

Abstract

We report the discovery of a new potent allosteric effector of sickle cell hemoglobin, GBT440 (36), that increases the affinity of hemoglobin for oxygen and consequently inhibits its polymerization when subjected to hypoxic conditions. Unlike earlier allosteric activators that bind covalently to hemoglobin in a 2:1 stoichiometry, 36 binds with a 1:1 stoichiometry. Compound 36 is orally bioavailable and partitions highly and favorably into the red blood cell with a RBC/plasma ratio of ∼150. This partitioning onto the target protein is anticipated to allow therapeutic concentrations to be achieved in the red blood cell at low plasma concentrations. GBT440 (36) is in Phase 3 clinical trials for the treatment of sickle cell disease (NCT03036813).
Keywords: Sickle cell disease, sickle cell hemoglobin, allosteric modulator, aldehyde, Schiff-base formation, oxygen affinity, red blood cell partitioning
Sickle cell disease (SCD) is caused by a single mutation in the β chain of hemoglobin (Hb) where a hydrophilic βGlu6 has been exchanged for a hydrophobic βVal6. Under low oxygen conditions the mutant hemoglobin (HbS) polymerizes via the mutated βVal6 from one Hb tetramer and a hydrophobic cavity formed by β1Ala70, β1Phe85, and β1Leu88 from a laterally located Hb tetramer. These polymers result in the red blood cell (RBC) losing its deformability properties and taking on a sickle-like shape. These sickle cells are unable to pass through narrow capillaries resulting in painful vaso-occlusive crises.1 The sickle cells undergo hemolysis leading to anemia and a shortened lifespan. The only approved therapy is a cytotoxic drug, hydroxyurea,2 that works via an unknown mechanism but involves inducing the expression of fetal hemoglobin, which is protective against sickling. There is variable patient response and compliance to hydroxyurea.2
An approach to therapy would be to maintain the HbS in the oxygenated state, as polymerization occurs only in the deoxygenated state under hypoxic conditions. The natural product, 5-hydroxymethylfurfural (5HMF, 1, Figure 1a), was shown3 to bind to HbS via a reversible Schiff-base linkage to the-N-terminal valine of the α chain, and thereby prevent polymerization by inducing an allosteric conformational change that increases the oxygen affinity of the HbS. It was suggested that 1 preferentially stabilizes the R2 state,3 which has higher affinity for oxygen,4 thus preventing polymerization under hypoxic conditions. More potent synthetic aldehyde analogues of 5HMF such as BW12C795,6 and Tucaresol7 (Figure 1a) were developed earlier and shown clinically to reduce hemolysis when 20–30% of the HbS is modified. Neither BW12C79 (2) nor Tucaresol (3) was developed further although 5HMF (1) is reported to be in a clinical trial.8 Tucaresol was terminated apparently due to immunotoxicity issues. That a positive pharmacodynamic effect is observed at 20–30% modification of HbS is a key point as it implies a target modification to be attained for a desirable result. This proposed modification target results from studies in individuals with compound heterozygosity for HbS and pancellular hereditary persistence of fetal hemoglobin (S/HPFH) infers that the presence of 10–30% HbF provides clinical benefits with patients having few, if any, sickle cell-related events.9 As the concentration of Hb in the RBC is estimated at 5 mM, a positive allosteric modulator that binds in a reversible covalent manner would need to reach a minimum concentration of 1 mM to allow 20% occupancy in the RBC in order to be effective. This requirement poses a significant challenge with respect to preventing off-target effects while attaining such high concentrations in the target RBC.
Figure 1.

(a) Literature Hb binding aldehydes. (b) Structures of compounds 4, 5, and 6.
The X-ray structure of 5HMF in coordination with HbS is available.3 Furthermore, compound 4 (Figure 1b) and other synthetic analogues have been shown to have potency in hemoximetry assays.10 The increase in oxygen affinity of Hb is illustrated by the ability of the test compound to facilitate 50% oxygen saturation at lower oxygen pressure than the normal control, hence the term, left shifter. The X-ray structure of 4 with Hb has also been described.11 Analogously to 5HMF (1), compound 4 was proposed to bind in Schiff-base linkage to the N-terminal valines of each α chain of HbS with the aromatic rings of 4 protruding into the intradomain cavity and appearing to interact with one another. We proposed by molecular modeling that 5 (Figure 1b) and other fused bicyclic analogues would fill this cavity more efficiently than 4 by providing stronger interactions between the aromatic rings themselves, perhaps via increasing dipole interactions.
From the outset we were aware of the propensity for aldehydes to be metabolized rapidly. While we were also concerned with the possibility that Schiff-base linkages could be formed with exposed amines on any protein, the N-terminal valine on the α chain in Hb, having a pKa = 6.912 is primarily unprotonated at physiological pH and hence is more nucleophilic than protonated amines, such as the ω amino group of exposed lysine which has a pKa = 10.5 in polypeptides.13 This differential pKa of the N-terminal valine offers an in-built possibility for selectivity. We also hoped that the rapid reversibility of these Schiff-base linkages would result in the aldehyde moving from protein to protein until it arrived at the most stable situation, that being reversibly bound to Hb. This would require that we build in high affinity for Hb, compared to other proteins. In addition, the high concentration of Hb in the RBC would act as a sink for the aldehyde thus protecting it from being rapidly metabolized.
In our initial assays, allosteric modulation of purified HbS was determined using a Hemox Analyzer (TCS) to generate Oxygen Equilibrium Curves (OEC) and measuring the change in p50. The assay therefore is a measure of the test compound’s ability to maintain the oxygenated or R state of Hb. Data in Table 1 are reported as the % change in p50 from baseline (Δp50) when compounds were tested at 30 μM against purified Hb (a mixture of HbA and HbS) at 25 μM. Here, compound 4 had a Δp50 (%) of 34% at 30 μM, while 5 with a bicyclic A-ring was similarly active with a Δp50 (%) = 33%. However, in a time course study where the onset of oxygenation was measured as a surrogate for on-rate, 5 impacted the oxygenation with a much faster rate than 4, which in turn was faster than 5HMF (Figure 2). Thus, the maximum effect of 5 was seen several minutes after the first measurement was taken, while 1 and 4 took almost 1 h to do so.
Table 1. B-Ring SARs on Oxygen Affinity with Fused Bicyclics.
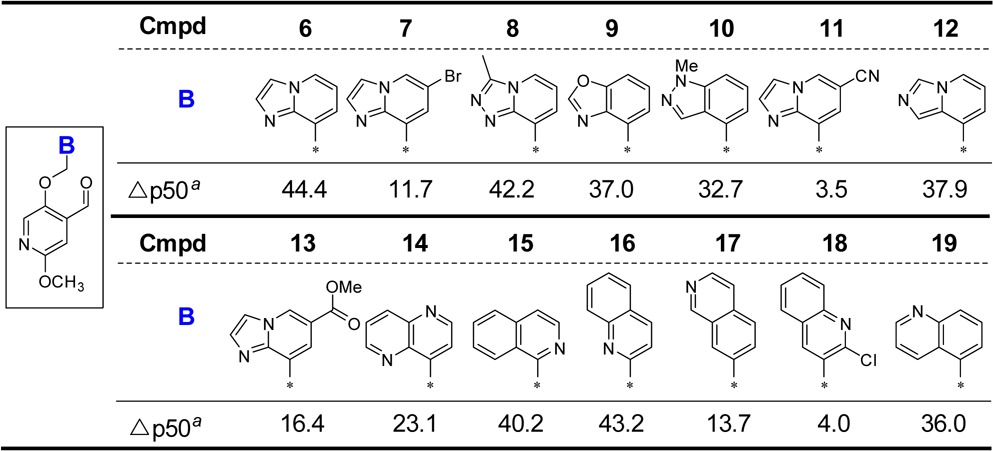
Data expressed as Δp50%: the % change from baseline of pO2 at which HbS (at 25 μM) is 50% saturated with O2 in the presence of test compound at 30 μM. Please see Supporting Information for standard deviation and number of tests.
Figure 2.
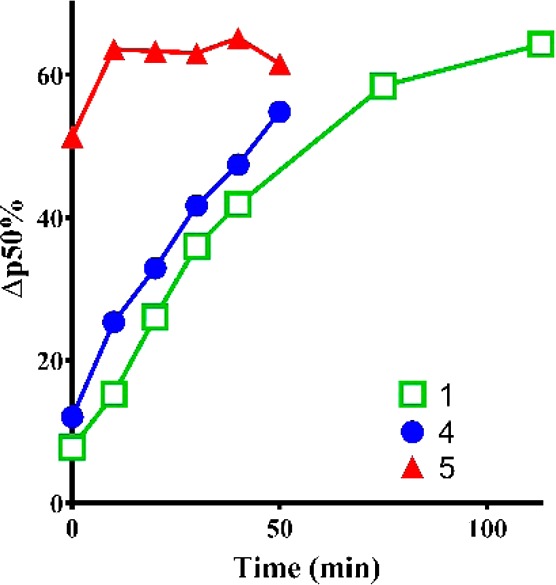
Time-dependent change in hemoglobin-oxygen affinity for 1, 4, and 5. Measurement was taken after a 20 min oxygenation/deoxygenation cycle in the Hemox Analyzer.
Consequently, as we anticipated that a fast on-rate would be an advantage, we elected to study further analogues bearing a bicyclic aromatic B-ring. In addition, with a concern that the para relationship of phenyl ethers, as in 4 and 5, may be a metabolic liability we prepared the methoxypyridine 6 (Figure 1b) that, to our surprise, elicited a Δp50 = 44%. This methoxypyridine A-ring was maintained through much of our early SAR program, while we concentrated on optimizing the aromatic B-ring that resides near the opening of the cavity. The X-ray crystal structure of 6 with HbS (Figure 3, PDB code 5UFJ) confirmed our molecular modeling hypothesis that 6 appears to form a Schiff-base linkage with the N-terminal valine of each α subunit, and the aromatic rings appear to interact with each other via π stacking. A representative set of the bicyclic B-ring analogues that we prepared is shown in Tables 1 and 2.
Figure 3.
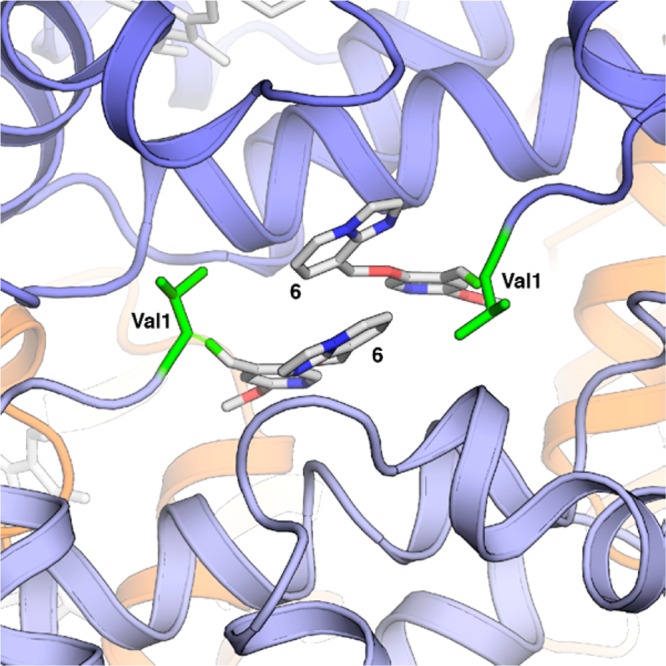
Cocrystal structure of 6 (in stick) with CO-ligand HbS.
Table 2. B-Ring SARs on Oxygen Affinity with Fused Bicyclic with Substituent.
Data expressed as Δp50%: the % change from baseline of pO2 at which HbS (at 25 μM) is 50% saturated with O2 in the presence of test compound at 30 μM. Please see Supporting Information for standard deviation and number of tests.
We hypothesized from our modeling that a heterocyclic ring attached to the bicyclic B-ring might benefit from further interactions with Hb protein; however, our initial efforts in this direction did not reveal compounds of greater potency (Table 2) despite greater MW and surface area, and we discontinued studies toward these tricyclic compounds.
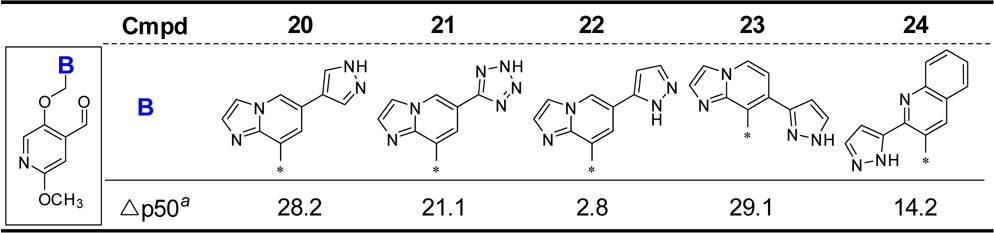
At this stage we elected to study the most potent compounds for their ability to increase Hb-oxygen affinity in whole blood from transfused SCD subjects, compared to the earlier protocol that used HbS/HbA protein purified from sickle disease blood. The impact of plasma protein binding became immediately apparent with even the most potent compounds described in Tables 1 and 2 losing a significant amount of their activity in whole blood, even at mM concentrations. However, biaryl structures, where one ring would be twisted with respect to the other, were found to be beneficial as shown by the compounds in Table 3. In particular, the combination of N at the 3-position and a pyrazole substituent at 2 with the pyrazole 1N-substituted with an isopropyl group proved among the most potent, increasing the Δp50 (%) in both the purified Hb and whole blood OEC. Compound 31 was outstanding as it maintained strong activity in the whole blood assay.
Table 3. B-Ring SARs on Oxygen Affinity with Purified HbS and Whole Blood and on the Change in Delay Time to the Onset of Polymerization.

Data expressed as Δp50%: the % change from baseline of pO2 at which HbS (at 25 μM) is 50% saturated with O2 in the presence of test compound at 30 μM.
Data expressed as Δp50%: the % change from baseline of pO2 at which whole blood (at 20% hematocrit, 1 mM) is 50% saturated with O2 in the presence of test compound at 1 mM.
Data expressed as ΔT%: the % change from baseline of delay time of HbS at 50 μM in the presence of test compound at 75 μM.
Not determined; please see Supporting Information for standard deviation and number of tests.
As our hypothesis was that compounds that improved the affinity of HbS for oxygen should prevent polymerization when HbS is subjected to hypoxic conditions, we developed a polymerization assay to confirm activity of the most potent compounds identified in the whole blood assay.14,15 Here purified HbS in a high concentration of KH2PO4 as a crowding agent induces polymerization by a temperature jump from 4 to 37 °C, and the impact of test compound on delay time to onset of polymerization is measured. Only key compounds from Table 3 were tested. The %Δ in the delay time (measured as the delay time in the presence of compound relative to the delay time of control) is presented in Table 3. At this stage, compound 31 remained the compound of most interest, as it was clearly superior in the critical whole blood OEC and confirmed by increasing the delay time in the polymerization assay.
Having arrived at an optimal B-ring substitution pattern as in 31, we began a brief survey of alternate A-ring aldehydes while maintaining the B-ring of 31 constant. Representative structures are shown in Table 4. We intended to survey the impact of changing the electronics of the aldehyde and hence the strength of the Schiff-base linkage (37, 38, 41, 43), and the consequences of a potential H bond from the phenolic OH to the imine of the Schiff-base (36 vs 44, 42 vs 43).16 Underlining the importance of the aldehyde itself, des-formyl compound 46, which lacks the aldehyde, is inactive.
Table 4. A-Ring SARs on Oxygen Affinity with Purified HbS and Whole Blood and on the Change in Delay Time to the Onset of Polymerization, and PK Properties in Rats.
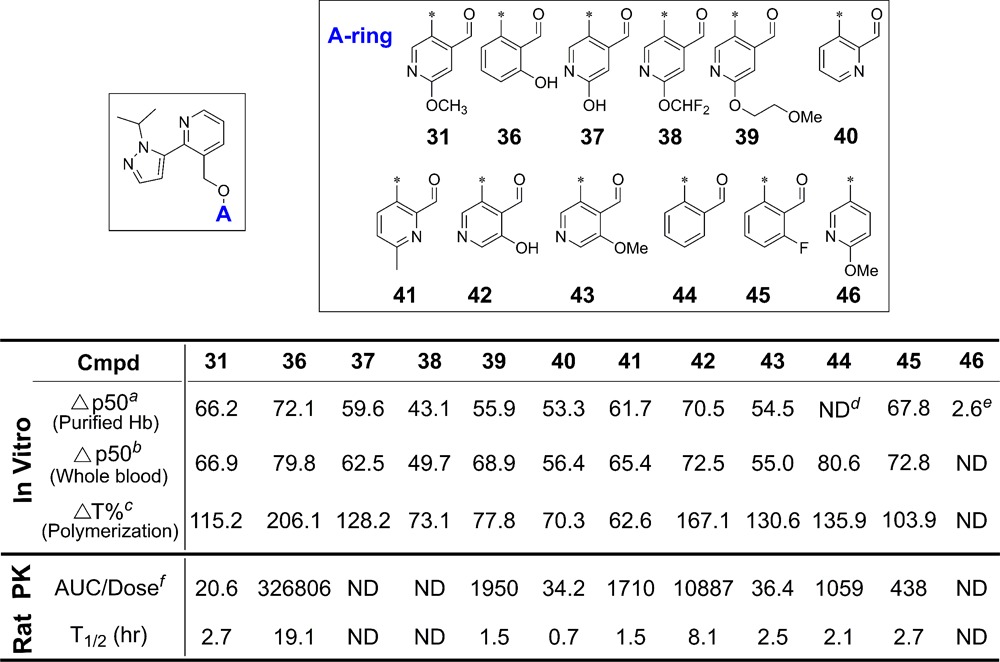
Data expressed as Δp50%: the % change from baseline of pO2 at which HbS (at 25 μM) is 50% saturated with O2 in the presence of test compound at 30 μM.
Data expressed as Δp50%: the % change from baseline of pO2 at which whole blood (at 20% hematocrit, 1 mM) is 50% saturated with O2 in the presence of test compound at 1 mM.
Data expressed as ΔT%: the % change from baseline of delay time of HbS at 50 μM in the presence of test compound at 75 μM.
Not determined.
Tested at 500 μM.
AUC(0-∞) value normalized by PO dose [(ng·h/mL)/(mg/kg)]. Please see Supporting Information for standard deviation and number of tests.
Several compounds from Table 4 were subjected to PK studies; however, compound 36 is clearly the superior compound when considering the hemox results with purified Hb and whole blood, polymerization assays, and initial PK screening. The impact of the phenolic hydroxyl in 36 is evidenced by the comparison in T1/2 in rat for 36 (19.1 h in blood) versus T1/2 for the des-hydroxy compound 44 (2.1 h). This difference may result from a stronger intramolecular H bond in the resultant Schiff-base of 36 with Hb compared to that from 44. These results prompted us to study 36 further in pharmacokinetic assays, to assess its oral bioavailability.
The pharmacokinetics of 36 was assessed in rats, dogs, and monkeys following i.v. and p.o. administration and has been noted in our earlier publication.17 Compound 36 has favorable oral bioavailability of 60, 37, and 36% in rats, dogs, and monkeys, respectively, with similar blood and plasma half-lives of approximately 20 h each. Of particular note is RBC/plasma ratio of ∼150 compared to ∼38 for 31. This high RBC/plasma ratio demonstrates that 36 is rapidly sequestered into RBCs in preference to plasma and implies that therapeutically effective blood concentrations in the target cell can be achieved at relatively low plasma concentrations. Thus, the possibility of off-target effects is diminished. T1/2 value of 36 in all animal species was significantly shorter than the T1/2 of red blood cells (∼20 days), which supports that binding of 36 to hemoglobin is a reversible process.
The ability of 36 to inhibit the major human liver CYP isozymes (CYP 1A2, 2C8, 2C9, 2C19, 2D6, and 3A4) was evaluated using pooled human liver microsomes and isozyme-specific probe substrates. While 36 inhibited these isozymes with IC50 ranging from 7.9 to 148 μM (Table 5), we feel that this level of inhibition is mitigated by the low plasma concentrations that are anticipated to allow therapeutic concentrations in the RBC as a consequence of the partitioning into the RBC. This high partitioning into RBC should protect against metabolism and against possible drug–drug interactions. There was no significant time-dependent inhibition of 36 against CYP2C19 and 3A4. Compound 36 is not a substrate for either P-gp or BCRP transporters.
Table 5. In Vitro CYP Inhibition with 36.
| Enzyme | Substrate | Control Inhibitor (IC50 (μM)) | Cmpd 36 IC50 (μM) |
|---|---|---|---|
| CYP1A2 | Phenacetin | Furafylline (6.29) | 58.6 |
| CYP2C8 | Paclitaxel | Quercetin (1.20) | 7.9 |
| CYP2C9 | Tolbutamide | Sulfaphenazole (0.205) | 8.5 |
| CYP2C19 | S-Mephentoin | Tranylcypromine (10.3) | 20.0 |
| CYP2D6 | Bufuralol | Quinidine (0.035) | 148 |
| CYP3A4 | Midazolam | Ketoconazole (0.018) | 81.9 |
| CYP3A4 | Testosterone | Ketoconazole (0.015) | 12.5 |
We followed the SAR with X-ray structures of key compounds. The fused bicyclic compounds as in Table 1 all presented similar poses to compound 6 with a 2:1 stoichiometry of allosteric modulator/HbS. However, the substituted pyridines such as compound 31 and 36 demonstrated a 1:1 stoichiometry (Figure 4 for 31, PDB code 5U3I; X-ray structure of 36 was published previously17).
Figure 4.
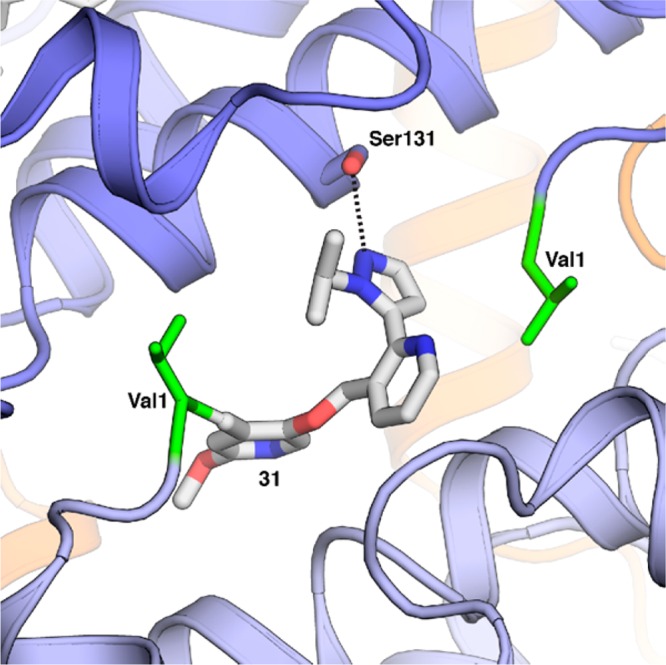
Zoomed image of the binding pocket for 31. The H-bond with Ser131 from the neighboring α chain appears to be a key interaction for maintaining the R-state of HbS.
A driving force for this pose and the 1:1 stoichiometry appeared to be the H bond from the pyrazole N to Ser131 on the second α chain, with the aldehyde forming the usual Schiff-base linkage to the N-valine of the first α chain (Figure 5). In this way 31 and other analogues filled the space normally taken up by two fused analogues or by two molecules of 5HMF (1). Not coincidentally those compounds that complexed Hb with a 1:1 stoichiometry performed better than compounds with a 2:1 stoichiometry in the whole blood assay and in the polymerization assay. As demonstrated in Figure 5, compounds 31 and 36 have similar orientations when bound to HbS. Compound 36 has favorable PK properties, and this difference is not immediately revealed by simply comparing how the compounds interact with HbS. We surmise that the PK of 36 is superior to that of 31 owing to an intramolecular H bond to the Schiff-base in the case of 36.
Figure 5.
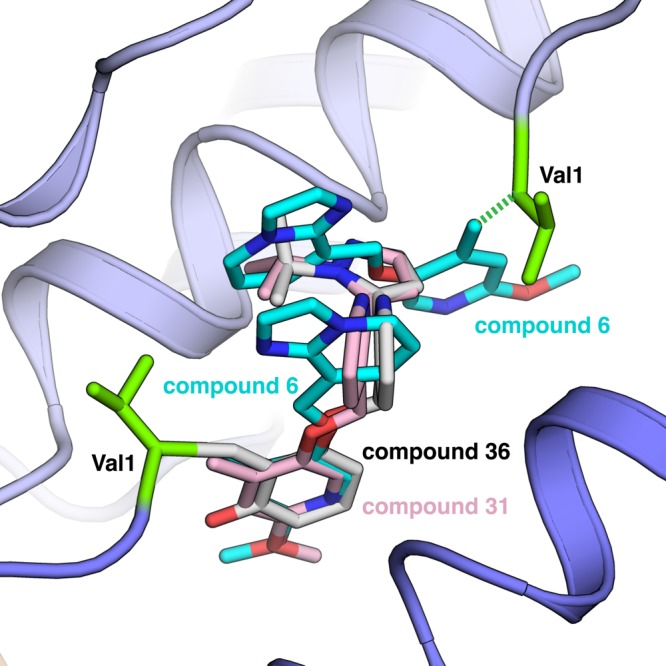
Overlay of compounds 6, 31, and 36. All compounds bind with Val1 (green). Compound 6 binds 2:1 per Hb tetramer, and both compounds 31 and 36 bind 1:1 per tetramer. This difference in stoichiometry is further demonstrated with the omit maps included in Figures S1 and S2.
The synthesis of this series of compounds is exemplified by the convergent synthesis of 36, shown in Scheme 1. The key coupling step of phenol 50 with the benzylic chloride 54 is affected using sodium carbonate as base in DMF at 65 °C for 1.5 h. Compound 55 was isolated in 81% yield by pouring the reaction mixture into ice water and filtering the solid. The protecting MOM group was removed using HCl (12 N) in THF. On neutralization the 36 free base was isolated by filtration. The key phenolic reactant, 50, was prepared from 2-bromoresorcinol (47) first by MOM protection of both hydroxyls, then lithiation via butyl lithium followed by quenching with DMF and HCl treatment. The second key reactant, the chloride 54, was prepared via a Suzuki coupling reaction between the pyrazole boronate ester 51 and the chloropyridine 52 to give the alcohol 53, which was chlorinated by thionyl chloride. Other aldehydes were prepared by varying the boronate ester and the phenol coupling agents (Supporting Information).
Scheme 1. Synthesis of 36.

Reagents and conditions: (a) MOMCl, DIEPA, DCM, 0 °C to rt 2 h, 90%; (b) nBuLi, DMF, THF, −78 to 0 °C, 94%; (c) 12 N HCl, THF, rt, 1.5 h, 81%; (d) Pd(dppf)Cl2, NaHCO3, H2O/dioxane, 100 °C, 12 h, 40%; (e) SOCl2, DCM, rt, 100%; (f) Na2CO3, DMF, 65 °C, 1.5 h, 81%; (g) 12 N HCl, THF, rt, 3 h, 96%.
In summary, we have developed a series of compounds that increase the oxygen affinity of HbS, both on the isolated protein and in whole blood from sickle cell patients. Key compounds increase the delay time to the onset of polymerization of HbS. These “left shifters” bind in a Schiff-base linkage to the amine of the N-terminal valine of the α chain of HbS. In comparison to those earlier reported such as 5HMF (1) and BW12C (2) that bind in a 2:1 stoichiometry, X-ray analysis reveals that key compounds such as 31 and GBT440 (36)17 bind to a single α chain in a 1:1 stoichiometry to the HbS tetramer. Compound 36 demonstrates favorable pharmacokinetics in the rat, dog, and monkey. As noted in the rat, oral bioavailability is 60%, the T1/2 is 19.1 h, and there is dramatic partitioning into blood with a RBC/plasma ratio of ∼150. It is anticipated that this unique partitioning into the target cell should allow therapeutic concentrations to be attained in the RBC at comparatively low plasma concentrations and thereby reduce the likelihood of off-target effects. Compound 36 (GBT440) is currently in Phase 3 clinical trials (NCT03036813) in SCD patients.
Acknowledgments
The authors thank Dr. Peter Rademacher for running HRMS for selected compounds. We wish to acknowledge the Hemoglobinopathy Center at the Children’s Hospital Oakland Research Institute (CHRCO, Oakland, CA) for providing blood from sickle cell patients. We thank J. Holton, G. Meigs, and ALS staff of beamline 8.3.1 for data collection and helpful discussions. Beamline 8.3.1 at the Advanced Light Source is operated by the University of California Office of the President, Multicampus Research Programs and Initiatives grant MR-15-328599 and Program for Breakthrough Biomedical Research, which is partially funded by the Sandler Foundation. Additional support comes from National Institutes of Health (GM105404, GM073210, GM082250, GM094625), National Science Foundation (1330685), Plexxikon Inc., and the M.D. Anderson Cancer Center. The Advanced Light Source (Berkeley, CA), a national user facility operated by Lawrence Berkeley National Laboratory on behalf of the US Department of Energy under contract number DE-AC02-05CH11231, Office of Basic Energy Sciences, through the Integrated Diffraction Analysis Technologies program, supported by the US Department of Energy Office of Biological and Environmental Research.
Glossary
ABBREVIATIONS
- 5HMF
5-hydroxymethylfurfural
- Cmax
the maximum concentration
- DIPEA
N,N-diisopropylethylamine
- Hb
hemoglobin
- HbA
adult hemoglobin
- HbS
sickle cell hemoglobin
- MOMCl
methoxymethyl chloride
- OEC
oxygen equilibrium curve
- Pd(dppf)Cl2
[1,1′-Bis(diphenylphosphino)ferrocene]palladium(II) dichloride
- RBC
red blood cell
- R and R2 states
relaxed or oxygenated states of hemoglobin
- SCD
sickle cell disease
- T1/2
half-life, the time required for Cmax to drop in half
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00491.
All experimental procedures and compounds characterization, material and methods about Hemox and polymerization assays, as well as DMPK and in vivo experiment protocols (PDF)
The authors declare the following competing financial interest(s): B.M., K.D., M.P., A.S., C.J., J.P., Q.X., S.G., C.Y., J.H., A.H., D.O., and Z.L. are employees of Global Blood Therapeutics, which has a commercial interest in hemoglobin modulators; M.P.J. owns GBT stock.
Notes
Carl Johnson: chagroth@stanford.edu. Lan Hua: hualan@nibs.ac.cn. Jason Harris: Jharris@jrhbiopharma.com. Joyce James: joycjames@gmail.com. Larysa Patskovska: larysa.patskovska@accentbiosystems.com. Stephen L. Gwaltney: sgwaltney@chrysalistherapeutics.com. Yury Patskovsky: yury.patskovsky@accentbiosystems.com.
Supplementary Material
References
- Rees D. C.; Williams T. N.; Gladwin M. T. Sickle-cell disease. Lancet 2010, 376, 2018–2031. 10.1016/S0140-6736(10)61029-X. [DOI] [PubMed] [Google Scholar]
- Charache S.; Terrin M. L.; Moore R. D.; Dover G. J.; Barton F. B.; Eckert S. V.; McMahon R. P.; Bonds D. R. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. N. Engl. J. Med. 1995, 332, 1317–1322. 10.1056/NEJM199505183322001. [DOI] [PubMed] [Google Scholar]
- Safo M. K.; Abdulmalik O.; Danso-danquah R.; Burnett J. C.; Nokuri S.; Joshi G. S.; Musayev F. N.; Asakura T.; Abraham D. J. Structural basis for the potent antisickling effect of a novel class of five-membered heterocyclic aldehydic compounds. J. Med. Chem. 2004, 47, 4665–4676. 10.1021/jm0498001. [DOI] [PubMed] [Google Scholar]
- Shibayama N.; Sugiyama K.; Park S. Y. Structures and Oxygen Affinities of Crystalline Human Hemoglobin C (β6 Glu→Lys) in the R and R2 Quaternary Structures. J. Biol. Chem. 2011, 286 (38), 33661–33668. 10.1074/jbc.M111.266056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrett M.; Stammers D. K.; White R. D.; Wootton R.; Kneen G. Characterization of the binding of the anti-sickling compound, BW12C79, to haemoglobin. Biochem. J. 1986, 239, 387–392. 10.1042/bj2390387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keidan A. J.; Franklin I.M.; White R. D.; Joy M.; Huehns E. R.; Stuart J. Effect of BW12C on Oxygen Affinity of Haemoglobin in Sickle-Cell Disease. Lancet 1986, 327, 831–834. 10.1016/S0140-6736(86)90941-4. [DOI] [PubMed] [Google Scholar]
- Rolan P. E.; Mercer A. J.; Wootton R.; Posner J. Pharmacokinetics and pharmacodynamics of tucaresol, an anti-sickling agent, in healthy volunteers. Br. J. Pharmacol. 1995, 39, 375–80. 10.1111/j.1365-2125.1995.tb04465.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato G. J.; Lawrence M. P.; Mendelsohn L. G.; Saiyed R.; Wang X.; Conrey A. K.; Starling J. M.; Grimes G.; Taylor J. G.; Mckew J.; Minniti C. P.; Stern W. Phase 1 clinical trial of the candidate anti-sickling agent Aes-103 in adults with sickle cell anemia. Blood 2013, 122, 1009. [Google Scholar]
- Akinsheye I.; Alsultan A.; Solovieff N.; Ngo D.; Baldwin C. T.; Sebastiani P.; Chui D. H. K.; Steinberg M. H. Fetal hemoglobin in sickle cell anemia. Blood 2011, 118, 19–27. 10.1182/blood-2011-03-325258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nnamani I. N.; Joshi G. S.; Danso-danquah R.; Abdulmalik O.; Asakura T.; Abrham D. J.; Safo M. K. Pyridyl derivatives of benzaldehyde as potential antisickling agents. Chem. Biodiversity 2008, 5, 1762–1769. 10.1002/cbdv.200890165. [DOI] [PubMed] [Google Scholar]
- Abdulmalik O.; Ghatge M. S.; Musayev F. N.; Parikh A.; Chen Q.; Yang J.; Nnamani I.; Danso-Danquah R.; Eseonu D. N.; Asakura T.; Abraham D. J.; Venitz J.; Safo M. K. Crystallographic analysis of human hemoglobin elucidates the structural basis of the potent and dual antisickling activity of pyridyl derivatives of vanillin. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, D67, 920–928. 10.1107/S0907444911036353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner M. H.; Bogardt R. A.; Gurd F. R. N. Determination of the pK values for the a-amino groups of human hemoglobin. J. Biol. Chem. 1975, 250, 4398–4404. [PubMed] [Google Scholar]
- Andre I.; Linse S.; Mulder F. A. A. Residue-Specific pKa Determination of Lysine and Arginine Side Chains by Indirect 15N and 13C NMR Spectroscopy: Application to apo Calmodulin. J. Am. Chem. Soc. 2007, 129, 15805–15813. 10.1021/ja0721824. [DOI] [PubMed] [Google Scholar]
- Adachi K.; Asakura T. Kinetics of the polymerization of hemoglobin in high and low phosphate buffers. Blood cells 1982, 8, 213–224. [PubMed] [Google Scholar]
- He Z.; Russell J. E. A high-throughput microplate method for assessing aggregation of deoxygenated hemoglobin s heterotetramers in vitro. Anal. Biochem. 2002, 306, 349–352. 10.1006/abio.2002.5683. [DOI] [PubMed] [Google Scholar]
- Wireko F. C.; Abraham D. J. X-ray diffraction study of the binding of the antisickling agent 12C79 to human hemoglobin. Proc. Natl. Acad. Sci. U. S. A. 1991, 88, 2209–2211. 10.1073/pnas.88.6.2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksenberg D.; Dufu K.; Patel M. P.; Chuang C. H.; Li Z.; Xu Q.; Silva-Garcia A.; Zhou C. J.; Hutchaleelaha A.; Patskovska L.; Patskovsky Y.; Almo S. C.; Sinha U.; Metcalf B. S. W.; Archer D. R. GBT440 increases haemoglobin oxygen affinity, reduces sickling and prolongs RBC half-life in a murine model of sickle cell disease. Br. J. Haematol. 2016, 175, 141–153. 10.1111/bjh.14214. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.