

Abstract
Aims
The aims of this study are to apply a theory‐based mechanistic model to describe the pharmacokinetics (PK) and pharmacodynamics (PD) of S‐ and R‐warfarin.
Methods
Clinical data were obtained from 264 patients. Total concentrations for S‐ and R‐warfarin were measured by ultra‐high performance liquid tandem mass spectrometry. Genotypes were measured using pyrosequencing. A sequential population PK parameter with data method was used to describe the international normalized ratio (INR) time course. Data were analyzed with NONMEM. Model evaluation was based on parameter plausibility and prediction‐corrected visual predictive checks.
Results
Warfarin PK was described using a one‐compartment model. CYP2C9 *1/*3 genotype had reduced clearance for S‐warfarin, but increased clearance for R‐warfarin. The in vitro parameters for the relationship between prothrombin complex activity (PCA) and INR were markedly different (A = 0.560, B = 0.386) from the theory‐based values (A = 1, B = 0). There was a small difference between healthy subjects and patients. A sigmoid Emax PD model inhibiting PCA synthesis as a function of S‐warfarin concentration predicted INR. Small R‐warfarin effects was described by competitive antagonism of S‐warfarin inhibition. Patients with VKORC1 AA and CYP4F2 CC or CT genotypes had lower C50 for S‐warfarin.
Conclusion
A theory‐based PKPD model describes warfarin concentrations and clinical response. Expected PK and PD genotype effects were confirmed. The role of predicted fat free mass with theory‐based allometric scaling of PK parameters was identified. R‐warfarin had a minor effect compared with S‐warfarin on PCA synthesis. INR is predictable from 1/PCA in vivo.
Keywords: body size, fat free mass, international normalized ratio, pharmacogenomics, turnover, warfarin
What Is Already Known about this Subject
A number of warfarin dosing algorithms have been reported using different methods for linking warfarin concentration to effect.
The link has often been described using empirical relationships without clear theoretical foundations.
What This Study Adds
In vivo, but not in vitro, observations are consistent with a simple theory‐based inverse relationship between international normalized ratio and prothrombin complex activity.
The relationship between warfarin pharmacokinetics and size is best described by fat free mass.
The relative quantitative contribution of genotypes has been evaluated.
R‐warfarin acts as a competitive antagonist of warfarin.
Tables of Links
| TARGETS | |
|---|---|
| Enzymes 2 | CYP2C9 |
| vitamin K epoxide reductase complex subunit 1 | CYP4F2 |
| LIGANDS | |
|---|---|
| warfarin | |
| amiodarone |
Introduction
Warfarin is a commonly prescribed oral anticoagulant used to prevent and treat thromboembolic events 3, 4, 5. Long‐term warfarin therapy is often required for cardiac surgery patients. Warfarin therapy is challenging because of a delayed onset of response, narrow therapeutic range and marked between‐subject variability 4. Warfarin is administered as a racemic mixture containing equal amounts of S‐ and R‐ enantiomers 3, 4, 6. The daily maintenance dose of warfarin may vary by more than 10‐fold 7, 8, 9, 10. The therapeutic effect and safety of warfarin are controlled by targeting the international normalized ratio (INR) within an acceptable range 5, 11, 12. The risk of thrombotic events will increase when the INR is lower than the lower limit of the acceptable range, whereas the risk of haemorrhagic events will increase when the INR is above the upper limit of the acceptable range 4, 5, 9, 11.
The factors that contribute to between and within‐subject variability in warfarin dose are numerous, including patients' demographic characteristics, dietary vitamin K intake, pathophysiological characteristics, and concomitant medications 7, 12. Genetic differences have been found through genome‐wide association studies that are strongly associated with the required dose of warfarin 9, 13, 14, 15. The genes that are involved in warfarin metabolism and action include CYP2C9, cytochrome P450 oxidoreductase (POR), CYP4F2, and vitamin K epoxide reductase complex subunit 1 (VKORC1). Numerous algorithms that are based on patient factors and genetic variants have been established by multivariate linear regression to predict warfarin daily dosage, but approximately 40% of individual variability remains unexplained 12, 16.
Modeling techniques can be valuable tools for individual dose predictions for drugs with considerable variability in individual requirements, including selection of the optimal initial dose and adjustment of subsequent doses 16, 17, 18, 19. Several studies of warfarin pharmacokinetics (PK) and pharmacodynamics (PD) models have been reported, but most have focused on mixed disease populations, such as patients with atrial fibrillation, deep vein thrombosis, pulmonary embolism 16, 18, 20 or healthy volunteers 21, 22. No reported studies have specifically focused on cardiac surgery patients.
The purposes of the present study were to apply a theory‐based mechanistic model to describe the PK and PD of S‐ and R‐warfarin on the turnover of clotting factors leading to a change in INR and to identify the contribution of body size, body composition, genotype and R‐warfarin to variability in INR response. The primary objective of this analysis is an integrated and principle based description of the clinical pharmacology of S‐ and R‐ warfarin.
Methods
Patients and sampling
The present study was approved by the Health Authority Ethics Committee of the First Affiliated Hospital of Soochow University and was in accordance with the Declaration of Helsinki. All patients gave written informed consent. Patients were administered warfarin once daily following cardiac surgery, including heart valve replacement, thoracic aorta vascular replacement, thoracic endovascular aortic repair, and followed‐up from the first administered warfarin dose. When artificial material comes in contact with blood, it increases the risk of thrombosis, so warfarin was used to reduce the risk. This was the only indication for warfarin. Demographic characteristics, genetic factors and concomitant medications were collected from the medical records.
Blood samples for INR measurements were performed according to the judgement of the treating physician. The acceptable INR was 1.5–2.5 23. The base INR value was the most recently measured INR value before cardiac surgery. Blood samples for S‐ and R‐warfarin samples were taken in addition to measurement of INR as part of clinical care of patients and preserved at −70°C.
Genotyping
Blood samples were used to extract genomic DNA. Genomic DNA was extracted from blood leucocytes with a genomic DNA purification kit (Promega, USA). The present study considered gene polymorphisms associated with warfarin metabolism and action that included CYP2C9 (rs1057910), POR*28 (rs1057868), PORrs286 (rs2868177), CYP4F2 (rs2108622) and VKORC1 (rs9923231). CYP2C9 and VKORC1 were performed as part of standard clinical care. The other genotypes were a research procedure. All genes were genotyped by pyrosequencing. Primers for pyrosequencing were designed with PyroMark Assay Design Software 2.0. One of the primers used for amplification of DNA for polymerase chain reaction (PCR) analysis was biotinylated. The PCR sequencing primers and the sequences of sites to analyze in the pyrosequencing assays are shown in Supplementary Table S1. Thermal cycling conditions for PCR were: 95°C for 5 min (for CYP2C9 and VKORC1) or 3 min (for CYP4F2, POR*28, and PORrs286); 35 cycles at 95°C for 30 s, at 60°C (for CYP2C9 and VKORC1) or 55°C (for CYP4F2, POR*28, and PORrs286) for 30 s and 72°C for 30 s; and a final extension step at 72°C for 7 min. The pyrosequencing genotyping assay for all genes was designed by Pyrosequencing Assay Design Software (Qiagen, Germany). Pyrosequencing was conducted according to the manufacturer's protocol.
Bioanalysis
Total (bound plus unbound) plasma concentrations of S‐ and R‐warfarin were measured simultaneously by ultra‐high performance liquid chromatography–tandem mass spectrometry (UPLC/MS–MS, Waters) 7. The linear range for S‐ and R‐warfarin concentration was 25–1000 ng ml–1. The precision of the assay, as indicated by the coefficient of variation, was 2.2% for S‐warfarin, and 9.63% for R‐warfarin at 25 ng ml–1. The intra‐ and interday precision at concentrations of 50, 250 and 800 ng ml–1, as indicated by the coefficient of variation, was between 0.7 and 5.5%, and the assay accuracy was within 15% of the actual quality control concentration for both S‐ and R‐warfarin (Supplementary Table S2).
The INR for prothrombin time was measured in a single hospital clinical laboratory. To ensure the result of INR, the hospital clinical laboratory participate several times in the quality control scheme of the National Center for Clinical Laboratories of China every year. The international sensitivity index of the hospital clinical laboratory was 1.05.
The PKPD and turnover model analysis used the population PK parameter and data sequential approach 24, 25. Data analysis was performed using NONMEM (version 7.3.0; ICON Development Solutions), and Wings for NONMEM (version wfn741, http://wfn.sourceforge.net/). All parameters were estimated using NONMEM's first‐order conditional estimation method with interaction. Computation was performed on the NeSI cluster at the University of Auckland using four parallel processors. The NONMEM license for use on a multicomputer cluster was granted to the Australasian Centre for Pharmacometrics by ICON Development Solutions.
PK model
One‐compartment PK models with first‐order absorption were considered to describe the PK of warfarin. The PK models were parameterized using clearance (CL in general, CLS for S‐, CLR for R‐warfarin), distribution volume (V in general, VS for S‐, VR for R‐warfarin), absorption half‐life, and assumed a population bioavailability (F) of 1. The PK models for S‐ and R‐warfarin were developed simultaneously using the NONMEM subroutine ADVAN 5.
PD and turnover model
The anticoagulant effect of warfarin is primarily due to inhibition of vitamin K epoxide reductase, thereby inhibiting the synthesis factors (factors II, VII, IX and X) determining prothrombin complex activity (PCA).
Warfarin concentrations were assumed to have an immediate effect on the synthesis of factors determining PCA. The PD model for the effects of S‐ and R‐warfarin on PCA synthesis was based on the sigmoid Emax model (Equation (1)) 26, 27. Two models were considered for the interaction of S‐ and R‐ warfarin. The first assumed that S‐ and R‐warfarin had different values of C50 (Equation (1)). The concentrations of S‐ (CS) and R‐warfarin (CR) were normalized by the concentrations producing 50% of Emax (C50S and C50R) if each enantiomer was given separately.
| (1) |
The second assumed R‐warfarin was a competitive antagonist of S‐warfarin (Equation (2)). RIC50 is the parameter describing competitive antagonism of R‐warfarin.
| (2) |
A turnover model was used to describe the kinetics of PCA synthesis and elimination (Equation (3)).
| (3) |
The initial condition for Equation (3) was PCA0 (baseline PCA assumed to be at steady state). Rsyn represents the zero‐order synthesis rate of PCA, and KPCA represents the first‐order elimination rate constant for PCA. The half‐life (T2PCA) corresponding to KPCA was estimated and KPCA calculated from ln(2)/T2PCA. Rsyn was predicted from KPCA multiplied by PCA0. The possibility of feedback of PCA on the synthesis of PCA was investigated with an empirical feedback function (Equation (4)) based on the PCA relative to the baseline PCA0 and a power parameter.
| (4) |
The INR is used as an index of anticoagulation factors 16. It is directly related to the time needed for plasma to coagulate under standardized conditions in vitro. Coagulation is assumed to occur when the factors described by PCA have produced a particular concentration of fibrin so that a clot can form. The theory of linear reaction kinetics predicts that the time to reach this concentration will be inversely proportional to the concentration of coagulation factors and thus to PCA 21, 28. The theory‐based relationship with A = 1 and B = 0 is shown in Equation (5).
| (5) |
An empirical extension of the theory‐based relationship has been used in earlier studies of warfarin using in vitro estimates of A and B 21, 28.
The relationship between INR and PCA was investigated in vitro. Blood was collected from 25 healthy subjects and 25 patients who needed warfarin before starting anticoagulant treatment. A 2.7‐ml sample of blood was draw from each subject into a plastic tube (Becton, Dickinson and Company), which included 0.3 ml 0.109 mol l–1 sodium citrate anticoagulation. Plasma was separated and then mixed with 0.9% saline to reach a final volume of 250 μl containing between 10 and 90% saline. The expected PCA was calculated from the dilution of plasma with saline. The INR was measured in each of the PCA dilutions using the same method used in clinical practice. The model for INR (Equation (5)) was used to estimate A and B with a proportional residual error model (Equation (12)) using NONMEM and $PRED.
The PKPD and turnover models for warfarin were developed using the NONMEM subroutine ADVAN 13.
Predictable sources of between‐ and within‐subject variability
The covariates included both continuous and categorical covariates. The continuous covariates included body size and age.
Fat‐free mass (FFMi) was calculated according to an individual patient's total body mass (TBMi), height, and sex 29. The individual normal fat mass (NFMi) was then calculated according to Equation (6).
| (6) |
where Ffat represents the fat mass (TBM – FFM) equivalent to FFM in terms of allometric size 30.
The standard fat free mass (FFMSTD) for a standard male subject with weight 70 kg (TBMSTD) and height 176 cm is 56.6 kg. Standard NFM is calculated using Equation (7):
| (7) |
The variability of CL and V was predicted using body size based on a theory‐based allometric model (Equation (8) and (9)).
| (8) |
| (9) |
where CLSTD is the CL in a standard individual and VSTD is the distribution volume in a standard individual.
The continuous covariate of age was divided into decades and the empirical association of age with each decade was estimated for CL, V, T2PCA, C50‐S and HILL relative to the 40–49 years decade.
The categorical covariates included sex, the CYP2C9, CYP4F2, POR*28, PORrs286, and VKORC1 genotypes, coadministration of amiodarone and fluconazole, the reason for using warfarin, and the type of heart valve replacement (mechanical, bioprosthetic valve or no valve replacement). Some patients were treated with intravenous vitamin K1 when the INR was considered too high. The effect of vitamin K1 was modelled by assuming the effect of warfarin (PD, Equation (1)) was zero during vitamin K1 treatment.
Random sources of between‐subject and between‐occasion variability
The between‐subject and between‐occasion variability of the parameters used an exponential model (Equation (10)).
| (10) |
where θij represents the ith individual value of the parameter on the jth occasion, θ represents the population value of the parameters, η represents the between subject variability, and κ represents the between occasion variability. η was normally distributed with a mean of 0 and a variance of ω2, κ was normally distributed with a mean of 0 and a variance of π2.
Between‐subject variability was estimated for the PK, PD and turnover model parameters. An occasion was defined by one or more concentration observations within a dosing interval. Between‐occasion variability was estimated for the PK parameters CL, V and F. It was assumed that between‐subject and between‐occasion variability were the same for S‐ and R‐ warfarin.
Residual unidentified variability
The residual unidentified variability (residual error) for the PK observations used a combined (proportional plus additive) model (Equation (11)).
| (11) |
where θPROP represents the parameter of the proportion residual error, θADD represents the parameter of addition residual error, CONC represents the individual predicted S‐ or R‐warfarin concentration, Y represents the observed concentrations, and ε is the residual error, assumed to be normally distributed with a mean of 0 and a variance of σ2.
The residual unidentified variability (residual error) model for INR (Equation (5)) used a proportional model (Equation (12)).
| (12) |
where Y represents the observed INR, INR the predicted INR and ε the random effect (Equation (12)).
Model selection
A nonparametric bootstrap was performed with a model including all covariate effects first for the PK model then for the PD and turnover model. The 95% bootstrap confidence interval was used to determine if the covariate explained parameter variability. Covariates were eliminated if this confidence interval included a value indicating no effect of the covariate as a predictor of individual differences. This method was used because of the large number of potential covariate effects and the potential problems of relying on multiple likelihood ratio tests 31, 32.
Certain models were selected based on a significant improvement in the goodness of fit using the likelihood ratio test assuming a chi‐square distribution and alpha = 0.05.
Model selection included a preference for models that were based on theory (e.g. theory‐based allometry) and mechanism (e.g. turnover of clotting factors) rather than empiricism (e.g. relationship between INR and PCA).
Model evaluation
Model evaluation was based on parameter plausibility and visual predictive checks (VPC). The prediction‐corrected VPC was preferred to mitigate the effect of dose adjustment based on INR 33.
Results
Patient demographic data
A total of 264 patients, with 2858 observed S‐ and R‐warfarin concentrations and 2163 observed INR values of S‐ and R‐warfarin, were enrolled. The median number of warfarin (S‐ and R‐) concentrations was 10 and the median number of INR values was eight per patient. The median INR at the end of follow up was 1.6 with a median follow up time of 8.8 weeks. The demographic characteristics of the patients are presented in Table 1. The frequency distributions of CYP2C9, VKORC1, CYP4F2, POR*28 and PORrs286 polymorphisms are described in Table 2. The distributions of the various genotypes, except for POR*28, were in accordance with Hardy–Weinberg equilibrium (P > 0.05). Amiodarone was administered to 113 patients (42.8%) for the prevention and treatment of atrial fibrillation. Seventeen patients (6.44%) were administered fluconazole because of postoperative infection. It was assumed that the enzyme inhibitory effect of each of the two drugs was maintained for 1 week after treatment was stopped. Seventy‐six patients (28.8%) were administered vitamin K1 by intravenous infusion because the INR value was high.
Table 1.
Demographic and disease characteristics of the patients
| Patient characteristic | Value, mean ± SD (95% interval, n) |
|---|---|
| Demographic characteristics | |
| Sex (male/female) | 148/116 |
| Age < 30 years | 24.7 ± 4.1 (16.8–29.0, 15) |
| 30–49 years | 35.4 ± 2.9 (30–39, 25) |
| 40–49 years | 45.2 ± 2.6 (40–49, 55) |
| 50–59 years | 55.2 ± 2.6 (50–59,74) |
| 60–69 years | 63.4 ± 2.8 (60–69,72) |
| Age ≥ 70 years | 73.9 ± 3.0 (70–79.8, 23) |
| Height (cm) | 164.2 ± 8.2 (150–180) |
| Weight (kg) | 61.8 ± 11.4 (40.0–86.0) |
| BMI (kg m–2) | 22.8 ± 3.38 (16.2–29.7) |
| BSA (m2) | 1.76 ± 0.18 (1.44–2.12) |
| Type of valve | |
| Mechanical valve prosthesis | 193 (73.1%) |
| Bioprosthetic heart valves | 59 (22.3%) |
| No valve replacement | 12 (4.5%) |
| Reason for surgery | |
| AVR (n, %) | 62 (23.5%) |
| MVR (n, %) | 113 (42.8%) |
| DVR (n, %) | 60 (22.7%) |
| TVR (n, %) | 3 (1.1%) |
| Bentall (n, %) | 14 (5.3%) |
| TEVAR (n, %) | 12 (4.5%) |
AVR, aortic valve replacement; MVR, mitral valve replacement; DVR, aortic and mitral valve replacement; SD, standard deviation; TVR, tricuspid valve replacement; TEVAR, thoracic endovascular aortic repair.
Table 2.
Frequency distributions of CYP2C9, VKORC1, CYP4F2, POR*28, and PORrs286 polymorphisms in the current patient population
| SNP | Allele frequencies | Genotype frequencies | Hardy–Weinberg equilibrium test (P value) | |||
|---|---|---|---|---|---|---|
| CYP2C9 (rs1057910) | *1 (97.2%) | *3 (2.8%) | *1/*1249 (94.3%) | *1/*3 15(5.7%) | *3/*3 0(0%) | 0.635 |
| VKORC1 (rs9923231) | G (8.9%) | A (91.1%) | GG 1(0.4%) | GA 45(17.0%) | AA 218 (82.6%) | 0.407 |
| CYP4F2 (rs2108622) | C (75.0%) | T (25.0%) | CC 148(56.1%) | CT 100(37.8%) | TT 16(6.1%) | 0.870 |
| POR*28 (rs1057868) | C (66.7%) | T (33.3%) | CC 108(40.9%) | CT 136(51.5%) | TT 20(7.6%) | 0.010 |
| PORrs286 (rs2868177) | G (44.5%) | A (55.5%) | GG 51(19.3%) | GA 133(50.4%) | AA 80 (30.3%) | 0.747 |
PK models
Because there were no observations during the absorption phase, the absorption half‐life was fixed to a value of 0.385 h corresponding to a first‐order absorption rate constant of 1.8 h−1. Other values (ranging from 1.6–2.0 h−1) were also tested according to previous reports but did not obviously improve the fit 16, 18, 20, 34.
The bootstrap confidence intervals for the full PK model (Supplementary Tables S3 and S4) were used to identify significant covariate effects. A minimal model was then obtained by removing all nonsignificant covariate effects. All the age covariate parameters were included because they may carry information associated with age such as impaired organ function. The parameter estimates for the minimal pharmacokinetic model are shown in Tables 3, 4. These intervals showed that the Ffat parameter were not different from zero for CL or V for either S‐ or R‐warfarin. Setting the Ffat parameters to 0 increased the objective function value (OFV) by 1.452 (chi‐square degrees of freedom [df] = 4, P = 0.835). Therefore, fat free mass was considered the most suitable descriptor of allometric size. Amiodarone (AMI) resulted in a 24.5% and 13.2% decrease in CL for S‐ and R‐warfarin, respectively. Coadministered fluconazole (FLU) resulted in a 43.8% decrease in CL for S‐warfarin without any significant effect on CL for R‐warfarin. Removal of the effect of fluconazole on CLR increased the OFV 1.827 (chi‐square df = 1, P = 0.176), which confirmed the bootstrap finding. CYP2C9 (rs1057910, A > C) *1/*3 genotype reduced CL for S‐ (16.7%) compared with *1/*1, but increased CL (23.4%) for R‐warfarin. Removal of the effect of CYP2C9 *1/*3 on CLR increased the OFV by 4.377, Chi‐square df = 1, P = 0.0364 which confirmed the bootstrap finding. A bioprosthetic type of valve was associated with a decrease in CL, Female sex was associated with an increase in VS and VR. There was no evidence for an effect of age on CLS, CLR, VS or VR in any decade. Removal of age from the model increased the OFV by 14.471, (chi‐square df = 10, P = 0.152; see Supplementary Figure S3 showing the trends with age). Female patients had a distribution volume 34.3% higher for S‐warfarin compared to male patients. There was a similar trend for R‐warfarin 23.3% higher.
Table 3.
Parameter estimates of the warfarin original minimal pharmacokinetic model and bootstrap average, 95% confidence interval (CI) and relative standard error (RSE). Only covariate effect parameters with 95% CI excluding 1 are shown
| Parameter | Original | Units | Average | 95% CI | RSE | |
|---|---|---|---|---|---|---|
| POP_CLS | 0.224 | l h–1 | 0.225 | 0.201 | 0.246 | 5% |
| POP_VS | 24.00 | l | 23.88 | 19.70 | 27.70 | 9% |
| POP_CLR | 0.124 | l h–1 | 0.124 | 0.111 | 0.135 | 5% |
| POP_VR | 16.20 | l | 16.11 | 13.29 | 18.61 | 9% |
| POP_TABS | 0.385 | FIXED | ||||
| FAMI_CLS | 0.760 | 0.755 | 0.666 | 0.851 | 6% | |
| FAMI_CLR | 0.872 | 0.868 | 0.789 | 0.954 | 5% | |
| FFLU_CLS | 0.549 | 0.562 | 0.284 | 0.882 | 27% | |
| F2C9_CLS | 0.830 | 0.833 | 0.689 | 0.991 | 10% | |
| F2C9_CLR | 1.230 | 1.234 | 1.020 | 1.470 | 9% | |
| FTYP2_CL | 0.775 | 0.761 | 0.625 | 0.905 | 9% | |
| FFEM_VS | 1.33 | 1.343 | 1.130 | 1.620 | 9% | |
| FFEM_VR | 1.22 | 1.233 | 1.080 | 1.410 | 7% | |
CLS, VS, CLR and VR are per 56.1 kg FFM. POP, population parameter; FAMI, effect of amiodarone on CLS and CLR; FFLU, effect of fluconazole on CLS; F2C9 is the effect of CYP2C9 *1/*3 genotype relative to *1/*1 on CLS and CLR; FTYP2, effect of bioprosthetic valve on CL (similar for both CLS and CLR); FFEM, effect of female sex on V.
Table 4.
Random effect parameter estimates of the warfarin original minimal pharmacokinetic model and bootstrap average, 95% confidence interval (CI) and relative standard error (RSE). Only parameters with 95% CI excluding 0.001 are shown
| Parameter | Original | Average | 95% CI | RSE | |
|---|---|---|---|---|---|
| BSV_CLS | 0.224 | 0.222 | 0.186 | 0.257 | 8% |
| BOV_VS1 | 0.123 | 0.111 | 0.001 | 0.178 | 37% |
| BSV_CLR | 0.073 | 0.052 | 0.001 | 0.134 | 83% |
| BSV_F | 0.156 | 0.148 | 0.092 | 0.195 | 18% |
| BOV_F1 | 0.284 | 0.287 | 0.235 | 0.344 | 10% |
| RUV_PROP_CS | 0.279 | 0.278 | 0.260 | 0.297 | 3% |
| RUV_ADD_CS | 4.57 | 4.297 | 0.046 | 7.071 | 39% |
| RUV_PROP_CR | 0.243 | 0.242 | 0.227 | 0.257 | 3% |
BSV, between‐subject variability; BOV, between‐occasion variability; PROP, proportional residual error; ADD, addition residual error; RSE, relative standard error.
Although several covariates had no statistically significant effect on PK parameters they were nevertheless included in the sequential PKPD model (fixed to the original data estimates shown in Supplementary Tables S3, S4 and S5) to improve the prediction of individual concentration profiles.
PD model
The in vitro estimates of A and B for prediction of INR (Equation (5)) were 0.482 and 0.409 for healthy subjects, and 0.560 and 0.386 for patients, respectively. The values of A and B were highly significantly different from the theory‐based values of A = 1 and B = 0 (OFV change 184.8, Chi‐square df = 2, P < 1e‐40). By contrast, there was no improvement of in vivo estimated values of A and B compared with theory‐based values (OFV change 0.672, Chi‐square df = 2, P = 0.714).
Emax was not significantly different from 1 and was therefore fixed to 1. The bootstrap confidence intervals for the full PD and turnover model (Supplementary Table S5) were used to identify significant covariate effects using the full PK model parameter estimates. A minimal model was then obtained by removing all nonsignificant PK and PD covariate effects. The minimal PD and turnover model parameter estimates are shown in Table 5. S‐warfarin concentrations predicted the inhibition of PCA synthesis. The C50 for S‐warfarin was 0.391 mg l–1. A lower C50 was estimated with VKORC1 (rs9923231,‐1639G > A) AA (72.2%) and CYP4F2 (rs2108622, C > T) CC or CT (76.6%) genotype. Combining the effect of CYP4F2 CC and CT genotypes did not worsen the fit significantly, indicating that the effects of these genotypes can be considered to be the same (OFV change 0.013, Chi‐square df = 1, P = 0.909). When the effect of R‐warfarin was described in terms of potency relative to S‐warfarin the potency estimate was −0.126. This does not have any good pharmacological interpretation so the effect of R‐warfarin was described as a competitive antagonist for S‐warfarin (Equation (2)). The R‐warfarin concentration producing a 50% increase in apparent C50 for S‐warfarin was 2.36 mg l–1.
Table 5.
Parameter estimates of the warfarin original minimal pharmacodynamic and turnover model and bootstrap average, 95% confidence interval (CI) and relative standard error (RSE)
| Parameter | Original | Units | Average | 95% CI | RSE | |
|---|---|---|---|---|---|---|
| POP_PCA0 | 0.979 | 0.979 | 0.964 | 0.992 | 1% | |
| POP_EMAX | 1 | FIXED | ||||
| POP_C50 | 0.378 | mg l–1 | 0.391 | 0.281 | 0.536 | 17% |
| POP_HILL | 2.720 | 2.723 | 2.330 | 3.320 | 10% | |
| POP_T2PCA | 11.400 | h | 11.5 | 10.1 | 13.0 | 7% |
| POP_RIC50 | 2.387 | mg l–1 | 2.357 | 0.935. | 238 | 72% |
| FVKOR_C50 | 0.725 | 0.722 | 0.632 | 0.826 | 7% | |
| F4F20/21_C50 | 0.770 | 0.766 | 0.668 | 0.869 | 7% | |
| PPV_PCA0 | 0 | FIXED | ||||
| PPV_EMAX | 0 | FIXED | ||||
| PPV_C50 | 0.26 | 0.258 | 0.199 | 0.318 | 12% | |
| PPV_HILL | 0.389 | 0.380 | 0.282 | 0.469 | 13% | |
| PPV_T2PCA | 0.341 | 0.335 | 0.213 | 0.464 | 21% | |
| PPV_RIC50 | 0 | FIXED | ||||
| RUV_PROP_INR | 0.177 | 0.176 | 0.166 | 0.187 | 3% | |
POP, population parameter; PPV, population parameter variability; T2PCA, prothrombin complex activity apparent turnover half‐life; FVKOR, effect of VKORC1 AA genotype relative to AG or GG on C50‐S; F4F20/21, effect of CYP4F2 CC or CT genotype relative to TT on C50‐S; PROP, proportional residual error.
The inclusion of an empirical feedback function did not improve the fit (OFV change 1.084, P = 0.298). The bootstrap confidence interval for the feedback parameter γ (−0.411 to 0.651) included zero indicating that there was no robust evidence for a feedback effect. The model with feedback worsened (OFV increase 7.2, Chi‐square df = 1, P = 0.007) when the competitive antagonist effect of R‐warfarin was removed.
Addition of an empirical age effect to the full model T2PCA, C50 and HILL parameters decreased the OFV by 23.3 but this was not statistically significant (Chi‐square df = 15, P = 0.078; see Supplementary Figure S4 showing the trends with age). There is no convincing evidence for an effect of age on T2PCA, C50‐S or HILL at any decade. There was no effect of sex associated with any of the turnover and PD model parameters (OFV decrease 0.013, chi‐square df = 6, P = 0.999).
Model evaluation
The goodness of fit of the minimal model for S‐ and R‐warfarin concentrations and effects on INR was evaluated using a prediction‐corrected VPC (Figures 1 and 2). The corresponding prediction‐corrected VPCs for the full model are shown in Supplementary Figures S1 and S2.
Figure 1.
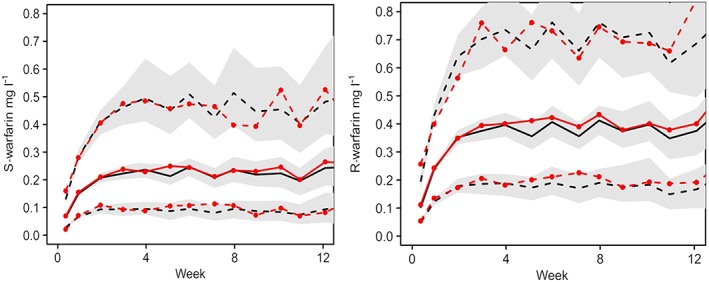
Minimal model Pred Corrected visual predictive checks of S‐ and R‐warfarin concentration time course over 12 weeks. Observed (with symbols) and predicted 5, 50 and 95 percentiles, with predicted 95% confidence intervals (shaded)
Figure 2.
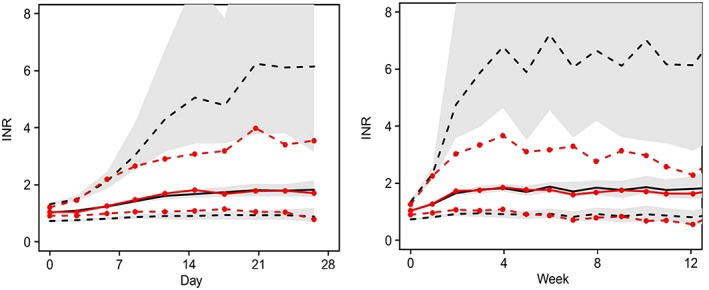
Minimal model Pred Corrected visual predictive checks of international normalized ratio time course over 28 days and 12 weeks. Observed (with symbols) and predicted 5, 50 and 95 percentiles, with predicted 95% confidence intervals (shaded)
Discussion
Theory driven models of warfarin PK, PD and clotting factor turnover were investigated with hospital‐based clinical data from 264 cardiac surgery patients. The relationship between body size and the PK parameters of CL and V was consistent with theory‐based allometry driven by fat‐free mass. The expected lower CL of S‐warfarin associated with CYP2C9 *1/*3 genotype was confirmed implying a lower dose is required to achieve a target INR. Unexpectedly the same genotype were associated with an increase in CL of R‐warfarin. Because R‐warfarin has only a minor effect on the effect of warfarin, this association has no implication for dosing.
As expected, the PD model showed that VKORC1 AA and CYP4F2 CC or CT haplotypes were associated with a lower C50 for S‐warfarin implying a lower dose is required to achieve a target INR.
Numerous algorithms based on patient factors and genetic variants have been established by multivariate linear regression to predict the warfarin daily dosage 12, 16. However, these models only predict warfarin dose at steady state. The PKPD models established here also provide guidance for initial warfarin dosage. Our results could be useful for clinicians in individualizing the dose of warfarin in the clinic. The use of this description of warfarin PKPD for dosing recommendations is a potential application of the model. Development of another dosing method was not the primary purpose of the analysis reported here. It would be possible to predict the first doses of warfarin, before INR measurements are available, using the model we describe. A limitation of this use of our model is the requirement to have available three specific genotypes (CYP2C9, CYP4F2 and VKORC1), which are not all routinely available (especially CYP4F2).
We had genotype information in all patients and so we were able to estimate genotype specific values for CL‐R, CL‐S and C50‐S and appropriate between‐subject variability for these parameters. The problem is how to use these values when the genotype information is not known. We fixed all the parameters except those that describe the genotype effect on CL‐S, CL‐R and C50‐S and estimated the effect that describes this study population when genotype is missing (Supplementary Table S6). These estimates may be used when genotype information is missing with the same model and parameters that are used when the genotype is known. We will use these estimates prospectively as an initial dosing method with a subsequent extension to predict the next dose when INR measurements are available. We plan to do this in the Chinese population because the distribution of genotypes should be similar to that observed in the current analysis. While the predictions may be less precise, we would not expect a systematic bias.
The bootstrap population standard values (fat‐free mass of 56.6 kg, e.g. 70 kg 176 cm male) of CL was 0.225 l h–1 and V was and23.88 L for S‐warfarin, 0.124 l h–1 and 16.11 L for R‐warfarin. Hamberg et al. 16 reported that CL was 0.314 l h–1 and V was 13.8 l for S‐warfarin, 0.139 l h–1 and 12.8 l for R‐warfarin. Yuen et al. 22 reported that CL was 0.276 l h–1 and V was 10.6 l for S‐warfarin, 0.130 l h–1 and 9.39 l for R‐warfarin. The CL values for S‐warfarin and R‐warfarin are consistent with those of previous reports. However, the V values for S‐ and R‐warfarin are higher than those reported. These discrepancies might be due in part to nonstandard reporting of PK parameters and regional or racial differences between the study populations 35, 36.
The bootstrap population values of C50S was 0.391 mg l–1, which is (perhaps) lower than those of previous reports. Hamberg et al. 16, 38 reported that C50S was 1.92 mg l–1 for VKORC1 AA, 3.01 mg l–1 for GA, and 4.10 mg l–1 for GG. Nagashima et al. 37 reported that C50S was 0.842 mg l–1. Wingard et al. 37 reported that C50S was 0.655 mg l–1. Powers et al. 37 reported that C50S was 0.562 mg l–1. Svec et al. 37 reported that C50S was 0.655 mg l–1. Murray et al. 37 reported that C50S was 0.468 mg l–1. Chan et al. 37 reported that C50S was 0.6 mg l–1.
Warfarin is considered an inhibitor of VKOR and as such its inhibitory potency was evaluated under the assumption that both S‐ and R‐warfarin were both capable of 100% inhibition but had different potencies. This model produced a negative estimate for R‐warfarin potency. We therefore used the analogy of partial agonism, where a partial agonist will appear to be a competitive antagonist of an agonist with greater efficacy. Under the assumption that S‐warfarin can completely inhibit VKOR and that R‐warfarin is less effective as an inhibitor, it was possible to describe the effect of R‐warfarin as if it was a competitive antagonist of S‐warfarin. The worsening of the feedback model fit when the R‐warfarin competitive inhibition effect was removed indicates that the R‐warfarin effect, which tends to reduce the apparent potency of warfarin with time, is not due to misspecification by not including a feedback mechanism.
The bootstrap average value of RIC50 was 2.36 mg l–1, which predicts that the effect of R‐warfarin would increase the apparent C50 of S‐warfarin to 0.530 mg l–1 (36% increase) with a dose of 5 mg day–1 in a standard size patient. This could explain why the estimate of C50S is lower than previous reports. The increased CL of R‐warfarin predicted in patients with the CYP2C9 *1/*3 genotype would make the effect of R‐warfarin on S‐warfarin smaller.
The PKPD and turnover model for PCA was used to predict INR. The in vitro parameters for the relationship between PCA and INR were markedly different from the theory‐based values (A = 1, B = 0). In contrast, the in vivo relationship between PCA and INR was consistent with a simple inverse relationship as expected from theory. The fit of in vivo INR values was no better (P = 0.715) using the empirical extension of the model for INR compared with the theory‐based model. In view of the empirical nature of the extension and the lack of correlation with in vitro estimates we recommend using the theory‐based model to predict INR from PCA.
The CL decrease with amiodarone and fluconazole is expected because both are potent inhibitors of cytochrome P450 enzymes 39, 40, 41, 42. The higher volume of distribution in women presumably reflects differences in body composition unrelated to fat free mass between men and women. Bioprosthetic valve patients had lower CL by about 23.9% than other patients’ CL. These patients tended to be older and may have worse cardiac function, which would be expected to impair warfarin CL.
Warfarin is extensively metabolized through specific cytochrome P450 enzymes in the liver. For example, CYP2C9 plays a major role in S‐warfarin metabolism, and CYP2C8, CYP2C18 and CYP2C19 play minor roles. Whereas R‐warfarin is mainly metabolized by CYP1A1, CYP1A2 and CYP3A4, the enzymes CYP2C8, CYP2C18, CYP2C19 and CYP3A4/5 only have a minor role 3. CYP4F2 has been demonstrated to be responsible for the metabolism of vitamin K1 to hydroxyvitamin K1 43, 44. POR is essential for electron donor to microsomal cytochrome P450‐mediated monooxygenation, which plays major roles in the metabolism of most drugs 15, 45. The distribution of the POR*28 genotypes were not in accordance with the Hardy–Weinberg equilibrium (P < 0.05). The percentage of POR*28 CT genotype was higher than expected. Some pharmacogenomics studies have demonstrated that genetic differences in POR and CYP4F2 are also involved in warfarin metabolism and action, and they have contributed to the between‐subject variability of warfarin 15.
Genetic differences in CYP2C9 were confirmed as having a significant impact on PK (CL) parameters for warfarin in the present study. The decrease in S‐warfarin CL with the CYP2C9*1/*3 genotype was expected, however, the increase in R‐warfarin CL with this genotype was not.
Warfarin exerts its anticoagulant effect by interfering with the synthesis of vitamin K‐dependent clotting factors by the inhibition of VKORC1 14, 46. Vitamin K1 is metabolized by CYP4F2 9, 47. Genetic differences in VKORC1, and CYP4F2 both had an effect to decrease the C50 for S‐warfarin.
Figure 3 shows the predicted time course of S‐ and R‐warfarin concentrations and with and without the most frequent genotypes observed in our study. Maintenance dose rates were chosen to achieve a target INR of about 2. Loading doses were chosen to approach steady state concentrations. It is of interest that the transient initial overshoot of the INR and slow decrease towards steady state as R‐warfarin concentrations slowly rise and decrease the effect of S‐warfarin when a loading dose is used with the most frequent genotype combination (Figure 3, upper right plot).
Figure 3.
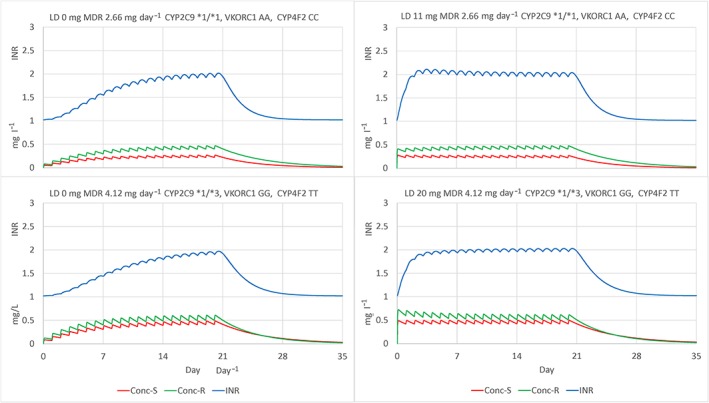
Predicted time course of S‐ and R‐warfarin concentrations and international normalized ratio in a standard size patient (FFM 56.1 kg) with the most frequent genotypes CYP2C9 *1/*1, VKORC1 AA, CYP4F2 CC (upper plots), and least frequent genotypes CYP2C9 *1/*3, VKORC1 GG, CYP4F2 TT (lower plots). Maintenance dose rates (MDR) chosen to achieve target INR of about 2 (left plots). Loading dose (LD) chosen to approach steady state concentrations plus MDR (right plots)
The frequency of the genotypes reported here may be different in other racial groups. For example, VKORC1 A alleles were 91% in this Chinese population, which is similar to Asian–Americans (89%) but higher than Caucasian–Americans (37%) and African–Americans (14%) 48. These racial associations are a well‐known finding for many genotypes that may affect the action of drugs. The differing distribution of genotypes in other racial groups would be expected to produce different effects if these genotypes were not accounted for.
Patients who undergo cardiac surgery are more likely to have other disease factors (e.g. heart failure). Initial dosing and adjustment of dose using INR may occur in a pre‐ and/or postoperative setting that is different from patients who need anticoagulation in a different setting. However, the parameter estimates we obtained are not particularly different from those reported in the literature for other populations of patients. Thus, these results obtained in the cardiac surgery population are likely to be applicable to others.
A negative feedback homeostatic mechanism related to PCA might be expected because of the need for close control of PCA. The nonsignificant improvement of the model fit and the inclusion of zero in bootstrap confidence interval for γ means that there is no support for a feedback mechanism.
A Bayesian dosing method has been developed using the warfarin model described here. It is part of the web based NextDose tool, which is implemented for use in a clinical environment. Instructions for access to NextDose are available at www.nextdose.org.
In summary, a theory‐based PKPD of warfarin was established. Fat‐free mass is the best predictor of body size influencing CL and V. Expected genotype effects were confirmed on CL and C50. R‐warfarin behaves more like a competitive antagonist of S‐warfarin.
Competing Interests
All authors have completed the Unified Competing Interest form and declare that L.X., N.H., X.L.D., C.R.H., H.Z., J.J.Z., Z.N.G., C.X., L.Z., Z.Y.C. and Z.Y.S. have received no support from any external organization for the submitted work. L.S.L. had support from the National Nature Science Foundation of China (grant number: 81503140) for the submitted work. L.Y.M. had support from the Jiangsu Province 333 High‐level Talents Engineering Training program for the submitted work. All authors had no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years. N.H. had salary support from University of Auckland.
Z.N.G. is a student of the College of Pharmaceutical Sciences, Soochow University. Others had salary support from the First Affiliated Hospital of Soochow University. None of the authors has other relationships or activities that could appear to have influenced the submitted work.
The authors would like to acknowledge Chinese individualized drug therapy group of International Network for the Rational Use of Drugs (INRUD), and the department of cardiovascular surgery of the First Affiliated Hospital of Soochow University.
Contributors
L.X.: data collection, concentration determination, data analysis, draft manuscript; N.H.: data analysis, revised manuscript; X.‐l.D.: concentration determination, draft manuscript; Z.‐y.S.: participating investigators, provided and cared for study patients; C.‐r.H.: concentration determination; H.Z.: concentration determination; J.‐j.Z.: gene detection; Z.‐n.G.: gene detection; C.X.: data collection; L.Z.: data collection; Z.‐y.C.: gene detection; L.‐s.L.: concentration determination; L.‐y.M.: principal investigator; revised manuscript.
Supporting information
Figure S1 Full model Pred Corrected visual predictive check S‐ and R‐warfarin concentration time course over 12 weeks. Observed (with symbols) and predicted 5, 50, 95 percentiles, with predicted 95% confidence intervals (shaded)
Figure S2 Full model Pred Corrected visual predictive check international normalized ratio time course over 28 days and 12 weeks. Observed (with symbols) and predicted 5, 50, 95 percentiles, with predicted 95% confidence intervals (shaded)
Figure S3 Empirical association of clearance and volume with age decades
Figure S4 Empirical association of T2PCA, C50 and HILL with age decades
Table S1 Primer sequence for CYP2C9, CYP4F2, POR*28, PORrs286, and VKORC1 single‐nucleotide polymorphism analysis
Table S2 The intra‐ and interday precision for replicate measurements of S‐ and R‐warfarin quality control concentrations
Table S3 Fixed effect parameter estimates of the warfarin original full pharmacokinetic model and bootstrap average, 95% confidence interval and relative standard error (RSE). Nonsignificant covariate parameters are marked NS
Table S4 Random effect parameter estimates of the warfarin original full pharmacokinetic model and bootstrap average, 95% confidence interval and relative standard error (RSE). Nonsignificant parameters are marked NS
Table S5 Random effect parameter estimates of the warfarin original full pharmacodynamic and turnover model and bootstrap average, 95% confidence interval and relative standard error (RSE). Nonsignificant parameters are marked NS
Table S6 Genotype effects (FCYP2C9‐S on CL‐S, FCYP2C9‐R on CL‐R, FVKORC1 and FCYP4F2 on C50‐S) estimated when known genotypes (CYP2C9, VKORC1, CYP4F2) were considered missing in all combinations. All other parameters were fixed to the minimal model estimates
Supporting info item
Supporting info item
Xue, L. , Holford, N. , Ding, X. , Shen, Z. , Huang, C. , Zhang, H. , Zhang, J. , Guo, Z. , Xie, C. , Zhou, L. , Chen, Z. , Liu, L. , and Miao, L. (2017) Theory‐based pharmacokinetics and pharmacodynamics of S‐ and R‐warfarin and effects on international normalized ratio: influence of body size, composition and genotype in cardiac surgery patients. Br J Clin Pharmacol, 83: 823–835. doi: 10.1111/bcp.13157.
References
- 1. Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP, et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 2016; 44 (D1): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alexander SP, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE, et al. The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 2015; 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Limdi NA, Veenstra DL. Warfarin pharmacogenetics. Pharmacotherapy 2008; 28: 1084–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sinxadi P, Blockman M. Warfarin resistance. Cardiovasc J Afr 2008; 19: 215–217. [PubMed] [Google Scholar]
- 5. Gong IY, Tirona RG, Schwarz UI, Crown N, Dresser GK, Larue S, et al. Prospective evaluation of a pharmacogenetics‐guided warfarin loading and maintenance dose regimen for initiation of therapy. Blood 2011; 118: 3163–3171. [DOI] [PubMed] [Google Scholar]
- 6. Muszkat M, Blotnik S, Elami A, Krasilnikov I, Caraco Y. Warfarin metabolism and anticoagulant effect: a prospective, observational study of the impact of CYP2C9 genetic polymorphism in the presence of drug‐disease and drug–drug interactions. Clin Ther 2007; 29: 427–437. [DOI] [PubMed] [Google Scholar]
- 7. Maddison J, Somogyi AA, Jensen BP, James HM, Gentgall M, Rolan PE. The pharmacokinetics and pharmacodynamics of single dose (R)‐ and (S)‐warfarin administered separately and together: relationship to VKORC1 genotype. Br J Clin Pharmacol 2013; 75: 208–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rettie AE, Tai G. The pharmocogenomics of warfarin: closing in on personalized medicine. Mol Interv 2006; 6: 223–227. [DOI] [PubMed] [Google Scholar]
- 9. Takeuchi F, McGinnis R, Bourgeois S, Barnes C, Eriksson N, Soranzo N, et al. A genome‐wide association study confirms VKORC1, CYP2C9, and CYP4F2 as principal genetic determinants of warfarin dose. PLoS Genet 2009; 5: e1000433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Verstuyft C, Delavenne X, Rousseau A, Robert A, Tod M, Diquet B, et al. A pharmacokinetic‐pharmacodynamic model for predicting the impact of CYP2C9 and VKORC1 polymorphisms on fluindione and acenocoumarol during induction therapy. Clin Pharmacokinet 2012; 51: 41–53. [DOI] [PubMed] [Google Scholar]
- 11. Sconce EA, Khan TI, Wynne HA, Avery P, Monkhouse L, King BP, et al. The impact of CYP2C9 and VKORC1 genetic polymorphism and patient characteristics upon warfarin dose requirements: proposal for a new dosing regimen. Blood 2005; 106: 2329–2333. [DOI] [PubMed] [Google Scholar]
- 12. Zhao L, Chen C, Li B, Dong L, Guo Y, Xiao X, et al. Verification of pharmacogenetics‐based warfarin dosing algorithms in Han‐Chinese patients undertaking mechanic heart valve replacement. PLoS One 2014; 9: e94573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Perera MA, Cavallari LH, Limdi NA, Gamazon ER, Konkashbaev A, Daneshjou R, et al. Genetic variants associated with warfarin dose in African‐American individuals: a genome‐wide association study. Lancet 2013; 382: 790–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yuan HY, Chen JJ, Lee MT, Wung JC, Chen YF, Charng MJ, et al. A novel functional VKORC1 promoter polymorphism is associated with inter‐individual and inter‐ethnic differences in warfarin sensitivity. Hum Mol Genet 2005; 14: 1745–1751. [DOI] [PubMed] [Google Scholar]
- 15. Zhang X, Li L, Ding X, Kaminsky LS. Identification of cytochrome P450 oxidoreductase gene variants that are significantly associated with the interindividual variations in warfarin maintenance dose. Drug Metab Dispos 2011; 39: 1433–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hamberg AK, Dahl ML, Barban M, Scordo MG, Wadelius M, Pengo V, et al. A PK‐PD model for predicting the impact of age, CYP2C9, and VKORC1 genotype on individualization of warfarin therapy. Clin Pharmacol Ther 2007; 81: 529–538. [DOI] [PubMed] [Google Scholar]
- 17. Minto C, Schnider T. Expanding clinical applications of population pharmacodynamic modelling. Br J Clin Pharmacol 1998; 46: 321–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sasaki T, Tabuchi H, Higuchi S, Ieiri I. Warfarin‐dosing algorithm based on a population pharmacokinetic/pharmacodynamic model combined with Bayesian forecasting. Pharmacogenomics 2009; 10: 1257–1266. [DOI] [PubMed] [Google Scholar]
- 19. Sheiner LB. Computer‐aided long‐term anticoagulation therapy. Comput Biomed Res 1969; 2: 507–518. [DOI] [PubMed] [Google Scholar]
- 20. Ohara M, Takahashi H, Lee MT, Wen MS, Lee TH, Chuang HP, et al. Determinants of the over‐anticoagulation response during warfarin initiation therapy in Asian patients based on population pharmacokinetic‐pharmacodynamic analyses. PLoS One 2014; 9: e105891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jiang X, Blair EY, McLachlan AJ. Investigation of the effects of herbal medicines on warfarin response in healthy subjects: a population pharmacokinetic‐pharmacodynamic modeling approach. J Clin Pharmacol 2006; 46: 1370–1378. [DOI] [PubMed] [Google Scholar]
- 22. Yuen E, Gueorguieva I, Wise S, Soon D, Aarons L. Ethnic differences in the population pharmacokinetics and pharmacodynamics of warfarin. J Pharmacokinet Pharmacodyn 2010; 37: 3–24. [DOI] [PubMed] [Google Scholar]
- 23. Xu JP, Shi YK, Dong L, Wei Y, Fu B, Liu RF. Low intensity anticoagulation therapy for Chinese population with heart valve replacement‐‐3 000 cases follow‐up. J Sichuan Univ Med Sci Ed 2016; 47: 90–92. [PubMed] [Google Scholar]
- 24. Zhang L, Beal SL, Sheiner LB. Simultaneous vs. sequential analysis for population PK/PD data I: best‐case performance. J Pharmacokinet Pharmacodyn 2003b; 30: 387–404. [DOI] [PubMed] [Google Scholar]
- 25. Zhang L, Beal SL, Sheinerz LB. Simultaneous vs. sequential analysis for population PK/PD data II: robustness of methods. J Pharmacokinet Pharmacodyn 2003a; 30: 405–416. [DOI] [PubMed] [Google Scholar]
- 26. Hill AV. The possible effects of the aggregation of the molecules of hemoglobin on its dissociation curves. J Physiol (Lond) 1910; 40: iv–vii. [Google Scholar]
- 27. Hill AV. The mode of action of nicotine and curari, determined by the form of the contraction curve and the method of temperature coefficients. J Physiol 1909; 39: 361–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chan E, Mclachlan A, Oreilly R, Rowland M. Stereochemical aspects of warfarin drug‐interactions‐use of a combined pharmacokinetic‐pharmacodynamic model. Clin Pharmacol Ther 1994; 56: 286–294. [DOI] [PubMed] [Google Scholar]
- 29. Janmahasatian S, Duffull SB, Ash S, Ward LC, Byrne NM, Green B. Quantification of lean bodyweight. Clin Pharmacokinet 2005; 44: 1051–1065. [DOI] [PubMed] [Google Scholar]
- 30. Anderson BJ, Holford NHG. Mechanistic basis of using body size and maturation to predict clearance in humans. Drug Metab Pharmacokinet 2009; 24: 25–36. [DOI] [PubMed] [Google Scholar]
- 31. Ravva P, Gastonguay MR, Tensfeldt TG, Faessel HM. Population pharmacokinetic analysis of varenicline in adult smokers. Br J Clin Pharmacol 2009; 68: 669–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yafune A, Ishiguro M. Bootstrap approach for constructing confidence intervals for population pharmacokinetic parameters. I: a use of bootstrap standard error. Stat Med 1999; 18: 581–599. [DOI] [PubMed] [Google Scholar]
- 33. Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. Prediction‐corrected visual predictive checks for diagnosing nonlinear mixed‐effects models. AAPS J 2011; 13: 143–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lane S, Al‐Zubiedi S, Hatch E, Matthews I, Jorgensen AL, Deloukas P, et al. The population pharmacokinetics of R‐ and S‐warfarin: effect of genetic and clinical factors. Br J Clin Pharmacol 2012; 73: 66–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Klein TE, Altman RB, Eriksson N, Gage BF, Kimmel SE, Lee MT, et al. Estimation of the warfarin dose with clinical and pharmacogenetic data. N Engl J Med 2009; 360: 753–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Holford N, Heo YA, Anderson B. A pharmacokinetic standard for babies and adults. J Pharm Sci‐Us 2013; 102: 2941–2952. [DOI] [PubMed] [Google Scholar]
- 37. Holford NH. Clinical pharmacokinetics and pharmacodynamics of warfarin. Understanding the dose‐effect relationship. Clin Pharmacokinet 1986; 11: 483–504. [DOI] [PubMed] [Google Scholar]
- 38. Hamberg AK, Wadelius M, Lindh JD, Dahl ML, Padrini R, Deloukas P, et al. A pharmacometric model describing the relationship between warfarin dose and INR response with respect to variations in CYP2C9, VKORC1, and age. Clin Pharmacol Ther 2010; 87: 727–734. [DOI] [PubMed] [Google Scholar]
- 39. Heimark LD, Wienkers L, Kunze K, Gibaldi M, Eddy AC, Trager WF, et al. The mechanism of the interaction between amiodarone and warfarin in humans. Clin Pharmacol Ther 1992; 51: 398–407. [DOI] [PubMed] [Google Scholar]
- 40. Katoh M, Nakajima M, Yamazaki H, Yokoi T. Inhibitory effects of CYP3A4 substrates and their metabolites on P‐glycoprotein‐mediated transport. Eur J Pharm Sci 2001; 12: 505–513. [DOI] [PubMed] [Google Scholar]
- 41. Miners JO, Birkett DJ. Cytochrome P4502C9: an enzyme of major importance in human drug metabolism. Br J Clin Pharmacol 1998; 45: 525–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pal D, Mitra AK. MDR‐ and CYP3A4‐mediated drug–drug interactions. J Neuroimmune Pharmacol 2006; 1: 323–339. [DOI] [PubMed] [Google Scholar]
- 43. Hirai K, Hayashi H, Ono Y, Izumiya K, Tanaka M, Suzuki T, et al. Influence of CYP4F2 polymorphisms and plasma vitamin K levels on warfarin sensitivity in Japanese pediatric patients. Drug Metab Pharmacokinet 2013; 28: 132–137. [DOI] [PubMed] [Google Scholar]
- 44. Ma C, Zhang Y, Xu Q, Yang J, Gao L, Xu B, et al. Influence of warfarin dose‐associated genotypes on the risk of hemorrhagic complications in Chinese patients on warfarin. Int J Hematol 2012; 96: 719–728. [DOI] [PubMed] [Google Scholar]
- 45. Zhang JJ, Zhang H, Ding XL, Ma S, Miao LY. Effect of the P450 oxidoreductase 28 polymorphism on the pharmacokinetics of tacrolimus in Chinese healthy male volunteers. Eur J Clin Pharmacol 2013; 69: 807–812. [DOI] [PubMed] [Google Scholar]
- 46. Miao L, Yang J, Huang C, Shen Z. Contribution of age, body weight, and CYP2C9 and VKORC1 genotype to the anticoagulant response to warfarin: proposal for a new dosing regimen in Chinese patients. Eur J Clin Pharmacol 2007; 63: 1135–1141. [DOI] [PubMed] [Google Scholar]
- 47. McDonald MG, Rieder MJ, Nakano M, Hsia CK, Rettie AE. CYP4F2 is a vitamin K1 oxidase: an explanation for altered warfarin dose in carriers of the V433 M Variant. Mol Pharmacol 2009; 75: 1337–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rieder MJ, Reiner AP, Gage BF, Nickerson DA, Eby CS, McLeod HL, et al. Effect of VKORC1 haplotypes on transcriptional regulation and warfarin dose. N Engl J Med 2005; 352: 2285–2293. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Full model Pred Corrected visual predictive check S‐ and R‐warfarin concentration time course over 12 weeks. Observed (with symbols) and predicted 5, 50, 95 percentiles, with predicted 95% confidence intervals (shaded)
Figure S2 Full model Pred Corrected visual predictive check international normalized ratio time course over 28 days and 12 weeks. Observed (with symbols) and predicted 5, 50, 95 percentiles, with predicted 95% confidence intervals (shaded)
Figure S3 Empirical association of clearance and volume with age decades
Figure S4 Empirical association of T2PCA, C50 and HILL with age decades
Table S1 Primer sequence for CYP2C9, CYP4F2, POR*28, PORrs286, and VKORC1 single‐nucleotide polymorphism analysis
Table S2 The intra‐ and interday precision for replicate measurements of S‐ and R‐warfarin quality control concentrations
Table S3 Fixed effect parameter estimates of the warfarin original full pharmacokinetic model and bootstrap average, 95% confidence interval and relative standard error (RSE). Nonsignificant covariate parameters are marked NS
Table S4 Random effect parameter estimates of the warfarin original full pharmacokinetic model and bootstrap average, 95% confidence interval and relative standard error (RSE). Nonsignificant parameters are marked NS
Table S5 Random effect parameter estimates of the warfarin original full pharmacodynamic and turnover model and bootstrap average, 95% confidence interval and relative standard error (RSE). Nonsignificant parameters are marked NS
Table S6 Genotype effects (FCYP2C9‐S on CL‐S, FCYP2C9‐R on CL‐R, FVKORC1 and FCYP4F2 on C50‐S) estimated when known genotypes (CYP2C9, VKORC1, CYP4F2) were considered missing in all combinations. All other parameters were fixed to the minimal model estimates
Supporting info item
Supporting info item