.17]), AUC0–tlast (0.98 [0.94–1.02]) and AUC0–inf (0.96 [0.93–1.00]) were within the predefined bioequivalence range of 0.80–1.25. The geometric mean ratios (90% confidence interval) of AI/PFS for Cmax (1.01 [0.94–1.08]), AUC0–tlast (1.01 [0.95–1.07]) and AUC0–inf (1.01 [0.96–1.07]) were also within the range 0.80–1.25. No new safety issues were reported. Three subjects had low titres of non‐neutralising anti‐drug antibodies during a follow‐up visit in the bioequivalence study.
Conclusions
The PK of GP2015 was similar to ETN, demonstrating bioequivalence. The safety profile of GP2015 was consistent with previous reports for ETN. The GP2015 AI provided equivalent dosing and tolerability to the GP2015 PFS.
Keywords: autoinjector, bioequivalence, biosimilar, GP2015, pharmacokinetics, subcutaneous administration, etanercept
What is Already Known about this Subject
Etanercept, an anti‐tumour necrosis factor agent, is indicated for the treatment of a wide range of inflammatory diseases via subcutaneous injection.
A biosimilar is a biological agent that is developed to be essentially the same as an already authorized biological medicinal product (originator).
Many patients have difficulties in operating conventional self‐injection devices due to impaired dexterity.
What this Study Adds
GP2015, a proposed etanercept biosimilar, demonstrates pharmacokinetic bioequivalence to the etanercept originator. There are no relevant differences in safety.
Administration of GP2015 by an autoinjector provided equivalent dosing and tolerability to that of the prefilled syringe, and may offer advantages in terms of convenience for treatment with GP2015.
Tables of Links
| TARGETS |
|---|
| Tumour necrosis factor |
| LIGANDS |
|---|
| Etanercept |
Introduction
Etanercept (Enbrel), an anti‐tumour necrosis factor (TNF) agent, is a fusion protein consisting of the extracellular ligand‐binding domains of the 75‐kDa TNF‐α receptor 2 linked to the Fc region of human immunoglobulin G1 (IgG1). Etanercept binds to and neutralizes the biological activity of TNF‐α 3. Etanercept, first licensed in the USA in 1998 for the treatment of rheumatoid arthritis (RA) under brand name Enbrel, has since been approved for other indications, including plaque psoriasis, psoriatic arthritis, ankylosing spondylitis and juvenile idiopathic arthritis 4. In the European Union (EU), etanercept is further indicated for the treatment of nonradiographic axial spondyloarthritis and paediatric plaque psoriasis 5. Depending on the indication, adults may be administered a 25 mg or 50 mg dose as subcutaneous (s.c.) injection, once or twice per week. Children weighing under 62.5 kg are dosed on a mg/kg basis 3, 4.
The term biosimilar refers to a biological product that is ‘essentially the same’ (EU) or ‘highly similar’ (USA) to a reference biological medicinal product that is previously approved in the respective region, in the following referred to as the originator product. Regulatory decisions for approval of biosimilars are based on data generated from a stepwise approach, starting with comparative structural and functional characterization of the originator and the proposed biosimilar, proceeding to comparing non‐clinical data (toxicity, pharmacokinetics [PK], pharmacodynamics [PD]), followed by clinical studies demonstrating similar PK, PD, immunogenicity and confirming similarity of the sameness of the molecules via evaluating clinical safety and efficacy 6. These data analyzed altogether provide the proper assessment of biosimilarity often referred to as the ‘totality‐of‐the‐evidence’ concept 7.
GP2015 is a proposed etanercept biosimilar whose development, in accordance with the ‘totality‐of‐the‐evidence’ concept, involved extensive analytical testing comparing structural, physicochemical properties and biological functions between GP2015 and the etanercept originator (EU authorized [EU‐ETN] as well as US‐licensed [Enbrel US‐ETN]; (Figure 1).
Figure 1.
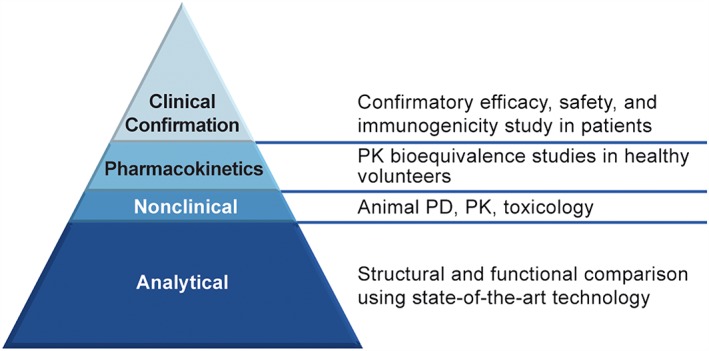
Assessment pathway of bioequivalence using the ‘totality of the evidence’ concept 4. Adapted from reference 6; PK, pharmacokinetics; PD, pharmacodynamics
Patients with advanced stages of RA may have difficulties in operating conventional or prefilled syringes (PFS) due to impaired dexterity 8. Therefore, it is important to provide patients with a mode of self‐administration that combines ease of use, comfort and convenience to maximize patient adherence to therapy and to improve disease management 9. The use of an autoinjector (AI) for drug delivery has been shown to increase patient adherence 9 by making self‐administration of subcutaneous drugs easier. AIs have been shown to increase patient acceptability and convenience as well as to reduce injection site pain 8, 10, 11. Therefore, in addition to the PFS, a ready to use, fixed dose, disposable AI has been developed for GP2015.
Two, randomized, two‐sequence, two‐period cross over studies, reported herein, were conducted in healthy subjects to compare the PK and safety of GP2015 with ETN (bioequivalence study) and to compare the administration of GP2015 by AI or PFS (delivery study).
Methods
Study designs
Bioequivalence study
The bioequivalence study was a Phase 1, single‐centre, randomized, double‐blind, two‐way, crossover study with two treatment periods (EudraCT number 2013–004 902‐25). Subjects were screened for eligibility from 2 to 28 days before dosing on Day 1. Healthy subjects were randomized to receive a single 50 mg s.c. injection of GP2015 or ETN (treatment period 1). Following a wash‐out period of at least 35 days after treatment period 1, subjects were crossed over and received a single 50 mg s.c. injection of the opposite treatment in treatment period 1, either GP2015 or ETN (treatment period 2). Subjects were discharged 48 h after postdose assessments on Day 3 of treatment periods 1 and 2, and outpatient visits were scheduled up to Day 19 for PK and safety assessments. Subsequent follow‐up was 28 days with a total study duration of up to 3 months.
Delivery study
The delivery study was a separately conducted open‐label, randomized, two‐way, cross over study with two treatment periods (EudraCT number 2013–004 901‐24). Subjects were screened for eligibility from 2 to 28 days before dosing on Day 1. Subjects were randomized to receive a single 50 mg s.c. dose of GP2015 via AI or PFS (treatment period 1). Following a wash‐out period of at least 35 days after treatment period 1, subjects were crossed over and received a single 50 mg s.c. injection of GP2015 via PFS or AI (opposite of what was used in treatment period 1; treatment period 2). Subjects were discharged 120 h postdose on Day 6 of periods 1 and 2, and outpatient visits up to Day 19 were carried out for PK and safety assessments. A follow‐up visit was carried out 28 days after the investigational medicinal product administration in treatment period 2.
Subjects
In the bioequivalence study, eligible subjects were healthy men aged 18–49 years, with body weight of 50–99.9 kg and body mass index (BMI) of 19.0 to 29.9 kg m–2. In the delivery study, healthy male subjects aged between 18 and 55 years, with body weight 50–140 kg and BMI 18.5–49.9 kg m2 were enrolled and randomization was stratified into three body weight categories (low: 50.0–79.9, medium: 80.0–99.9 and high: 100.0–140.0 kg). The weight stratification was used to confirm that the delivery of GP2015 with an AI or with a PFS is the same across a large spectrum of body weights representative of the patient population. Subjects were not allowed to enter either study if they had previously received a TNF‐α inhibitor or if they had active infections within the last 4 weeks.
The study protocols were approved by the Institutional Review Board of National Health Research Authority, London, UK (bioequivalence study) and the Independent Ethics Committee of the Foundation Evaluation of the Ethics of Biomedical Research, Assen, the Netherlands (delivery study). Both studies were conducted according to the guidelines for Good Clinical Practice and the Declaration of Helsinki. All subjects provided written informed consent.
Objectives
Bioequivalence study
The primary objective was to determine the bioequivalence of GP2015 and ETN in terms of the following PK parameters: maximum observed serum concentration (Cmax), area under the serum concentration–time curve measured from the time of dosing and extrapolated to infinity (AUC0–inf) and AUC measured from the time of dosing to the last measurable concentration (AUC0–tlast). Secondary objectives included comparison of other PK parameters, time to the maximum observed serum concentration (tmax), elimination rate constant (kel) and the apparent terminal half‐life of elimination phase (t½), potential immunogenicity, and overall safety and tolerability of GP2015 and ETN.
Delivery study
The primary objective was to show bioequivalence of GP2015 when administered by an AI or PFS in terms of the PK parameters AUC0–tlast, AUC0–inf and Cmax and to confirm that the delivery of GP2015 with the AI is the same as the delivery with the PFS in healthy subjects across the spectrum of body weights by comparing PK parameters within each body weight category. Secondary objectives included comparison of other PK parameters, such as tmax, kel, and t½ between AI and PFS in total as well as by weight category and to evaluate and compare the overall safety, tolerability, and local tolerance of GP2015.
Investigational medicinal product
In the bioequivalence study, a single batch of GP2015 and EU‐licensed ETN were used. In the delivery study, a single GP2015 drug substance batch was administered by either PFS or AI. In both studies, single s.c. doses of 50 mg GP2015 (PFS/AI) or Enbrel/EU (PFS) were administered by investigators.
Assessments
Pharmacokinetic assessments
Blood samples (3.5 ml) for PK assessment were drawn at 0, 6, 12, 24, 36, 48, 60, 72, 84, 96, 120, 168, 216, 264, 336 and 432 h after dosing in each treatment period. Blood was collected (BD vacutainer) and allowed to clot after standing for at least 30 min at room temperature. The blood samples were centrifuged (Micro Centrifuge 2D, Roth) at room temperature for 10 min at 1100–1300 g. The serum samples were frozen and stored at –70°C until being shipped in dry ice for analysis to Hexal AG (Oberhaching, Germany). GP2015 as well as Enbrel/EU concentrations in the serum were quantified using a validated enzyme‐linked immunosorbent assay. Anti‐human soluble TNF receptor 2 (TNFR2) antibodies were used as capture and detection reagents. Serum containing GP2015 or Enbrel/EU was then added to the plate and detected using a second anti‐human TNFR2 antibody. The assay read‐out was performed by an enzyme substrate reaction and etanercept concentrations were measured using a microplate reader. During method validation, selectivity and comparability experiments demonstrated that the assay detects GP2015 and Enbrel/EU identically. Consequently, one and the same assay was used during subsequent sample analysis for both determination of GP2015 and Enbrel/EU serum concentrations. The range of the assay was 6.7–800 ng ml–1 with 6.7 ng ml–1 defined as the lower limit of quantification; samples containing GP2015 or Enbrel/EU concentrations above the assay range were diluted appropriately. The inter‐run assay accuracy was expressed as the percent bias for quality control samples and ranged from –9 to –1% and –14 to –4% for the delivery study and the bioequivalence study, respectively. The interrun assay precision was expressed as the coefficient of variation for quality control samples and ranged from 6 to 19% and 5 to 18% for the delivery study and the bioequivalence study respectively.
The concentration–time data were analyzed by a noncompartment method using WinNonlin 6.3 (Pharsight Corp, Mountain View, California, USA). Cmax and tmax were obtained directly from the observed values. The kel was estimated at the terminal phase by linear regression after log‐transformation of the concentrations. The t½ was calculated as ln(2)/kel. The linear up/log down trapezoidal rule was used to obtain the area under the concentration–time curve from time zero to the last quantifiable concentration (AUClast). AUC extrapolated to infinity (AUCinf) was calculated as AUClast + Clast/kel (Clast: the last quantifiable concentration).
Safety assessments
Safety was assessed by collecting all treatment emergent adverse events (TEAEs) and serious adverse events (SAEs) with their severity and relationship to study drug. Vital signs, including blood pressure (BP) and pulse rate measurements, were assessed at the screening visit, in the morning on Day 1 predose, at Days 2, 3, 4, 8 and 15 of each treatment period and at the follow‐up visit on Day 29. An electrocardiogram (ECG) was recorded at screening, Day –1, and Day 29 of treatment period 2. Blood and urine samples for laboratory haematology, clinical chemistry, coagulation and urinalysis were collected at the screening visit, Days –1, 2, 4, 6, 8 and 15 of each treatment period and at the follow‐up visit on Day 29.
Immunogenicity assessments
For immunogenicity assessments, blood samples (5 ml) were collected into serum tubes (BD vacutainer) at –0.5 h predose on Day 1 of each period and at the follow‐up visit on Day 29 of treatment period 2. Serum preparation and handling was identical to samples used for pharmacokinetic assessment. The anti‐drug antibody (ADA) assessment and analysis followed a tiered approach. Study samples were first analyzed in an electrochemiluminescence (ECL) screening assay in which antibodies binding to etanercept were detected. Samples that were positive in the screening assay were subsequently analyzed in an ECL confirmatory assay which evaluated the specificity of the antibodies by preincubation of the samples with excess of etanercept leading to a reduction of the assay signal. In addition, the titre of confirmed positive ADA samples was determined, and the neutralising capacity of ADAs was assessed in a competitive ligand binding neutralising assay.
The applied method for the detection of ADAs is based on a bridging ECL assay format including acid dissociations steps. The validated assay sensitivity (defined as the concentration of ADAs at the screening cut‐point level) was determined to be 21 ng ml–1 by using a polyclonal positive control antibody. In both studies, ADA samples were drawn at a late timepoint, i.e. at Day 29 of treatment period 2, to prevent interference of ADA detection with either GP2015 or ETN.
Statistical analysis
The PK analysis set comprised all subjects who completed the study. Previous Phase I studies of Enbrel in healthy volunteers have shown that the coefficient of variation (CV) for intrasubject variability varied between 21.6 and 30.85% for AUC and Cmax 12. Based on this estimate of variability, a CV of 28% was considered adequate for sample size estimation. In the bioequivalence study, it was planned to randomize a total of 54 subjects, with the aim of obtaining 48 evaluable subjects, which would provide at least 90% power to show bioequivalence within the predefined range of 0.80–1.25. In the delivery study, 42 evaluable subjects were required to provide 90% power to show bioequivalence within the predefined range of 0.80–1.25. The sample size calculations were performed with nQuery Advisor 7.0 (Statistical Solutions Ltd, Cork, Ireland).
The bioequivalence of primary PK parameters was considered to have been demonstrated for a given test‐to‐reference comparison if the 90% confidence intervals (CIs) for the geometric mean ratios were completely contained within the predefined bioequivalence range of 0.80–1.25. PK parameters AUC0–tlast, AUC0–inf and Cmax were separately analyzed by ANOVA in the bioequivalence study and by an analysis of covariance (ANCOVA) using the weight of the subjects as a covariate in the delivery study. The effects due to sequence, period and treatment were included as fixed effects and subject nested within sequence as a random effect.
Results
Subject disposition
Bioequivalence study
A total of 54 subjects were randomized and all completed the study (27 subjects in the treatment sequence GP2015/ETN and 27 subjects in ETN/GP2015 treatment sequence). The majority of subjects were white (53.7%) with a mean (standard deviation) age of 32.9 (8.27) years, and a mean BMI of 24.85 (range: 19.0–29.4) kg m–2 (Table 1).
Table 1.
Demographic and baseline characteristics
| Bioequivalence study | GP2015/ETN | ETN/GP2015 | Total n = 54 |
|---|---|---|---|
| Age, years, mean (SD) | 35.2 (8.45) | 30.6 (7.55) | 32.9 (8.27) |
| Race, n (%) | |||
| White | 15 (55.6) | 14 (51.9) | 29 (53.7) |
| Asian | 7 (25.9) | 6 (22.2) | 13 (24.1) |
| Black/African American | 3 (11.1) | 5 (18.5) | 8 (14.8) |
| Other | 2 (7.4) | 2 (7.4) | 4 (7.4) |
| Weight, kg, (mean SD) | 75.51 (10.08) | 76.71 (9.48) | 76.11 (9.7) |
| BMI, kg m–2, (mean, range) | 24.58 (19.0–29.4) | 25.11 (20.5–29.4) | 24.85 (19.0–29.4) |
| Delivery study | AI/PFS | PFS/AI | Total (n = 51) |
|---|---|---|---|
| Age, years, mean (SD) | 33.8 (10.02) | 34.3 (10.29) | 34.1 (10.06) |
| Race, n (%) | |||
| White | 22 (88) | 18 (69) | 40 (78) |
| Black | 3 (12) | 6 (23) | 9 (18) |
| American Indian/Alaska native | 0 | 1 (4) | 1 (2) |
| Other | 0 (0) | 1 (4) | 1 (2) |
| Body weight group, n (%) | |||
| 50–79.9 kg | 9 (36) | 8 (31) | 17 (33) |
| 80–99.9 kg | 8 (32) | 9 (35) | 17 (33) |
| 100–140 kg | 8 (32) | 9 (35) | 17 (33) |
| BMI, kg m–2, mean (range) | 27.20 (19.3–39.0) | 28.22 (20.1–37.0) | 27.72 (19.3–39.0) |
AI, autoinjector; BMI, body mass index; ETN, etanercept originator; SD, PFS, pre‐filled syringe; standard deviation
Delivery study
Fifty‐one subjects were randomized, 25 subjects randomized to the treatment sequence of AI/PFS and 26 to PFS/AI; 49 subjects completed the study. There were two premature discontinuations, one due to a protocol violation (treatment sequence PFS/AI) and one due to an adverse event (AE; treatment sequence AI/PFS). The majority of subjects were white (78%), with a mean (standard deviation) age of 34 (10.06) years and a mean BMI of 27.72 (range: 19.3–39.0) kg m–2 (Table 1).
PK results
Bioequivalence study
In the bioequivalence study, all 54 randomized subjects who received study medication in both treatment periods were included in the PK analysis set, as there were no major protocol deviations. The mean serum concentration time profiles were similar between GP2015 and ETN (Figure 2). The geometric mean ratios (90%CI) of GP2015/ETN for Cmax –(1.11 [1.05–1.17]), AUC0–tlast (0.98 [0.94–1.02]), AUC0–inf (0.96 [0.93–1.00]) – were within the predefined bioequivalence range of 0.80–1.25 (Table 2). Among the secondary endpoints, the mean t½ for GP2015 and ETN were 104.7 h and 110.7 h, respectively and median tmax was 58.3 h and 59.8 h, respectively.
Figure 2.
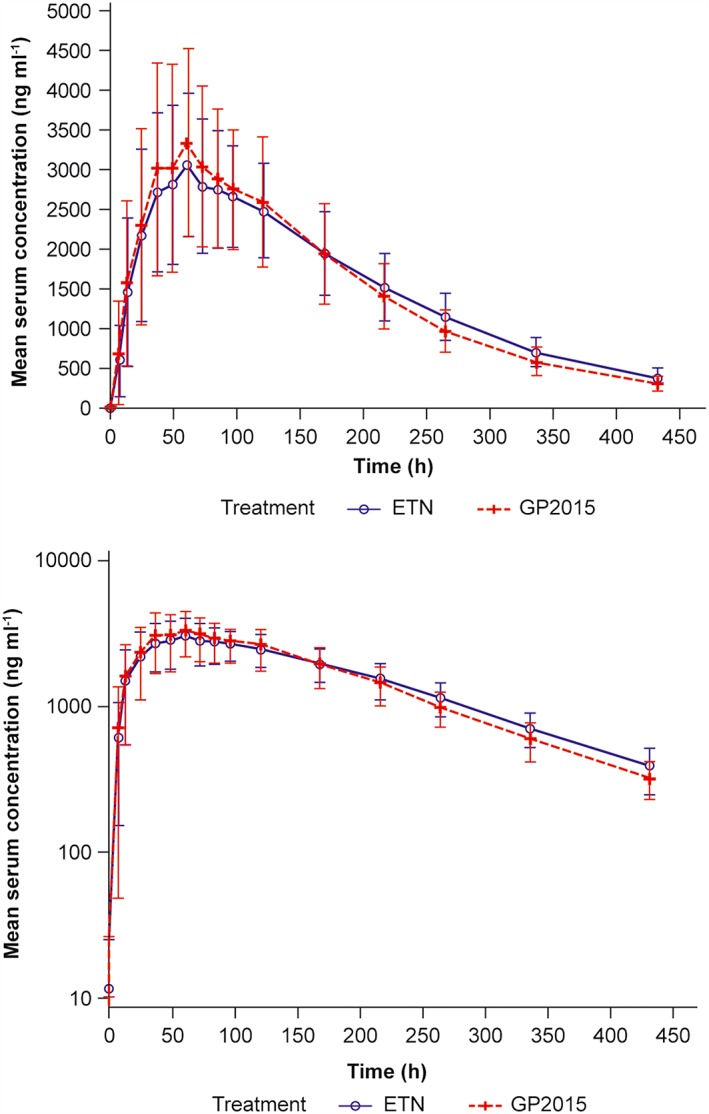
Mean serum concentration–time profiles of GP2015 and ETN (PK set) in the bioequivalence study (linear and semilogarithmic). The error bars depict the standard deviation (SD). ETN, etanercept originator; PK, pharmacokinetic
Table 2.
Primary PK parameters and statistical analysis following a single dose of GP2015 or etanercept originator (both studies)
| Bioequivalence study | Geometric means | Mean ratio (%) | 90% CI of ratio | Intraindividual CV (%) | |
|---|---|---|---|---|---|
| GP2015 | ETN | ||||
| Cmax (μg ml–1) | 3.4 | 3.1 | 1.11 | 1.05–1.17 | 16.4 |
| AUC0–tlast (h μg ml–1) | 630 | 642 | 0.98 | 0.94–1.02 | 12.1 |
| AUC0–inf (h μg ml–1) | 679 | 705 | 0.96 | 0.93–1.00 | 12.3 |
| Delivery study | Geometric means | Mean ratio (%) | 90% CI of ratio | Intraindividual CV (%) | |
|---|---|---|---|---|---|
| AI | PFS | ||||
| Cmax (μg ml–1) | 3.7 | 3.6 | 1.01 | 0.94–1.08 | ‐ |
| AUC0–tlast (h μg ml–1) | 684.1 | 678.4 | 1.01 | 0.95–1.07 | ‐ |
| AUC0–inf (h μg ml–1) | 745.2 | 737.4 | 1.01 | 0.96–1.07 | ‐ |
AI autoinjector; AUC0–inf, area under the serum concentration‐time curve measured from the time of dosing and extrapolated to infinity; AUC0–tlast, measured from the time of dosing to the last measurable concentration; Cmax, maximum observed serum concentration; CI, confidence interval; CV, coefficient of variation; ETN, etanercept originator; PK, pharmacokinetic; PFS, prefilled syringe
Delivery study
In the delivery study, the PK analysis set included 48 subjects. One subject was found to have a predose etanercept concentration of >5% of Cmax in treatment period 2, indicating carry‐over from treatment period 1 and was therefore excluded from the PK analysis set. Mean serum concentration‐time profiles were similar between both AI and PFS treatment administrations (Figure 3). Also, the mean Cmax, AUC0–tlast and AUC0–inf were similar for both treatment administrations. The 90% CIs for the ratio of the geometric means (AI/ PFS) for Cmax (1.01 [0.94–1.08]), AUC0–tlast (1.01 [0.95–1.07]), and AUC0–inf (1.01 [0.96–1.07]) were within the predefined bioequivalence range of 0.80–1.25 (Table 2]. The serum concentration profiles of GP2015 were similar across the body weight categories (Figure 4). The PK parameters of GP2015 administered by AI and PFS in the different weight groups are listed in Table 3. The mean t½ were identical (109 h) for both AI and PFS treatment administrations as expected when using the identical drug substance batch in both devices.
Figure 3.
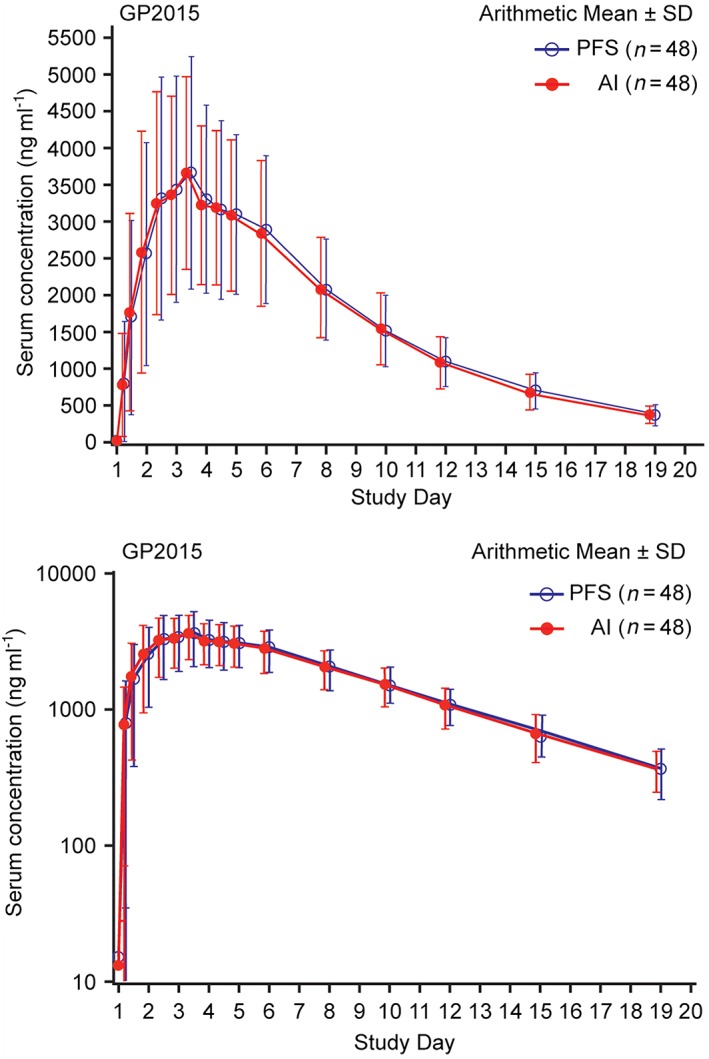
Mean serum concentration–time profiles of GP2015 AI and PFS in the delivery study (linear and semilogarithmic). The error bars depict the standard deviation (SD). AI, autoinjector; PFS, prefilled syringes
Figure 4.
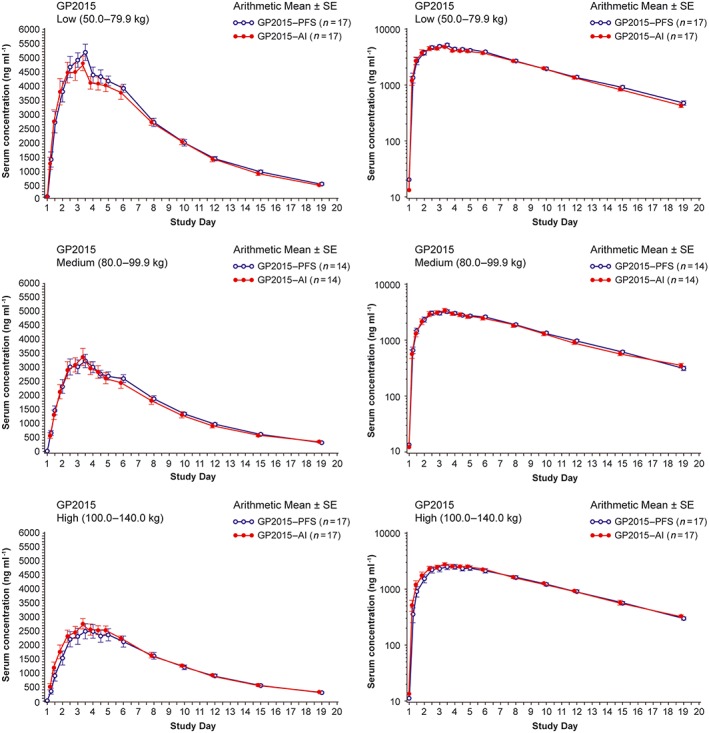
Mean serum concentration–time profiles of GP2015 AI and PFS by body weight category in the delivery study (linear and semilogarithmic). AI, autoinjector; PFS, prefilled syringes; SE, standard error
Table 3.
Primary PK parameters within each weight category in the delivery study
| Mean (SD) | |||
|---|---|---|---|
| Weight categories | PK Parameter | AI | PFS |
| Low (50.0–79.9 kg) n = 17 | Cmax (μg ml–1) | 5.21 (1.4) | 5.55 (1.3) |
| tmax (h) | 50.83 (15.0) | 57.18 (13.8) | |
| AUC0–tlast (h μg ml–1) | 941 (199) | 975 (199) | |
| AUC0–inf (h μg ml–1) | 1006 (213) | 1049 (224) | |
| t1/2 (h) | 101 (13.5) | 104 (13.3) | |
| Medium (80.0–99.9 kg) n = 14 | Cmax (μg ml–1) | 3.52 (1.1) | 3.48 (0.87) |
| tmax (h) | 64.3 (13.0) | 60.0 (24.9) | |
| AUC0–tlast (h μg ml–1) | 629 (174) | 647 (119) | |
| AUC0–inf (h μg ml–1) | 686 (180) | 695 (122) | |
| t1/2 (h) | 109 (18.2) | 104 (16.1) | |
| High (100.0–140.0 kg) n = 17 | Cmax (μg ml–1) | 2.97 (0.79) | 2.84 (1.08) |
| tmax (h) | 72.7 (21.9) | 72.8 (33.4) | |
| AUC0–tlast (h μg ml–1) | 571 (97.5) | 539 (171) | |
| AUC0–inf (h μg ml–1) | 629 (92.7) | 592 (173) | |
| t1/2 (h) | 117 (31.6) | 118 (33.8) | |
AI autoinjector; AUC0–inf, area under the serum concentration–time curve measured from the time of dosing and extrapolated to infinity; AUC0–tlast, measured from the time of dosing to the last measurable concentration; Cmax, maximum observed serum concentration; PK, pharmacokinetic; SD, standard deviation; tmax, time to the maximum observed serum concentration; t½, the apparent terminal half‐life of elimination phase; PFS, prefilled syringe
Safety
Bioequivalence study
At least one TEAE was reported in 23 (42.6%) subjects in the GP2015 group and 20 (37%) subjects in the ETN group. The most frequently reported TEAEs were neutropenia (GP2015, n = 7 [13%]; ETN, n = 8 [14.8%]), headache (GP2015, n = 5 [9.3%]; ETN, n = 5 [9.3%]) and nasopharyngitis (GP2015, n = 4 [7.4%]; ETN, n = 4 [7.4%]; Table 4). All TEAEs were mild or moderate in severity, and TEAEs considered related to the study drug were reported in 10 (18.5%) and 13 (24.1%) subjects for GP2015 and ETN, respectively. No SAEs or deaths occurred during the study.
Table 4.
Most frequent treatment emergent adverse events (TEAEs) regardless of relationship to study treatment by preferred term
| Bioequivalence study Preferred term | GP2015 n (%) | ETN n (%) | Overall n (%) |
|---|---|---|---|
| Neutropenia | 7 (13) | 8 (14.8) | 10 (18.5) |
| Headache | 5 (9.3) | 5 (9.3) | 9 (16.7) |
| Nasopharyngitis | 4 (7.4) | 4 (7.4) | 8 (14.8) |
| Oropharyngeal pain | 3 (5.6) | 4 (7.4) | 7 (13.0) |
| Feeling hot | 0 | 3 (5.6) | 3 (5.6) |
| Cough | 3 (5.6) | 0 | 3 (5.6) |
| Fatigue | 1 (1.9) | 1 (1.9) | 2 (3.7) |
| Back pain | 2 (3.7) | 0 | 2 (3.7) |
| Musculoskeletal chest pain | 1 (1.9) | 1 (1.9) | 2 (3.7) |
| Delivery study Preferred term | GP2015 AI n (%) | GP2015 PFS n (%) | Overall n (%) |
|---|---|---|---|
| Headache | 8 (16) | 5 (10) | 10 (20) |
| Neutropenia | 5 (10) | 5 (10) | 6 (12) |
| Hematoma | 1 (2) | 3 (6) | 4 (8) |
| Rhinitis | 1 (2) | 3 (6) | 4 (8) |
| Nausea | 3 (6) | 1 (2) | 4 (8) |
| Pollakiuria | 2 (4) | 3 (6) | 3 (6) |
| Back pain | 1 (2) | 2 (4) | 3 (6) |
| Neck pain | 1 (2) | 2 (4) | 3 (6) |
| Pain in extremity | 2 (4) | 1 (2) | 3 (6) |
| Vessel puncture site pain | 1 (2) | 2 (4) | 3 (6) |
| Cough | 2 (4) | 1 (2) | 2 (4) |
| Flatulence | 2 (4) | 1 (2) | 2 (4) |
| Myalgia | 0 | 2 (4) | 2 (4) |
| Erythema | 0 | 2 (4) | 2 (4) |
| Gamma‐glutamyltransferase increased | 1 (2) | 1 (2) | 2 (4) |
| Vomiting | 1 (2) | 1 (2) | 2 (4) |
TEAEs are presented for at least two subjects for overall and all TEAEs are presented in descending order in the overall group. Events were coded using MedDRA (Version 17.0)
%, percentage of subjects that experienced the TEAE per treatment administration ([n/N] × 100%), where n is the number of subjects (percentage) with at least one TEAE and N is the total number of subjects studied; AI, autoinjector; ETN, etanercept originator; PFS, prefilled syringe; TEAE, treatment‐emergent adverse event
Delivery study
The incidence of TEAEs was identical (25 subjects each) in the AI and PFS groups. The most frequent TEAEs were headache (GP2015‐AI, n = 8 [16%]; GP2015‐PFS, n = 5 [10%]), neutropenia (GP2015‐AI, n = 5 [10%]; GP2015‐PFS, n = 5 [10%]) and rhinitis (GP2015‐AI, n = 1 [2%]; GP2015‐PFS, n = 3 [6%]; Table 4). One subject was discontinued by the Principal Investigator due to an AE (animal bite). All treatment related AEs were of mild severity and resolved during the study. TEAEs considered related to the study drug were reported in 11 (22%) and nine (18%) subjects for AI and PFS group, respectively. None of the neutropenia cases (range: 1.1–1.7 × 109 l–1; reference range: 1.8–7.9 × 109 l–1) were considered clinically significant. Three subjects had a mild injection site reaction (GP2015‐AI, n = 1 [2%] and GP2015‐PFS, n = 2 [4%]). No SAEs or deaths occurred during the study.
In both studies, no clinically relevant changes were observed in clinical laboratory parameters, vital signs, ECGs, physical examination findings or local tolerance at the injection site.
Immunogenicity
In the bioequivalence study, three subjects (treatment sequence GP2015/ETN) had a confirmed positive non‐neutralising ADA response at the follow‐up visit with a low titre close to the detection limit, while all samples from the predose (Day 1) of each period were ADA negative. In the delivery study, none of the subjects developed ADAs after treatment with GP2015 administered by AI or PFS during both treatment periods and during the follow‐up visit.
Discussion
The development of GP2015, a proposed etanercept biosimilar, was geared towards the ‘totality of the evidence’ concept 7, involving an extensive analytical and nonclinical characterization to support bioequivalence. The bioequivalence study presented here expands the evidence for GP2015 being biosimilar to etanercept.
PK bioequivalence studies are often carried out in healthy volunteers as they represent the most sensitive population for evaluation of product similarity because they are likely to produce less PK variability compared with patients harbouring potentially confounding factors, such as underlying and/or concomitant disease and concomitant medications. Furthermore, healthy volunteers represent an immunocompetent population to compare the safety and immunogenicity of GP2015. Given the elimination half‐life of approximately 100 h and the low incidence of ADAs (5%) reported for the originator etanercept 12, 13, the study was designed as a crossover study in keeping with regulatory guidelines 14 requiring a smaller sample size than a parallel group design. In this sensitive experimental setting, GP2015 PK was shown to be bioequivalent to Enbrel/EU, further expanding the clinical evidence for GP2015 being biosimilar to or essentially the same as etanercept.
As patients with advanced stages of RA may have compromised dexterity 8 and have previously been shown to be more comfortable with etanercept being administered with an AI 11, a single use, pre‐filled AI of GP2015 was developed in addition to the PFS. The PK evaluations demonstrated bioequivalence between GP2015 administered by AI or PFS. Similar results were observed in both groups, providing clear evidence that the new device delivers drug in an equivalent way to PFS suggesting that GP2015 can be dosed via AI or PFS interchangeably. Such clinical evidence is required by regulatory agencies outside Europe.
The AI was designed to be preferred by patients owing to its ergonomic contours for ease of patient use, thereby reducing usability challenges. It was shown to be a convenient, easy to use device for s.c. self‐administration of secukinumab in another study 15.
The bioequivalence between GP2015 administered by AI and PFS was not affected by recruiting subjects across a broad range of body weights. This demonstration of PK similarity across a broad weight range is commonly excluded from PK studies, since healthy volunteer studies are commonly restricted to subjects with a BMI of less than 30 kg m–2 by excluding obese subjects. However, subcutaneous administration is influenced by the thickness of the hypodermis which is strongly correlated with body weight 16. Therefore, subjects with a BMI ranging from 19.3–39.0 including obese subjects, were included in the delivery study to compare the performance of both devices in subjects with varying thickness of the subcutaneous adipose tissue. We have shown that GP2015 delivery from PFS was similar to delivery from the AI. No difference was observed in mean Cmax and tmax for both AI and PFS treatment administrations across the different weight groups, providing additional evidence for a similar delivery/absorption rate of GP2015 from the two devices independent of body weight and submucosal thickness. In addition, the PK data obtained in the device study reproduces earlier findings on the influence of low body weight on the exposure to etanercept that were derived from population PK analysis with the ETN in healthy volunteers and ankylosing spondylitis patients 17.
GP2015 was well tolerated, with no serious AEs reported in any of the subjects. TEAEs frequently noted in these studies are known and documented side effects of ETN 5. No new or unexpected safety issues were observed during the study and the safety profile of GP2015 was consistent with that previously reported for the ETN 5.
Both studies were carried out in healthy subjects, i.e. an immunocompetent population. The majority of the subjects tested negative for ADAs in the bioequivalence study and no confirmed ADA‐positive samples were detected in the delivery study. In the bioequivalence study, three subjects who had been assigned to the GP2015/ETN treatment sequence showed a positive ADA response at the follow‐up visit subsequent to treatment period 2. Therefore, no direct association between the occurrence of ADA in these subjects and exposure to one of the two drugs administered in this treatment sequence having caused this effect could be made. ADA titres in all three subjects were very low, i.e. near the detection limit of the highly sensitive binding ADA assay (21 ng ml–1). None of the subjects developed injection site reactions or clinically significant changes in protocol‐defined lab parameters. One of the subjects reported an AE of mild intensity (‘feeling hot’) on day 8 of treatment period 2. The positive ADA results were considered not to be clinically meaningful, given the low incidence and non‐neutralizing nature of ADAs in both studies, their low titres which were approximately three‐fold of the lower limit of quantitation of the bioanalytical PK assay, and the fact that no clinically significant AE occurred. This is in keeping with previous reports that have shown that etanercept is associated with a low incidence of non‐neutralizing ADA in different patient populations 4, 18, 19, 20 as well as in healthy volunteers 12.
Overall, the results reported here strengthen the ‘totality of the evidence’ that GP2015 is essentially the same as etanercept demonstrating PK bioequivalence in humans. The AI device provided dosing and tolerability equivalent to the PFS across subjects with a large range of body weights and may offer advantages in terms of convenience and ease of use to patients.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author). All authors except Andrej Skerjanec are employees of Hexal AG. Andrej Skerjanec is an employee of Sandoz AG. None of the authors have any other relationships or activities that could appear to have influenced the submitted work.
The study was sponsored by Hexal AG, a Sandoz company. The authors thank Tina Patrick (Novartis Ireland Limited, Dublin, Ireland) and Divya Chandrasekhar (Novartis Healthcare Pvt. Ltd., Hyderabad, India) for medical writing and editorial assistance.
Contributors
All of the authors contributed to the design and conduct of the study, analysis and interpretation of data, preparation, review and approval of the manuscript.
von Richter, O. , Skerjanec, A. , Afonso, M. , Sanguino Heinrich, S. , Poetzl, J. , Woehling, H. , Velinova, M. , Koch, A. , Kollins, D. , Macke, L. , and Wuerth, G. (2017) GP2015, a proposed etanercept biosimilar: Pharmacokinetic similarity to its reference product and comparison of its autoinjector device with prefilled syringes. Br J Clin Pharmacol, 83: 732–741. doi: 10.1111/bcp.13170.
References
- 1. Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP, et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 2016; 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E, et al. The Concise Guide to PHARMACOLOGY 2015/16: Overview. Br J Pharmacol 2015; 172: 5729–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Goffe B, Cather JC. Etanercept: an overview. J Am Acad Dermatol 2003; 49: S105–S111. [DOI] [PubMed] [Google Scholar]
- 4. Amgen . Enbrel US Prescribing Information, 1998. Available at http://pi.amgen.com/united_states/enbrel/derm/enbrel_pi.pdf (last accessed 16 March 2016).
- 5. European Medicines Agency. Enbrel . Summary of Product Characteristics. Available at http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Product_Information/human/000262/WC500027361.pdf (last accessed 18 March 2016).
- 6. McCamish M, Woollett G. The state of the art in the development of biosimilars. Clin Pharmacol Ther 2012; 91: 405–417. [DOI] [PubMed] [Google Scholar]
- 7. Holzmann J, Balser S, Windisch J. Totality of the evidence at work: the first U.S. biosimilar. Expert Opin Biol Ther 2016; 16: 137–142. [DOI] [PubMed] [Google Scholar]
- 8. Kivitz A, Cohen S, Dowd JE, Edwards W, Thakker S, Wellborne FR, et al. Clinical assessment of pain, tolerability, and preference of an autoinjection pen versus a prefilled syringe for patient self‐administration of the fully human, monoclonal antibody adalimumab: the TOUCH trial. Clin Ther 2006; 28: 1619–1629. [DOI] [PubMed] [Google Scholar]
- 9. Bewley A, Page B. Maximizing patient adherence for optimal outcomes in psoriasis. J Eur Acad Dermatol Venereol 2011; 25: 9–14. [DOI] [PubMed] [Google Scholar]
- 10. Paul C, Stalder JF, Thaci D, Vincendon P, Brault Y, Kielar D, et al. Patient satisfaction with injection devices: a randomized controlled study comparing two different etanercept delivery systems in moderate to severe psoriasis. J Eur Acad Dermatol Venereol 2012; 26: 448–455. [DOI] [PubMed] [Google Scholar]
- 11. Müller‐Ladner U, Flipo RM, Vincendon P, Brault Y, Kielar D. Comparison of patient satisfaction with two different etanercept delivery systems. A randomised controlled study in patients with rheumatoid arthritis. Z Rheumatol 2012; 71: 890–899. [DOI] [PubMed] [Google Scholar]
- 12. Sullivan JT, Ni L, Sheelo C, Salfi M, Peloso PM. Bioequivalence of liquid and reconstituted lyophilized etanercept subcutaneous injections. J Clin Pharmacol 2006; 46: 654–661. [DOI] [PubMed] [Google Scholar]
- 13. Zhou H. Clinical Pharmacokinetics of Etanercept: A fully humanized soluble recombinant tumor necrosis factor receptor fusion protein. J Clin Pharmacol 2005; 45: 490–497. [DOI] [PubMed] [Google Scholar]
- 14. FDA draft guidance for industry: Clinical Pharmacology Data to Support a Demonstration of Biosimilarity to a Reference Product, 2014. Available at http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm397017.pdf.
- 15. Paul C, Lacour JP, Tedremets L, Kreutzer K, Jazayeri S, Adams S, et al. Efficacy, safety and usability of secukinumab administration by autoinjector/pen in psoriasis: a randomized, controlled trial (JUNCTURE). J Eur Acad Dermatol Venereol 2015; 29: 1082–1090. [DOI] [PubMed] [Google Scholar]
- 16. Ludescher B, Rommel M, Willmer T, Fritsche A, Schick F, Machann J. Subcutaneous adipose tissue thickness in adults – correlation with BMI and recommendations for pen needle lengths for subcutaneous self‐injection. Clin Endocrinol (Oxf) 2011; 75: 786–790. [DOI] [PubMed] [Google Scholar]
- 17. Zhou H, Buckwalter M, Boni J, Mayer P, Raible D, Wajdula J, et al. Population‐based pharmacokinetics of the soluble TNFr etanercept: a clinical study in 43 patients with ankylosing spondylitis compared with post hoc data from patients with rheumatoid arthritis. Int J Clin Pharmacol Ther 2004; 42: 267–276. [DOI] [PubMed] [Google Scholar]
- 18. Dore RK, Mathews S, Schechtman J, Surbeck W, Mandel D, Patel A, et al. The immunogenicity, safety and efficacy of etanercept liquid administered once weekly in patients with rheumatoid arthritis. Clin Exp Rheumatol 2007; 25: 40–46. [PubMed] [Google Scholar]
- 19. Hoshino M, Yoshio T, Onishi S, Minota S. Influence of antibodies against infliximab and etanercept on the treatment effectiveness of these agents in Japanese patients with rheumatoid arthritis. Mod Rheumatol 2012; 22: 532–540. [DOI] [PubMed] [Google Scholar]
- 20. Mazilu D, Opriş D, Gainaru C, Iliuta M, Apetrei N, Luca G, et al. Monitoring drug and antidrug levels: a rational approach in rheumatoid arthritis patients treated with biologic agents who experience inadequate response while being on a stable biologic treatment. Biomed Res Int 2014; 2014: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]