

Abstract

Endoplasmic reticulum aminopeptidase 2 assists with the generation of antigenic peptides for presentation onto Major Histocompatibility Class I molecules in humans. Recent evidence has suggested that the activity of ERAP2 may contribute to the generation of autoimmunity, thus making ERAP2 a possible pharmacological target for the regulation of adaptive immune responses. To better understand the structural elements of inhibitors that govern their binding affinity to the ERAP2 active site, we cocrystallized ERAP2 with a medium activity 3,4-diaminobenzoic acid inhibitor and a poorly active hydroxamic acid derivative. Comparison of these two crystal structures with a previously solved structure of ERAP2 in complex with a potent phosphinic pseudopeptide inhibitor suggests that engaging the substrate N-terminus recognition properties of the active site is crucial for inhibitor binding even in the absence of a potent zinc-binding group. Proper utilization of all five major pharmacophores is necessary, however, to optimize inhibitor potency.
Keywords: Aminopeptidase, inhibitors, X-ray crystallography, antigen
Endoplasmic reticulum aminopeptidase 2 (ERAP2) is an intracellular enzyme that belongs to the M1 family of zinc aminopeptidases and is tasked with processing antigenic peptide precursors to generate (antigenic) peptides for presentation by Major Histocompatibility Class I molecules.1 With this function, ERAP2 can regulate adaptive immune responses in humans and influence cytotoxic responses against healthy or aberrant cells. ERAP2 has distinct specificity to the homologous ERAP1 and has been proposed to assist the trimming function of ERAP1 possibly through the formation of a functional dimer.2,3 ERAP2 acts in synergy with ERAP1 to help determine the repertoire of peptides presented by MHCI, while retaining a unique role in shaping that repertoire.4 Additionally, ERAP2 deficiency can elicit the unfolded protein response pathway in cells, contributing to inflammatory responses.5 Common single-nucleotide polymorphisms in the ERAP2 gene have been associated with the development of autoimmunity, preeclampsia, immune evasion by cancer, and HIV infection resistance, by affecting either protein expression or enzymatic activity.6−9 In particular, in a recent study, the expression levels of ERAP2 have been associated with predisposition to birdshot chorioretinopathy, an inflammatory disease with autoimmune etiology, suggesting that ERAP2 inhibition may have therapeutic value for treating autoimmunity.10 Furthermore, ERAP1 down-regulation has been shown to be sufficient to elicit potent antitumor responses in murine experimental models making ERAP1 a tractable target for cancer immunotherapy.11,12 Unlike humans, mice do not express ERAP2, and as a result, the tractability of ERAP2 inhibition for cancer immunotherapy has not been evaluated yet, but given the variable expression of ERAP2 in human tumors, the possibility that ERAP2 inhibition can lead to enhancement of antitumor responses has been proposed.13,14 The proposed secondary role of ERAP2 in generating antigenic peptides compared to ERAP1, however, possibly makes ERAP2 a more attractive target for pharmacological intervention since its inhibition is less likely to elicit unwanted side-effects.
Recently developed ERAP2 inhibitors include phosphinic pseudotripeptides and 3,4-diaminobenzoic acid derivatives.15,16 Hydroxamic acid derivatives have also been described to be potent inhibitors of aminopeptidases, but not yet thoroughly investigated as ERAP2 inhibitors.17 A crystal structure of a phosphinic pseudotripeptide (DG013A) bound into the active site of ERAP2 revealed five distinct protein elements that were hypothesized to drive inhibitor potency.18 These are (i) the zinc cation, (ii) the binding site of primary amine (shown in red in Figure 1, mimicking the N-terminus of the peptide substrates), and (iii) at least three specificity pockets that determine substrate selectivity (S1, S1′, and S2′) (Figure 1). Despite achieving nanomolar potency for ERAP2, DG013A demonstrated very limited selectivity for homologous enzymes ERAP1 and IRAP, raising questions whether utilization of all five identified potential groups in the ligand that may interact with the protein (pharmacophores) is necessary for further inhibitor-selectivity optimization. To address this question we solved two crystal structures of ERAP2 with two bound ligands that have only some of the five pharmacophores and correlated structural features with in vitro inhibitory potency. One of the ligands is a 3,4-diaminobenzoic acid derivative described before (compound 1(16)) that has been hypothesized to lack a zinc binding group (ZBG). The other ligand is a hydroxamic acid derivative previously described to be a nanomolar inhibitor of aminopeptidase PfAM1 (compound 2(17)). Both compounds carry side chains that should be near-optimal for the S1 and S2′ specificity pockets of the enzyme.3 The IC50 of 1 for ERAP2 was measured at 2 μM, whereas the hydroxamic acid derivative 2 was much weaker, with an IC50 value >100 μM (Figure 2). The structures of ERAP2 with compounds 1 and 2 were compared to the previously published structure of ERAP2 with DG013A. The wide range of inhibitory potencies for ERAP2 (ranging from 50 nM to >100 μM) allow us to suggest conclusions about the importance of distinct pharmacophores and ligand affinity.
Figure 1.
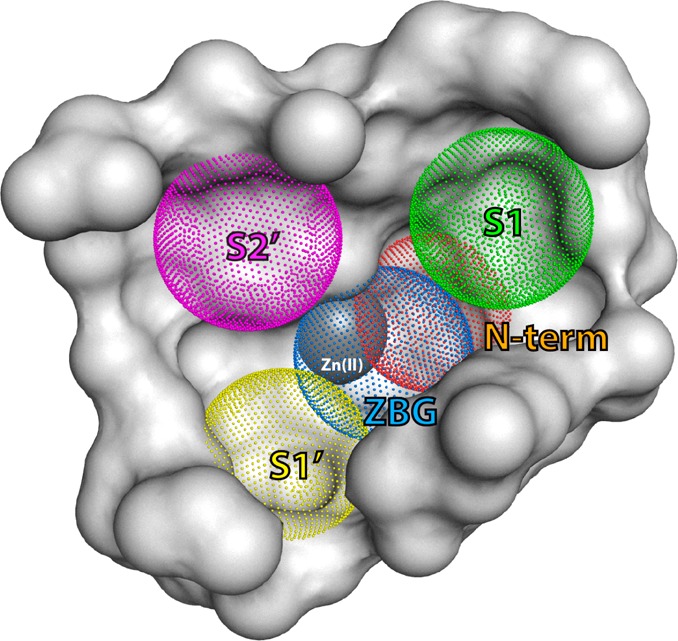
Schematic representation of the ERAP2 catalytic site in the “closed” conformation. Key known sites are indicated with dotted spheres and labeled to indicate the three first specificity pockets of the enzyme (S1 in green, S1′ in yellow, and S2′ in magenta) as well as the location of zinc-binding groups of putative inhibitors (blue) and the location of recognition of the free NH2 functionality of the N-terminus of the substrate (orange). The Zn(II) atom is indicated as a gray sphere.
Figure 2.
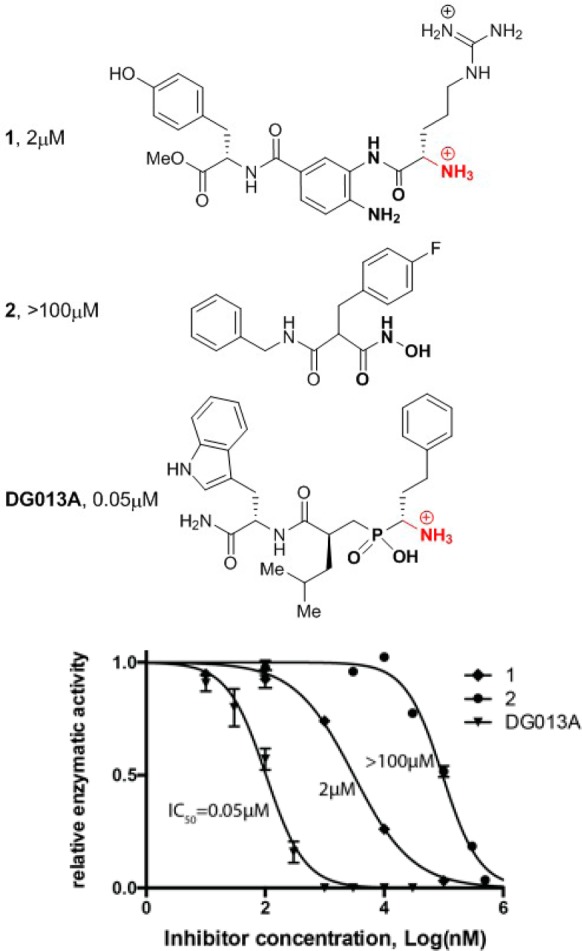
(Top) Chemical structures of inhibitors cocrystallized with ERAP2. (Bottom) Representative titrations showing the relative potency of tested ligands.
Cocrystals of ERAP2 with 1 and 2 were obtained with experimental approaches similar to those previously described19 (for more details see the Supporting Information). Data collection and refinement statistics are shown in Table 1. Atomic coordinates and structure factors have been deposited in the Protein Data Bank (www.pdb.org), with PDB codes 5K1V (for 1) and 5J6S (for 2).
Table 1. Data Collection and Refinement Statistics.
| ERAP2/2 | ERAP2/1 | |
|---|---|---|
| Data Collection | ||
| space group | P21 | P21 |
| a, b, c (Å) | 73.5, 134.3, 127.6 | 74.4, 135.2, 127.5 |
| β (deg) | 91.7 | 90.1 |
| resolution (Å) | 73.51–2.80 (2.95–2.80)a | 48.35–2.90 (3.05–2.90)a |
| Rmerge(%) | 11.2 (111)a | 6.2 (67)a |
| I/σ(I) | 8.6 (1.7)a | 17.8 (2.3)a |
| Refinement | ||
| resolution (Å) | 73.51–2.80 (2.84–2.80)a | 48.35–2.90 (2.93–2.90)a |
| no. reflections (all/used) | 60772/60732 | 108521/108499 |
| Rwork/Rfree(%) | 20.5/27.6 (30.2/39.7)a | 20.5/27.3 (35.2/44.2)a |
| no. atoms (per asymmetric unit) | 15067 (47 alternate) | 14637 (41 alternate) |
| nonsolvent | 15018 (47 alternate) | 14593 (41 alternate) |
| average B overall (Å2) | 74.0 | 76.5 |
| RMSD bond lengths (Å) | 0.011 | 0.010 |
| RMSD bond angles (deg) | 1.387 | 1.379 |
| Ramachandran | ||
| preferred (%) | 86.5 | 87.0 |
| allowed (%) | 10.9 | 11.6 |
| outliers (%) | 2.6 | 1.4 |
Values for the highest resolution shell.
For the crystal structure of ERAP2 in complex with the 3,4-diaminobenzoic acid derivative 1, one ligand molecule was modeled at the catalytic site of chain A (Figure 3A and Supporting Figure 1A). Some patches of negative electron density, after refining with the model, probably indicate less than full occupancy, which is further corroborated by the lack of assignable ligand density at one of the two structurally identical protein chains in the crystal asymmetric unit (referred to as chains A and B in what follows).
Figure 3.
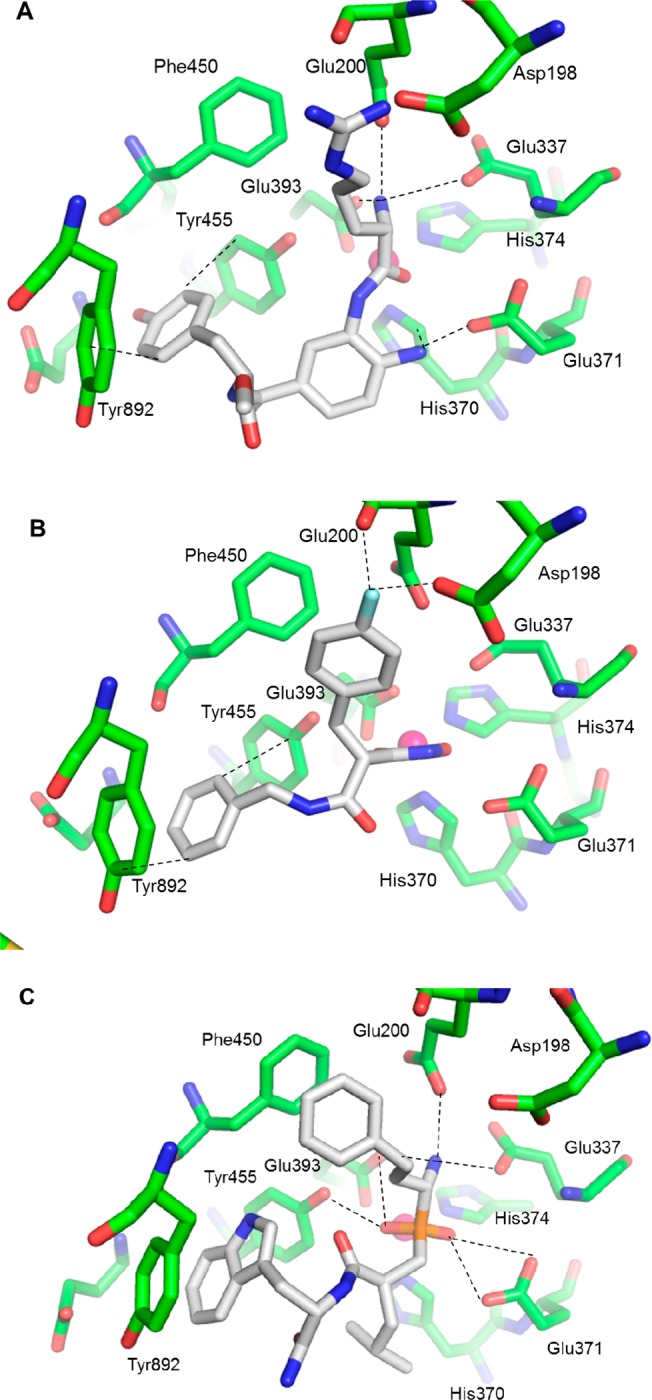
Schematic representation of the crystal structures of (a) compound 1, (b) compound 2 (chain B), and (c) DG013A (chain A), bound inside the ERAP2 catalytic site. Oxygen atoms are shown in red, nitrogen in blue, phosphorus in orange, fluorine in cyan, and the Zn(II) ion in magenta. Bonding interactions that stabilize the bound inhibitor are shown as dashed lines. The structures were rendered in PyMOL.21
The amide carboxylic oxygen of Arg coordinates the active site Zn(II) atom (Zn···O distance 2.0 Å) and is further stabilized by interacting with the OE2 carboxylic oxygen of Glu371 at the catalytic site (distance 3.0 Å), a conserved residue between ERAP1, ERAP2, and IRAP. The free terminal NH2 of 1 (shown in red in Figure 1) is engaged in electrostatic interactions with the side chains of Glu200 (3.2 Å), Glu337 (3.2 Å), and Glu393 (2.5 Å), the latter also being a zinc-coordinating residue. As expected, Arg penetrates deep into the S1 specificity pocket of the enzyme, stabilized by interactions with the Asp198 side chain (distances between the Asp198 carboxyl oxygens and a guanidinium nitrogen of Arg is 2.8 Å), as well as with Glu200 (closest distance 2.7 Å). The Tyr side chain of 1 is stacked between (not parallel with) the phenyl rings of Tyr455 (closest distance 3.3 Å) and Tyr892 (3.1 Å), which form the S2′ specificity pocket; it occupies the same position as the third residue (Trp) of DG013A. The stabilizing role of Glu200, Glu337, Glu371, Glu393, Tyr455, and Tyr892 had already been identified for the binding of the phosphinic pseudotripeptide.18 An additional residue, Asp198, that stabilizes the present complex was not involved in DG013A stabilization although it had been involved in two previously reported ERAP2 structures.9,20 However, Phe450, which lines the base of the S1 specificity pocket, is not here involved in a hydrophobic interaction (shortest distance 4.6 Å) as was the case for DG013A (shortest DG013A homophenylalanine (hPhe); Phe450 distance is 3.7 Å).
For the structure of ERAP2 in complex with the hydroxamic acid derivative 2, two alternative conformations (α and β), both at the catalytic site, were modeled based on the difference electron density map for chain A (Supporting Figure 2), whereas a unique conformation was found at chain B (Figure 3B and Supporting Figure 1B). The latter is fairly close to but not the same as one of the alternative conformations (β) of chain A. Residual electron density at both ERAP2 chains indicates some dynamic disorder in the structure and therefore a flexible ligand; more alternative binding positions at lower occupancies in the catalytic site cannot be excluded, in line with poor binding and thus the observed poor inhibition. The oxygens of the hydroxamic group coordinate the active site Zn in a canonical fashion for conformations α and β at chain A and the unique conformation at chain B. In chain A and conformation α, the fluorine atom of the fluorobenzene moiety (first residue) of the ligand lays close to a carboxyl oxygen of Asp198 (2.7 Å) and to the main chain oxygen of Glu200 (3.1 Å), both inside specificity pocket S1. In contrast, in conformation β the fluorine atom is found at longer distances from both residues but is stabilized by Phe450 (π-stacking with closest approach 3.4 Å). Tyr892 presents an interesting structural feature: its aromatic ring can be modeled at two distinct positions (T1 and T2) in ERAP2 chain A though not in chain B. For steric reasons, Tyr892 at position T2 is only compatible with the ligand being in conformation β. It appears therefore that position T1 corresponds uniquely to conformation α of the ligand and T2 to β (both Tyr892 positions and both ligand conformations freely refine to occupancies of approximately 0.5). Position T1 of Tyr892/A comes fairly close to the fluorobenzene in conformation α (3.2 Å), whereas T2 is far from its corresponding fluorobenzene. The phenyl ring (third residue) of 2 in conformation α is involved in a T-shaped aromatic interaction with the Tyr892/T1 side chain. In position β, the ligand phenyl ring is sandwiched between Tyr455 and Tyr892, in what had been previously identified as specificity pocket S2′ (although it is too far (5.9 Å) from Tyr892 to be involved in a proper π-stacking interaction). Note that, contrary to most other residues involved in the stabilization of the ligands which are conserved between ERAP1, ERAP2, and IRAP, Tyr892 is only found in ERAP2.
In chain B, there is no disorder at Tyr892, the position of which corresponds to position T2 of chain A. The fluorobenzene moiety (first residue) is now at a position midway between conformations α and β of chain A, which might be explained by the absence of either a steric clash or an interaction with Tyr892. The fluorine comes, however, a lot closer to the main chain oxygen of Glu200 (2.3 Å) and remains rather close to Asp198 (3.1 Å). The phenyl ring is again neatly stacked between the Tyr455 and Tyr892 rings in specificity pocket S2′. In 2, the fluorobenzene in all three conformations corresponds to Arg of 1 (and to equivalent residues of other known ligands), inside specificity pocket S1. The phenyl ring (third residue) of 2 in conformation α does not coincide with the tyrosine ring of the third residue of 1, but with its main chain carboxyl group, whereas in conformation β it does coincide with the Tyr side chain, inside specificity pocket S2′. This also applies for the phenyl ring of 2 at chain B.
Comparison of the crystal structures of ERAP2 in complex with 1 and 2 to the previously determined crystal structure in complex with DG013A can be useful in establishing correlations between the importance of distinct structural components and inhibitor potency (Figure 3C). In all three inhibitor complexes, the compounds assume a highly similar orientation, with the side chains occupying the S1 and S2′ specificity pockets of the enzyme. Proper orientation is determined by interactions of the hydroxamic acid or phosphinic acid ZBGs or the carbonyl group of the first peptidic bond for the aminobenzoic acid derivative. DG013A also features a leucine side chain that occupies a shallow S1′ pocket, whereas the hydroxamic acid derivative lacks such a feature. However, the diaminobenzoic ring is itself found near the S1′ pocket. The arginine side chain of 1 is highly optimized for the S1 pocket, making electrostatic interactions with both Asp198 and Glu200. Similarly, the hPhe side chain of DG013A makes strong π-stacking interactions with Phe450. The fluorophenyl side chain of 2 is involved in interactions with acidic residues and in π-stacking interactions in the S1 pocket, although neither orientation appears optimal. The tyrosine, phenyl, and tryptophan side chains of the three inhibitors bind to the S2′ pocket of the enzyme, making π-stacking interactions with Tyr892 and Tyr455, although again the orientation of the tryptophan of DG013A is more optimal.
Overall, DG013A interacts with all five sites shown in Figure 1, whereas compound 1 interacts with four since it lacks a ZBG. Compound 2 interacts with only three sites, having a strong ZBG and side chains for the S1 and S2′ pockets. Accordingly, there appears to be a clear correlation between potency and number of pharmacophores in the ligand. Although compound 2 employs a strong ZBG binding the zinc in a canonical fashion, a strong ZBG is neither necessary (as shown with 1) nor sufficient. It is the lack of utilization of multiple pharmacophores and of optimization of the S1 side chain that result in low potency.
In conclusion, we analyzed two new cocrystal structures of ERAP2, an enzyme important for the regulation of immune responses in humans, with ligands of various potencies. Our results suggest that utilization of all five pharmacophores is crucial for achieving potency and that no single component is dominant including the ZBG. Our results provide a useful structural framework for generation of potent and selective ERAP2 inhibitors.
Acknowledgments
We thank Efthalia Zervoudi for help with preparing recombinant ERAP2.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00505.
Experimental details regarding recombinant protein production, enzymatic assays, crystallization, data collection, and structure determination (PDF)
Author Contributions
A.M. prepared recombinant ERAP2, characterized inhibitors with help from D.K., and solved the crystal structures in collaboration with Em.S and I.M. P.G. analyzed ERAP2 crystals and helped with data processing. R.D-P., R.G., A.P., S.T. and D.V. synthesized the inhibitors. Ef.S. and Em.S. interpreted data and wrote the manuscript with contributions from all authors. All authors have given approval to the final version of the manuscript.
This work has been funded by the EU (European Regional Development Fund) and the Greek Secretariat for Research and Technology (Bilateral Greece-France collaboration action - grant no. HIAP), the French Ministry of Foreign Affairs (PHC Hubert Curien Grant – PLATON N° 30375YL) and the Region Nord-Pas de Calais (grant to R.G.). It has also received funding from the European Community’s Seventh Framework Programme (FP7/2007–2013) under BioStruct-X (grant agreement no. 283570).
The authors declare no competing financial interest.
Supplementary Material
References
- Evnouchidou I.; Papakyriakou A.; Stratikos E. A New Role for Zn(II) Aminopeptidases: Antigenic Peptide Generation and Destruction. Curr. Pharm. Des. 2009, 15, 3656–3670. 10.2174/138161209789271816. [DOI] [PubMed] [Google Scholar]
- Saveanu L.; Carroll O.; Lindo V.; Del Val M.; Lopez D.; Lepelletier Y.; Greer F.; Schomburg L.; Fruci D.; Niedermann G.; van Endert P. M. Concerted peptide trimming by human ERAP1 and ERAP2 aminopeptidase complexes in the endoplasmic reticulum. Nat. Immunol. 2005, 6, 689–697. 10.1038/ni1208. [DOI] [PubMed] [Google Scholar]
- Zervoudi E.; Papakyriakou A.; Georgiadou D.; Evnouchidou I.; Gajda A.; Poreba M.; Salvesen G. S.; Drag M.; Hattori A.; Swevers L.; Vourloumis D.; Stratikos E. Probing the S1 specificity pocket of the aminopeptidases that generate antigenic peptides. Biochem. J. 2011, 435, 411–420. 10.1042/BJ20102049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Esteban A.; Sanz-Bravo A.; Guasp P.; Barnea E.; Admon A.; Lopez de Castro J. A. Separate effects of the ankylosing spondylitis associated ERAP1 and ERAP2 aminopeptidases determine the influence of their combined phenotype on the HLA-B*27 peptidome. J. Autoimmun. 2017, 10.1016/j.jaut.2016.12.008. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Ciccia F.; Zeng F.; Guggino G.; Yee K.; Abdullah H.; Silverberg M. S.; Alessandro R.; Triolo G.; Haroon N. Functional interaction of ERAP2 and HLA-B27 activates the unfolded protein response. Arthritis Rheumatol. 2016, 10.1002/art.40033. [DOI] [PubMed] [Google Scholar]
- Cagliani R.; Riva S.; Biasin M.; Fumagalli M.; Pozzoli U.; Lo Caputo S.; Mazzotta F.; Piacentini L.; Bresolin N.; Clerici M.; Sironi M. Genetic diversity at endoplasmic reticulum aminopeptidases is maintained by balancing selection and is associated with natural resistance to HIV-1 infection. Hum. Mol. Genet. 2010, 19, 4705–4714. 10.1093/hmg/ddq401. [DOI] [PubMed] [Google Scholar]
- Vanhille D. L.; Hill L. D.; Hilliard D. D.; Lee E. D.; Teves M. E.; Srinivas S.; Kusanovic J. P.; Gomez R.; Stratikos E.; Elovitz M. A.; Romero R.; Strauss J. F. 3rd A Novel Haplotype Structure in a Chilean Population: Implications for ERAP2 Protein Expression and Preeclampsia Risk. Mol. Genet. Genomic Med. 2013, 1, 98–107. 10.1002/mgg3.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruci D.; Giacomini P.; Nicotra M. R.; Forloni M.; Fraioli R.; Saveanu L.; van Endert P.; Natali P. G. Altered expression of endoplasmic reticulum aminopeptidases ERAP1 and ERAP2 in transformed non-lymphoid human tissues. J. Cell. Physiol. 2008, 216, 742–749. 10.1002/jcp.21454. [DOI] [PubMed] [Google Scholar]
- Evnouchidou I.; Birtley J.; Seregin S.; Papakyriakou A.; Zervoudi E.; Samiotaki M.; Panayotou G.; Giastas P.; Petrakis O.; Georgiadis D.; Amalfitano A.; Saridakis E.; Mavridis I. M.; Stratikos E. A common single nucleotide polymorphism in endoplasmic reticulum aminopeptidase 2 induces a specificity switch that leads to altered antigen processing. J. Immunol. 2012, 189, 2383–2392. 10.4049/jimmunol.1200918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuiper J. J.; Van Setten J.; Ripke S.; Van ’t Slot R.; Mulder F.; Missotten T.; Baarsma G. S.; Francioli L. C.; Pulit S. L.; De Kovel C. G.; Ten Dam-Van Loon N.; Den Hollander A. I.; Veld P. H.; Hoyng C. B.; Cordero-Coma M.; Martin J.; Llorenc V.; Arya B.; Thomas D.; Bakker S. C.; Ophoff R. A.; Rothova A.; De Bakker P. I.; Mutis T.; Koeleman B. P. A genome-wide association study identifies a functional ERAP2 haplotype associated with birdshot chorioretinopathy. Hum. Mol. Genet. 2014, 23, 6081–6087. 10.1093/hmg/ddu307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James E.; Bailey I.; Sugiyarto G.; Elliott T. Induction of Protective Antitumor Immunity through Attenuation of ERAAP Function. J. Immunol. 2013, 190, 5839–5846. 10.4049/jimmunol.1300220. [DOI] [PubMed] [Google Scholar]
- Cifaldi L.; Lo Monaco E.; Forloni M.; Giorda E.; Lorenzi S.; Petrini S.; Tremante E.; Pende D.; Locatelli F.; Giacomini P.; Fruci D. Natural killer cells efficiently reject lymphoma silenced for the endoplasmic reticulum aminopeptidase associated with antigen processing. Cancer Res. 2011, 71, 1597–1606. 10.1158/0008-5472.CAN-10-3326. [DOI] [PubMed] [Google Scholar]
- Stratikos E.; Stamogiannos A.; Zervoudi E.; Fruci D. A role for naturally occurring alleles of endoplasmic reticulum aminopeptidases in tumor immunity and cancer pre-disposition. Front. Oncol. 2014, 4, 363. 10.3389/fonc.2014.00363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratikos E. Regulating adaptive immune responses using small molecule modulators of aminopeptidases that process antigenic peptides. Curr. Opin. Chem. Biol. 2014, 23C, 1–7. 10.1016/j.cbpa.2014.08.007. [DOI] [PubMed] [Google Scholar]
- Kokkala P.; Mpakali A.; Mauvais F. X.; Papakyriakou A.; Daskalaki I.; Petropoulou I.; Kavvalou S.; Papathanasopoulou M.; Agrotis S.; Fonsou T. M.; van Endert P.; Stratikos E.; Georgiadis D. Optimization and Structure-Activity Relationships of Phosphinic Pseudotripeptide Inhibitors of Aminopeptidases that Generate Antigenic Peptides. J. Med. Chem. 2016, 59, 9107–9123. 10.1021/acs.jmedchem.6b01031. [DOI] [PubMed] [Google Scholar]
- Papakyriakou A.; Zervoudi E.; Tsoukalidou S.; Mauvais F. X.; Sfyroera G.; Mastellos D. C.; van Endert P.; Theodorakis E. A.; Vourloumis D.; Stratikos E. 3,4-diaminobenzoic acid derivatives as inhibitors of the oxytocinase subfamily of M1 aminopeptidases with immune-regulating properties. J. Med. Chem. 2015, 58, 1524–1543. 10.1021/jm501867s. [DOI] [PubMed] [Google Scholar]
- Deprez-Poulain R.; Flipo M.; Piveteau C.; Leroux F.; Dassonneville S.; Florent I.; Maes L.; Cos P.; Deprez B. Structure-activity relationships and blood distribution of antiplasmodial aminopeptidase-1 inhibitors. J. Med. Chem. 2012, 55, 10909–10917. 10.1021/jm301506h. [DOI] [PubMed] [Google Scholar]
- Zervoudi E.; Saridakis E.; Birtley J. R.; Seregin S. S.; Reeves E.; Kokkala P.; Aldhamen Y. A.; Amalfitano A.; Mavridis I. M.; James E.; Georgiadis D.; Stratikos E. Rationally designed inhibitor targeting antigen-trimming aminopeptidases enhances antigen presentation and cytotoxic T-cell responses. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 19890–19895. 10.1073/pnas.1309781110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mpakali A.; Giastas P.; Mathioudakis N.; Mavridis I. M.; Saridakis E.; Stratikos E. Structural Basis for Antigenic Peptide Recognition and Processing by Endoplasmic Reticulum (ER) Aminopeptidase 2. J. Biol. Chem. 2015, 290, 26021–26032. 10.1074/jbc.M115.685909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birtley J. R.; Saridakis E.; Stratikos E.; Mavridis I. M. The Crystal Structure of Human Endoplasmic Reticulum Aminopeptidase 2 Reveals the Atomic Basis for Distinct Roles in Antigen Processing. Biochemistry 2012, 51, 286–295. 10.1021/bi201230p. [DOI] [PubMed] [Google Scholar]
- Delano W. L.The PyMOL Molecular Graphics System; DeLano Scientific LLC: San Carlos, CA, 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.