

Abstract
Apoptosis signal-regulating kinase 1 (ASK1/MAP3K) is a mitogen-activated protein kinase family member shown to contribute to acute ischemia/reperfusion injury. Using structure-based drug design, deconstruction, and reoptimization of a known ASK1 inhibitor, a lead compound was identified. This compound displayed robust MAP3K pathway inhibition and reduction of infarct size in an isolated perfused heart model of cardiac injury.
Keywords: Apoptosis signal-regulating kinase 1 (ASK1), structure-based drug design (SBDD), cardiac injury
Apoptosis signal-regulating kinase 1 (ASK1) is a mitogen-activated protein kinase kinase kinase (MAP3K) family member residing upstream of both Jun N-terminal kinase (JNK) and p38. ASK1 is capable of activating JNK and P38 via the phosphorylation of intermediate kinases. ASK1 plays a role in the mammalian cell stress response and the induction of apoptotic cell death. It also contributes to a range of systemic diseases including heart failure1 and acute ischemia/reperfusion injury, by reducing structural and functional integrity of the mitochondria in cardiac cells.2−4 ASK1-deficient mice display reduced levels of cardiomyocyte apoptosis, hypertrophy, and interstitial fibrosis.5 Thus, selective inhibition of ASK1 represents an attractive strategy for slowing or potentially reversing harmful tissue changes associated with various forms of heart failure.
Using ASK1 structural information and deconstruction of known ASK1 inhibitors such as 1 and 2 (Figure 1), our research team generated a novel, potent, and orally bioavailable ASK1 inhibitor with favorable physicochemical properties to help further elucidate the role of ASK1 in cardiac injury. Compound 1 was an early lead for Takeda’s ASK1 inhibitor program.6 Compound 2 (GS-4997, Gilead Sciences) is a clinical stage ASK1 inhibitor, which has been evaluated as an experimental treatment for diabetic nephropathy and kidney fibrosis.7
Figure 1.
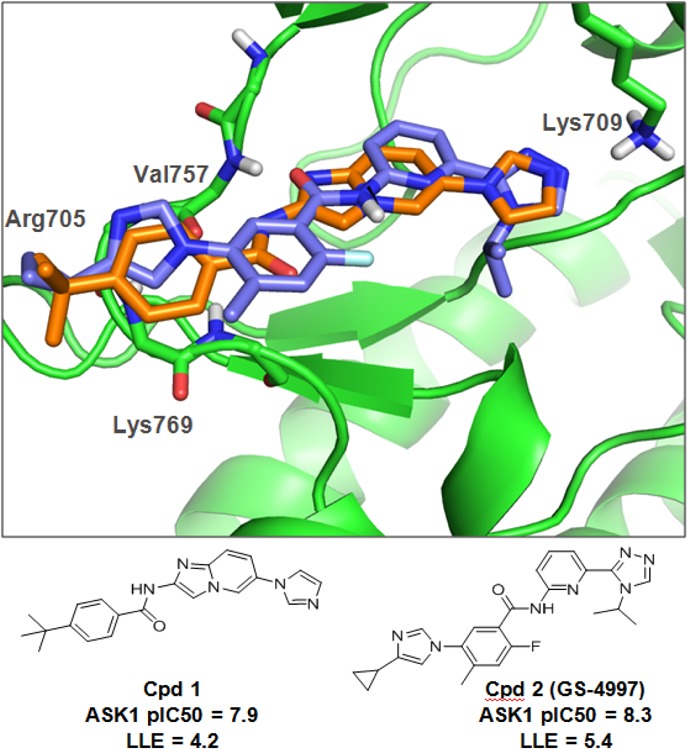
Known (labeled) key interactions between ASK1 protein and small molecule inhibitors. Overlay of the crystal structure of 1 (orange) and model of 2 (blue).
Key structural features with respect to ligand interactions within the ATP binding site were identified using our internal database as well as public domain crystal structures of small molecules in ASK1 and published pharmacophore models.8,9Figure 1 illustrates the cocrystal structure of 1 in hASK1 (PDB: 3VW6) and highlights the key interactions between 1 and the ASK1 ATP binding pocket. Binding to the kinase hinge is characterized by a hydrogen bonding interaction with the backbone NH and carbonyl of Val757. Furthermore, the imidazole of 1 reaches deep into the active site making an H-bond with the side-chain amine of the catalytic lysine (Lys709). The compound also extends toward the solvent exposed region with a t-butyl phenyl amide. The t-butyl group is sandwiched between Arg705 and Lys769 and the phenyl ring makes a Π–NH interaction with Gly759.6 These interactions recapitulate those highlighted in published pharmacophore models.8,9 Based on this information, an in-house model of 2 was generated (Figure 1). The triazole ring of 2 (synthesis described in the Supporting Information (SI) along with all other compounds mentioned in the text) is able to interact with Lys709, while the central benzamide moiety binds to the hinge through Val757. The cyclopropyl imidazole moiety at the opposite end likely makes the same interactions to that of the t-butyl phenyl group in 1.
In addition to identifying the key protein interactions for ASK1 inhibitors, we prepared three molecules (3, 4, 5) based on the deconstruction of 2 to evaluate the relative contributions of the different binding elements to potency. Efficiency metrics such as lipophilic ligand efficiency (LLE),10 ligand efficiency (LE),11 and group efficiency (GE)12 were calculated and are summarized in Table 1.
Table 1. ASK1 Potency and Efficiency Metrics for Compounds 2–5.

Geometric mean of at least two experiments unless otherwise stated. LLE = pIC50 −cLogP; LE = −RT ln(pIC50)/Nheavy; GE = Δ(ΔG)/ΔNheavy.
Removal of the cyclopropyl imidazole (3) from the parent 2 resulted in a 10-fold loss in potency and reduction in LLE from 5.3 to 3.8. It may be noted that, when comparing 3 to 2, although the imidazole ring system contributes approximately 10-fold in potency with little change in LE (0.4 compared to 0.35), the overall increase in LLE (3.8 to 5.3) indicates this interaction may play a key role in binding affinity. Removal of the methyl group and fluorine atom to give an unsubstituted benzamide (4) resulted in a concomitant 8-fold potency loss with a negligible reduction in LE or LLE. Finally, truncation of the isopropyl triazole ring (5) from the right-hand side of the molecule resulted in >1000-fold loss in potency and a significant drop in LLE. The calculated GE value for this ring system is 0.56, highlighting the value of this moiety on binding affinity for ASK1.
In light of the published data, structural analysis, and deconstruction data, subsequent efforts were directed toward the development of a novel and more efficient class of ASK1 inhibitor through the introduction of a more efficient hinge-binding moiety while maintaining vectors toward both Lys709 and Lys769 and/or Arg705.
The model and deconstruction analysis of 2 suggest that the benzamide group remains coplanar with the rest of the core in the active site and that the ortho-fluorine atom, in particular, shields the amide NH. A number of hinge binders bearing a similar motif while maintaining a planar character were explored (results not shown). From this effort, we identified compound 6 (Figure 2), which contains an isoindolinone core as the primary hinge binder but otherwise displays similar substitution to that seen in 4. The pIC50 (7.9) and LLE (5.9) values for 6 were higher than 4 (pIC50 = 6.4 and LLE = 3.6), indicating that the isoindolinone moiety makes a superior interaction with the protein relative to the simple benzamide of compound 4. The crystal structure of 6 bound to ASK1 was solved at 2.7 Å. Figure 2 illustrates a similar binding mode to 2. The carbonyl and aryl hydrogen of the isoindolinone core interact with the NH and carbonyl of Val757, respectively, while the triazole moiety interacts with Lys709 as expected. Finally, the 6-position of the isoindolinone ring provided a vector toward the solvent.
Figure 2.
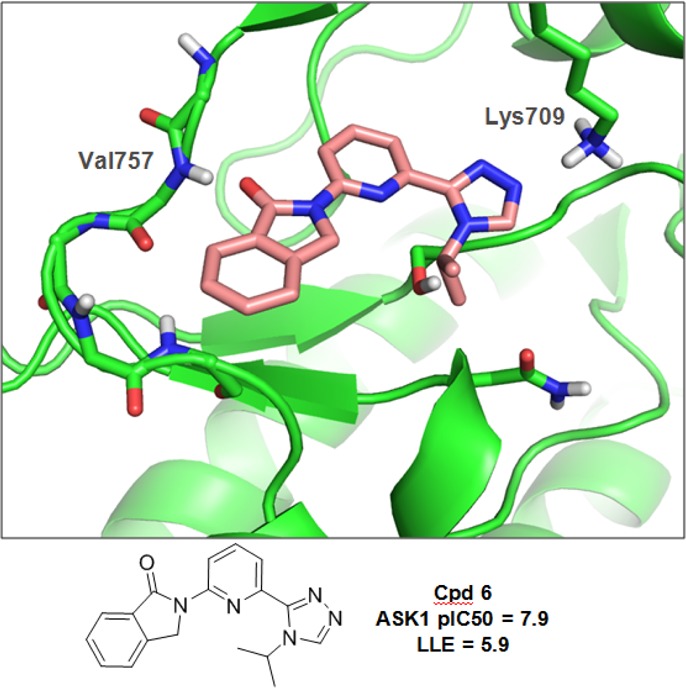
Crystal structure of 6 in ASK1.
Based on the structural information obtained for 6, we first sought to elaborate the isopropyl group on the triazole ring. This substituent projects deep in the kinase active site near the polar residues Asn808 and Ser821. Accordingly, we focused on introducing substituents capable of hydrogen bonding with those residues (summarized in Table 2). Most compounds from this exercise showed only a marginal increase in ASK1 inhibition. However, compound 9 illustrates that the addition of a terminal hydroxyl likely results in a positive interaction with the protein, translating to an increase in LLE from 5.9 to 6.6 (compounds 6 and 9, respectively). The 1-hydroxypropan-2-yl addition (9 and 10) resulted in the highest LLE and therefore considered the best group to get the chemotype in a desirable drug-like chemical space.
Table 2. ASK1 Potency and Efficiency Metrics for Compounds 7–14.
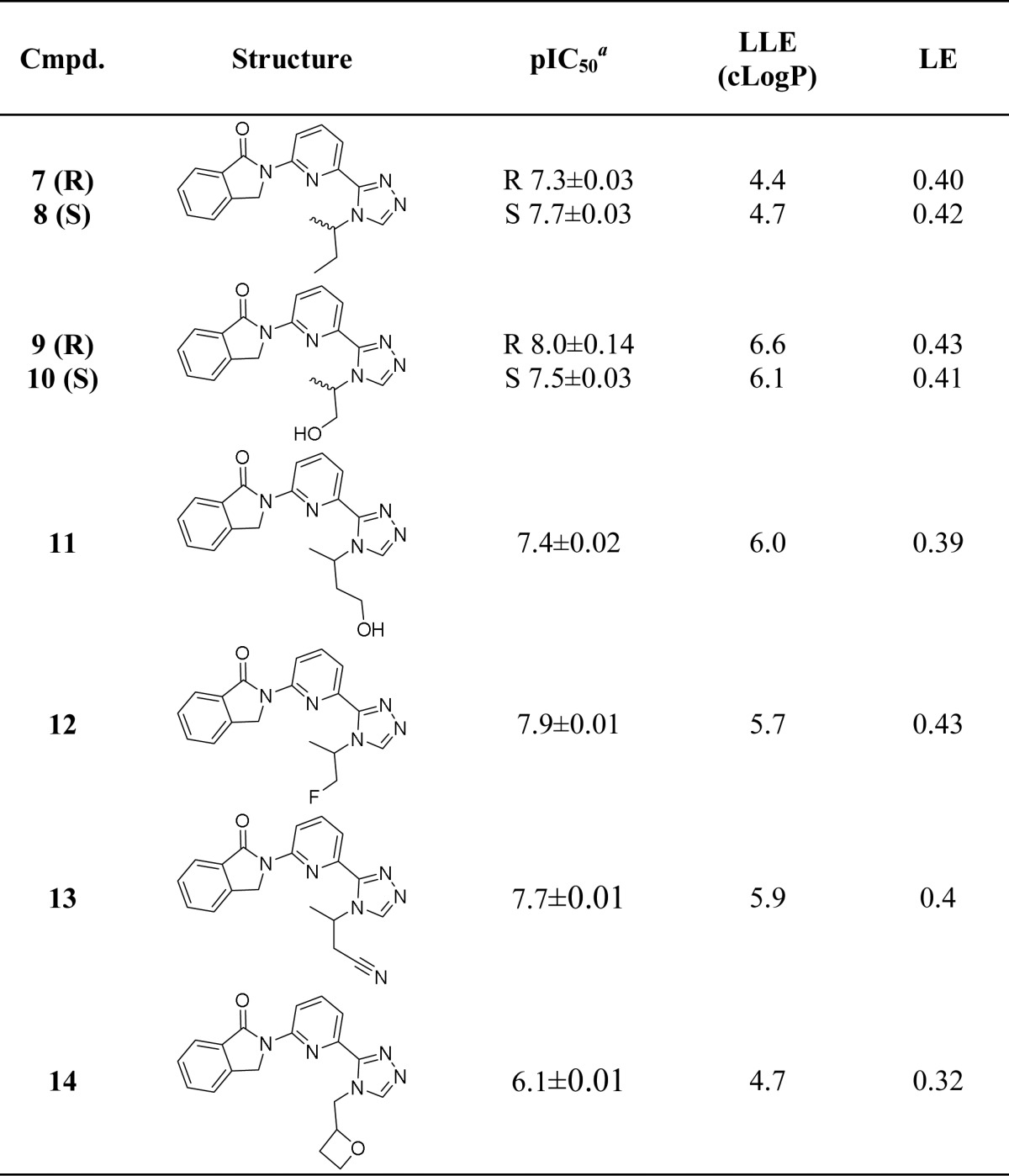
Geometric mean of at least two experiments unless otherwise stated.
In parallel, we made modifications at the 6-position of the isoindolinone ring to target the solvent region. In contrast to other known ASK1 inhibitors that contain aromatic rings in this portion of the active site, we targeted Gly759 by extending into the region with aliphatic groups bearing hydrogen bond-accepting substituents. It was considered that this would allow us to maintain a low LogP and good physicochemical properties.
A number of compounds were synthesized with a substitution at the 6-position and containing an ether oxygen atom as a hydrogen bond acceptor. Compounds were tested in an ASK1 enzyme activity assay as well as a cell-based assay, which monitors formation of phospho-JNK. Table 3 lists key compounds from this effort. Using this strategy, a number of compounds were identified that exhibited pIC50 > 8 and high LLE values in the ASK1 enzyme assay and pEC50 > 7.5 in the cellular assay.
Table 3. ASK1 Enzymatic and Cell Potency and LLE Values for Compounds 15–19.
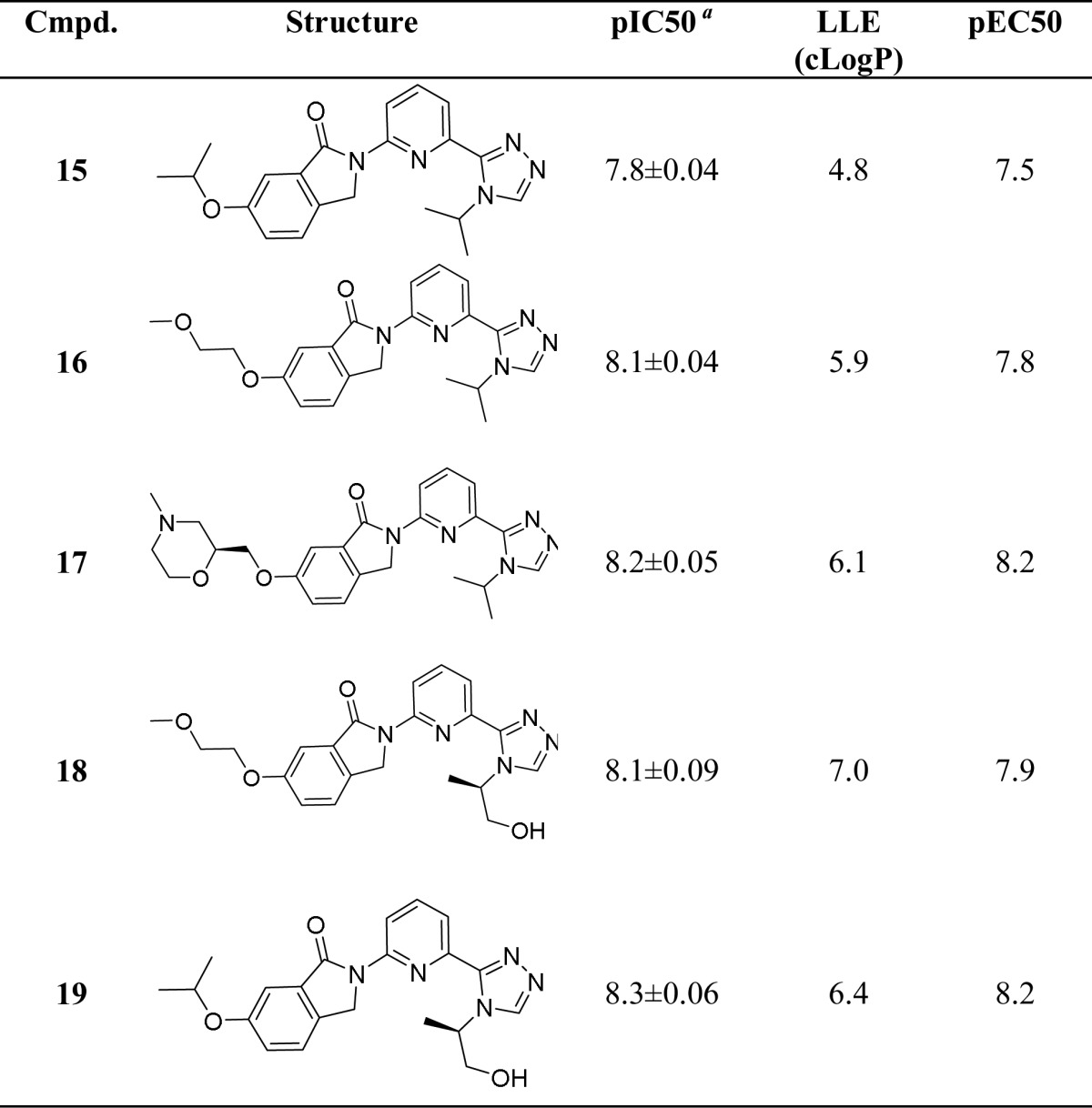
Geometric mean of at least two experiments unless otherwise stated.
Cocrystal structures with ASK1 were solved for two key compounds (17 and 19). The crystal structure of 17 (Figure 3a) recapitulates the general binding features of the truncated compound 6. The observed hydrogen bond between the morpholine oxygen and the backbone NH of Gly759 confirmed our design hypothesis. The structure of 19 illustrates an additional interaction between the terminal hydroxyl group and the side chains of Asn808 and Ser821. The increased potency and LLE value for 19 relative to the des-hydroxy analogue 15 can be attributed to this favorable interaction.
Figure 3.
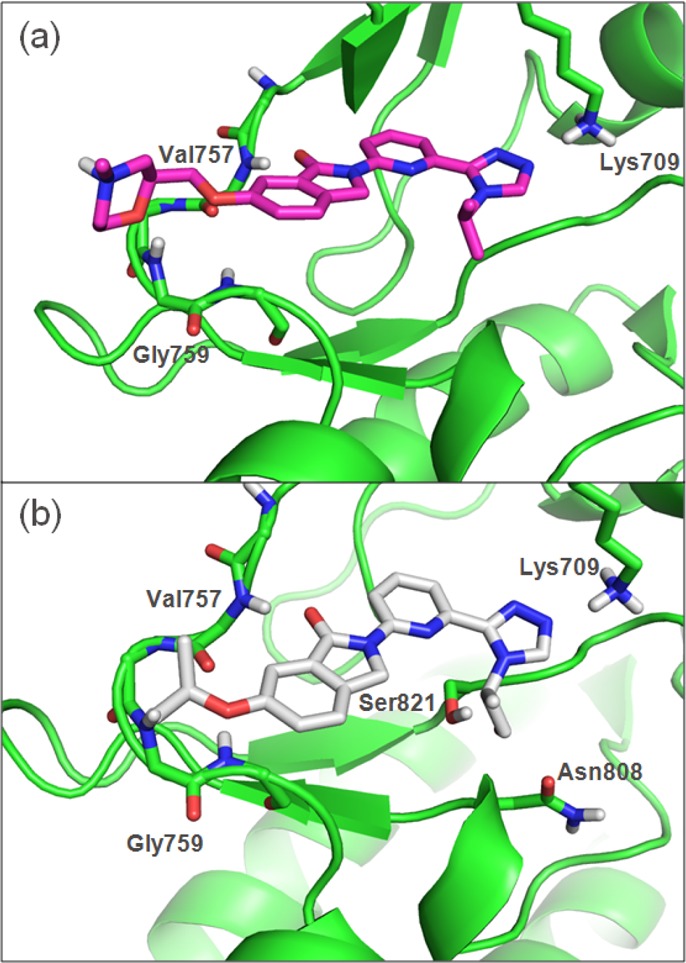
Crystal structures of compounds 17 (a) and 19 (b) bound to ASK1 kinase domain.
Compound 19 showed overall good in vitro ADME properties (human and rat liver microsome clearance <8.3 and <16.6 mL/min/kg, respectively; Papp (A-B)/(B-A) 54.9/262.3 nm/s at 10 μM in MDR1-LLC-PK1; soluble at 155 μM at pH 1, details in SI). To further evaluate 19, screening was conducted against 356 kinases (screened at 1 μM at Invitrogen then full dose response curves for kinases with inhibition greater than 95% at 1 μM). Good kinase selectivity was observed with only 32 kinases having less than 30-fold selectivity (Figure 4). None of them are highly expressed in the heart.
Figure 4.
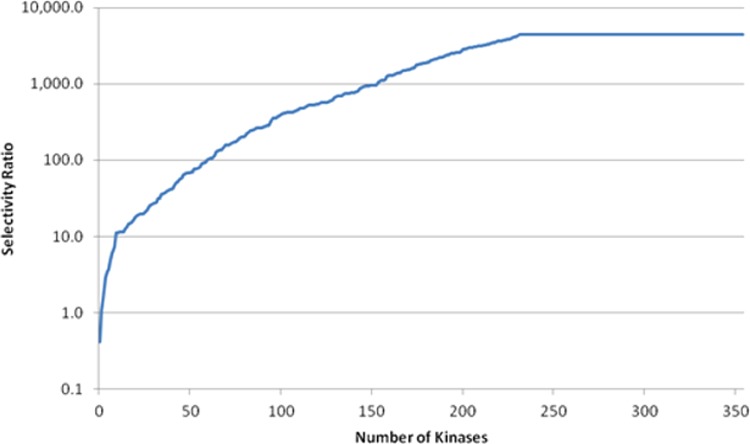
Overall kinase selectivity for compound 19.
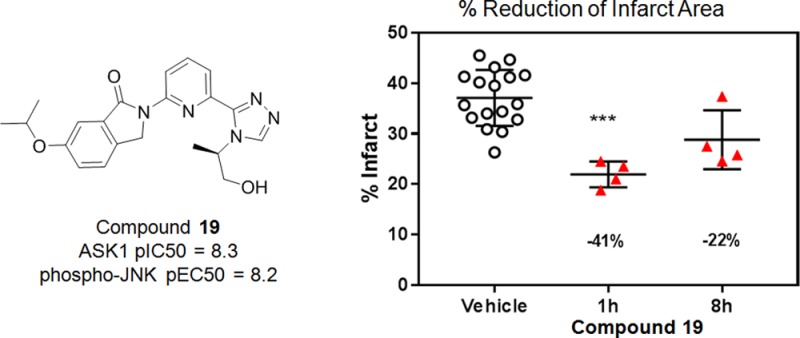
Compound 19 was subsequently evaluated in a low-dose rat pharmacokinetic (PK) study. From this, an estimated dose required to provide at least 8 h of >50% ASK1 inhibition in a rat PD model of cardiac ischemia was calculated (Figure 5a). Accordingly, compound 19 was tested at 2.25 mg/kg in the Langendorff perfused heart model to determine the effect of ASK1 inhibition on cardiac infarct size. Cardiac infarct size was measured in isolated heart preparations at 1 and 8 h after dosing (Figure 5b). Oral administration of 19 resulted in a 42% reduction in infarct size after 1 h and a 24% reduction after 8 h (Figure 5b) suggesting that sustained ASK1 inhibition may provide significant tissue protection against the effects of myocardial ischemia.
Figure 5.
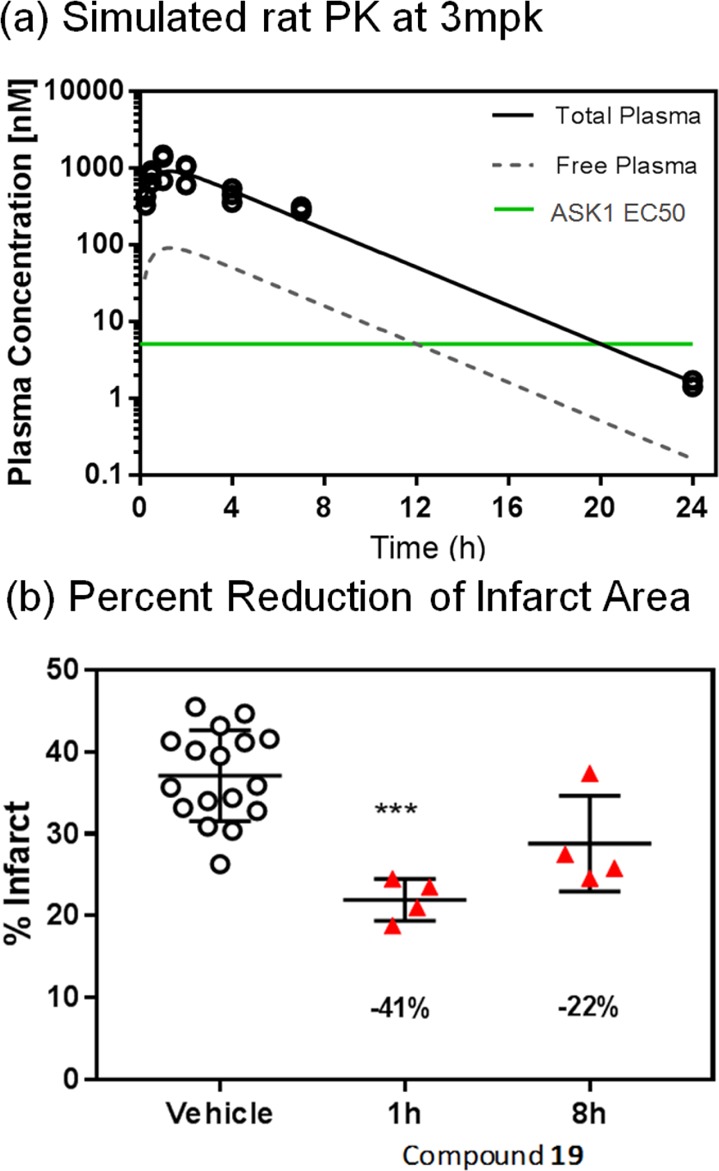
(a) Simulated rat pharmacokinetics and simulated ASK1 inhibition for 19 at 2.25 mpk. (b) Ex vivo profiling of 19 in cardiac ischemia/reperfusion injury.
In summary, a combined structure-based and compound deconstruction approach was used to rapidly produce a novel series of potent ASK1 inhibitors with ideal physiochemical properties and efficiencies. Following an initial deconstruction exercise, compound 6 was identified as a prototype for an efficient hinge-binding series. Subsequent elaboration led to compounds such as 19, which displayed suitable properties for broader pharmacological evaluation. Compound 19 was later shown to reduce infarct size in the Langendorff perfused ex vivo heart model.
With an appropriate tool molecule in hand it will be possible to test additional hypotheses surrounding ASK1 inhibition in relation to harmful tissue changes associated with chronic heart failure and fibrotic disease.
Acknowledgments
We would like to thank Steve Bowlin for running the HR-MS, and Georgia Kefala, Irena Levin, Weston Lane, and Gyorgy Snell for their help getting the crystal structures. The staff of the Berkeley Center for Structural Biology is gratefully acknowledged for support of beamline 5.0.3 at the Advanced Light Source. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. This research used also resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00481.
Synthetic procedures for all the compounds listed in the manuscript; protocols for in vitro assays (binding assay, functional assay, ADME assays); pharmacological evaluation (PK, Langendorff Perfused Heart Model); crystal structures information (ASK1_6 (5UP3), ASK1_17 (5UOR), ASK1_19 (5UOX)) (PDF)
Accession Codes
Coordinates and structure factors are available from the Protein Data Bank with accession codes 5UP3, 5UOR, 5UOX for ASK1_1, ASK1_6, ASK1_17, and ASK1_19 respectively.
The authors declare no competing financial interest.
This paper was published ASAP on February 16, 2017. An additional author was added and the corrected version was reposted on March 2, 2017.
Supplementary Material
References
- Izumiya Y.; Kim S.; Izumi Y.; Yoshida K.; Yoshiyama M.; Matsuzawa A.; Ichijo H.; Iwao H. Apoptosis Signal-Regulating Kinase 1 Plays a Pivotal Role in Angiotensin II–Induced Cardiac Hypertrophy and Remodeling. Circ. Res. 2003, 93, 874–883. 10.1161/01.RES.0000100665.67510.F5. [DOI] [PubMed] [Google Scholar]
- Saxena G.; Chen J.; Shalev A. Intracellular Shuttling and Mitochondrial Function of Thioredoxin-interacting Protein. J. Biol. Chem. 2010, 285, 3997–4005. 10.1074/jbc.M109.034421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiizaki S.; Naguro I.; Ichijo H. Activation mechanisms of ASK1 in response to various stresses and its significance in intracellular signaling. Adv. Biol. Regul. 2013, 53, 135–144. 10.1016/j.jbior.2012.09.006. [DOI] [PubMed] [Google Scholar]
- Tibbles L. A.; Woodgett J. R. The stress-activated protein kinase pathways. Cell. Mol. Life Sci. 1999, 55, 1230–1254. 10.1007/s000180050369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori K.; Naguro I.; Runchel C.; Ichijo H. The roles of ASK family proteins in stress responses and diseases. Cell Commun. Signaling 2009, 7, 9–19. 10.1186/1478-811X-7-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terao Y.; Suzuki H.; Yoshikawa M.; Yashiro H.; Takekawa S.; Fujitani Y.; Okada K.; Inoue Y.; Yamamoto Y.; Nakagawa H.; Yao S.; Kawamoto T.; Uchikawa O. Design and biological evaluation of imidazo[1,2-a] pyridines as novel and potent ASK1 inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 7326–7329. 10.1016/j.bmcl.2012.10.084. [DOI] [PubMed] [Google Scholar]
- Lin J. H.; Zhang J. J.; Lin S. L.; Chertow G. M. Design of a phase 2 clinical trial of an ASK1 inhibitor, GS-4997, in patients with diabetic kidney disease. Nephron. 2015, 129, 29–33. 10.1159/000369152. [DOI] [PubMed] [Google Scholar]
- Starosyla S. A.; Volynets G. P.; Bdzhola V. G.; Golub A. G.; Protopopov M. V.; Yarmoluk S. M. ASK1 pharmacophore model derived from diverse classes of inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 4418–4423. 10.1016/j.bmcl.2014.08.011. [DOI] [PubMed] [Google Scholar]
- Singh O.; Shillings A.; Craggs P.; Wall I.; Rowland P.; Skarzynski T.; Hobbs C. I.; Hardwick P.; Tanner R.; Blunt M.; Witty D. R.; Smith K. J. Crystal structures of ASK1-inhibtor complexes provide a platform for structure-based drug design. Protein Sci. 2013, 22, 1071–1077. 10.1002/pro.2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LLE = pEC50 – cLogP or pIC50 – cLogP.Leeson P. D.; Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discovery 2007, 6, 881–890. 10.1038/nrd2445. [DOI] [PubMed] [Google Scholar]
- LE = pIC50 × (−1.4)/HAC where HAC is heavy atom count.Hopkins A. L.; Groom C. R.; Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discovery Today 2004, 9, 430–431. 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- GE = ΔG/ΔHAC = Δ(−RT ln Ki)/ΔHAC where HAC is heavy atom count.Verdonk M. L.; Rees D. C. Group efficiency: a guideline for hits-to-leads chemistry. ChemMedChem 2008, 3, 1179–1180. 10.1002/cmdc.200800132. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.