

Abstract

Misdirected catalytic activity of histone methyltransferase Dot1L is believed to be causative for a subset of highly aggressive acute leukemias. Targeting the catalytic domain of Dot1L represents a potential therapeutic approach for these leukemias. In the context of a comprehensive Dot1L hit finding strategy, a knowledge-based virtual screen of the Dot1L SAM binding pocket led to the discovery of 2, a non-nucleoside fragment mimicking key interactions of SAM bound to Dot1L. Fragment linking of 2 and 3, an induced back pocket binder identified in earlier studies, followed by careful ligand optimization led to the identification of 7, a highly potent, selective and structurally novel Dot1L inhibitor.
Keywords: Dot1L, protein lysine methyltransferase, inhibitor, mixed lineage leukemia, fragment linking
Acute myeloid leukemia (AML) is a genetically heterogeneous disease associated with a wide range of chromosomal aberrations with strong prognostic power. Translocations affecting the chromatin modifying protein MLL (mixed lineage leukemia) are found in 5–10% of patients with AML (many of whom have contracted AML as a late effect after prior chemotherapy) and in 70% of infants with acute lymphoblastic leukemia (ALL). There is strong genetic evidence that Dot1L, a histone methyltransferase targeting H3K79, is required for leukemogenesis driven by the most common MLL translocations (e.g., MLL-AF4, MLL-AF9, MLL-AF10, MLL-AF6).1−7 Beyond MLL-translocated leukemia, recent literature reports describe Dot1L dependence in AML with MLL-PTD fusions,8 NUP98-NSD1 translocations,9 IDH mutations,10 and HoxA overexpression.9,11
Targeting Dot1L, either by inhibiting the catalytic domain or the interaction with its fusion protein complex partner, is a potential therapeutic approach for the above-mentioned leukemias. Epizyme’s pioneering work in discovering catalytic Dot1L inhibitors resulted in the identification of the clinical candidate EPZ-5676.12 We and other research groups have discovered alternative means to inhibit Dot1L.13−19
Herein, we describe the discovery of potent, selective, and structurally novel Dot1L inhibitors via fragment linking. Fragment linking is an attractive strategy due to the promise of additive binding potency. In theory, the entropic benefit from the restriction of translational freedom and the displacement of waters by the linker might even result in superadditive linking.20 In reality, though, this potential is rarely fulfilled due to the stringent requirements on binding geometry and mutually compatible binding conformations. There are few cases that approach additive binding potency, e.g., thrombin (pIC50: 3.5 + 4.9 ⇒ 8.9)21 and BCL2 (pIC50: 3.5 + 2.2 ⇒ 5.9).22 Superadditive linking is mostly restricted to fragmentation of highly potent ligands (for e.g. MMPs, avidin),20 and a case study showed that a favorable entropic component is indeed responsible for the high linking efficiency.23 Some of the most successful examples that used linking prospectively involve the dimerization of ligands binding to dimeric enzymes with proximal binding sites, e.g., FBPase (pIC50: 2 × 3.2 ⇒ 7.8)24 and XIAP.25
As described in earlier reports,18,19 our Dot1L hit finding campaigns by fragment-based screening and high-throughput screening led to the discovery of chemical matter binding to an induced pocket adjacent to the binding site of cofactor S-adenosylmethionine (SAM). Some of the identified ligands extended into the methionine pocket of the SAM binding site. However, to our surprise, chemical matter occupying the adenosine pocket of the SAM binding site was not discovered by the above-mentioned techniques. We decided to develop alternative hit finding approaches to close this potential opportunity gap.
During our mode of action studies with the earlier identified induced pocket binders, it was realized that all of these ligands function through a SAM-competitive mode of action. Interestingly, though, adenosine can bind to Dot1L in the presence of the induced pocket binders. This observation triggered the generation of a ternary X-ray crystal structure of adenosine (IC50 = 100 μM) and induced pocket binder 1, a potent Dot1L inhibitor (IC50 = 190 nM),19 complexed to Dot1L (Figures 1 and 2).
Figure 1.

Chemical structure and biochemical potency of adenosine and 1.
Figure 2.
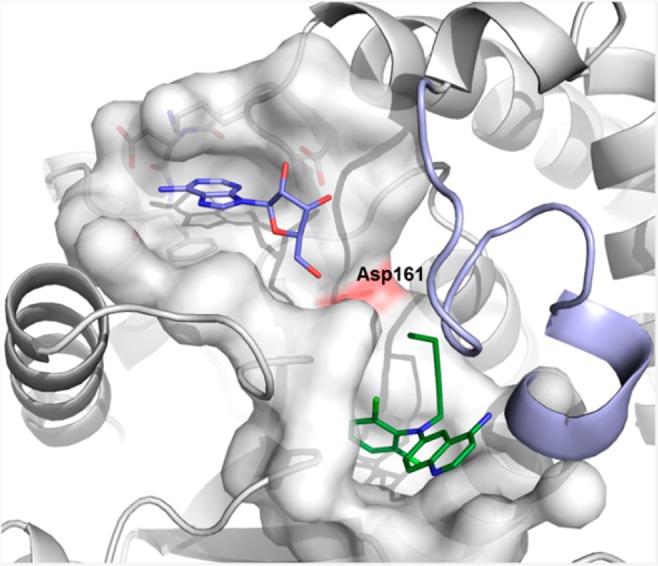
X-ray crystal structure of Dot1L with 1 and adenosine (pdb code 5mvs). Cartoon and surface representation of Dot1L (gray), flexible loop of Dot1L (126–140) (light blue), stick model of ligands 1 (green), and adenosine (blue). Asp161 is shown as red surface for orientation. The flexible loop has been omitted from the surface representation for clarity.
As observed for the Dot1L–compound 1 cocrystal structure, the flexible loop of Dot1L comprised amino acids 126–140, which forms the “lid” of the SAM binding pocket in the SAM-bound state, is folded over and collapsed onto ligand 1 (Figure 2). The interactions of 1 with Dot1L are identical in the binary versus ternary complex. Adenosine does not interact with the flexible loop in the ternary complex but otherwise forms the same contacts with Dot1L as in the binary complex.
With this information in hand, we chose to pursue two avenues to discover adenosine pocket binders. First, a fragment-based second-site screen by NMR was run with the first site (“the induced pocket”) being blocked with 1. Second, a knowledge-based virtual screen of the Novartis corporate archive for fragments resembling kinase hinge binders that satisfy an adenosine-based pharmacophore was conducted (Figure 3). This screen made use of our in house kinase inhibitor collection (mainly in the form of intermediates) and roughly 200 compounds were selected for a high concentration biochemical screen.
Figure 3.
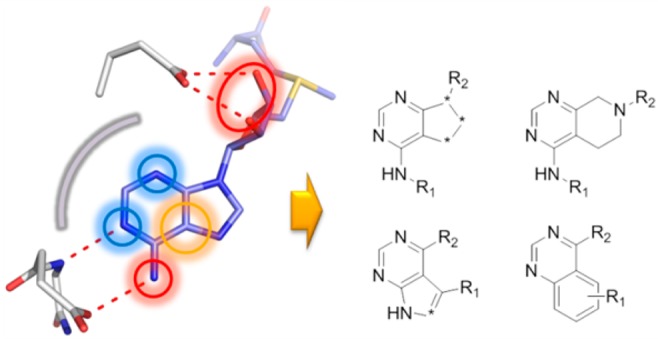
Virtual screen for SAM site binders was based on the SAM adenosine site pharmacophore (pdb code 5mvs; blue, acceptor; red, donor; orange, aromatic; gray, excluded volume), resulting in substructure and pharmacophore searches in which R2 was required to contain a suitable hydrogen bond donor.
All hits discovered from both approaches were submitted to cocrystallization experiments with Dot1L in the absence and presence of 1. Gratifyingly, the ternary X-ray structure for one of the virtual screen hits, pyrrolopyrimidine 2, a weak, fragment-like Dot1L inhibitor (IC50 = 240 μM, MW = 217 Da, clogP = 0.73, HBD = 2, HBA = 2), could be obtained (Figures 4 and 5).
Figure 4.

Chemical structure and biochemical potency of 2 and 3.
Figure 5.
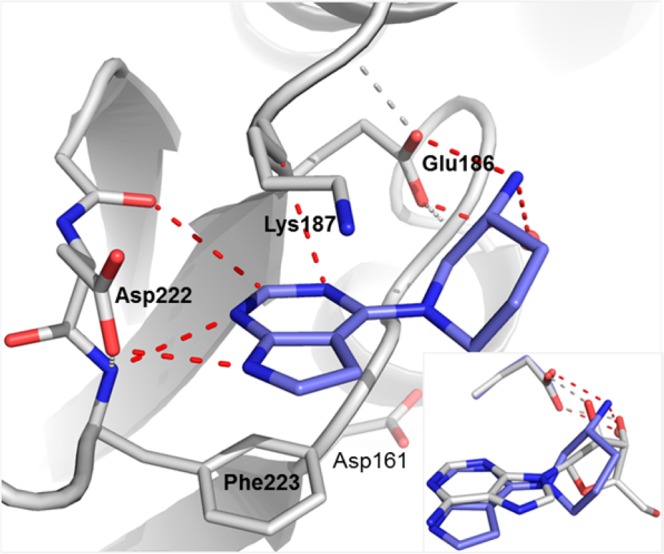
Detailed interactions of 2 with Dot1L from a ternary X-ray crystal structure of Dot1L (gray) with 2 (blue) and 1 (not displayed) (pdb code 5mw3). Amino acid side chains engaging in key interactions with the ligand illustrated as sticks and polar contacts highlighted as dotted red lines (protein hydrogen bonds in gray). Inset: Overlay of adenosine (gray) and 2 (blue) based on the ternary structures. Only subtle conformational changes were observed on the protein side between the two ternary complexes.
The overall protein conformation and the binding mode of ligand 1 are identical to the ternary complex discussed in Figure 2. As envisioned, 2 occupies the adenosine pocket of Dot1L. The NH of the pyrrolopyrimidine core serves as a hydrogen bond donor, interacting with the side chain of Asp222, while the adjacent nitrogen acts as a hydrogen bond acceptor for the backbone NH of Phe223 (Figure 5). Further, the primary amine of 2 is forming a salt bridge with Glu186 and coordinates a structural water, mimicking the bidentate coordination of Glu186 by the ribose moiety of SAM in the Dot1L–SAM complex. Importantly, the pyrrolopyrimidine core is nicely sandwiched between the side chains of Lys187 and Phe223 (Figure 5). Overall, the interactions of 2 with Dot1L are comparable to the adenosine–protein contacts in the SAM-bound Dot1L structure, validating the pharmacophore model used in the virtual screen (Figure 5 inset).
Two approaches to capitalize on the discovery of the adenosine replacement 2 were considered. Compound 2 could either be optimized by stepwise growing or by linking to fragment-like binders of the “induced pocket”. The latter approach was prioritized; however, ligand 1 was deemed to be not well suited for linking to 2 due to an unfavorable vector geometry/attachment point situation. In contrast, a different “induced pocket” binder, the previously discovered indole urea 3 (IC50 = 9.0 μM, MW = 314 Da) (Figure 4),18 seemed to be an attractive option for attempting fragment linking to 2.
A ternary model of 2 and 3 complexed to Dot1L was built based on the individual X-ray cocrystal structures of 2 and 3 (Figure 6). As indicated by the green arrow, it became apparent that the two ligands are perfectly lined up for linking via the primary amine of 2 and the amide NH2 of 3 by either a three- or four-carbon aliphatic chain.
Figure 6.
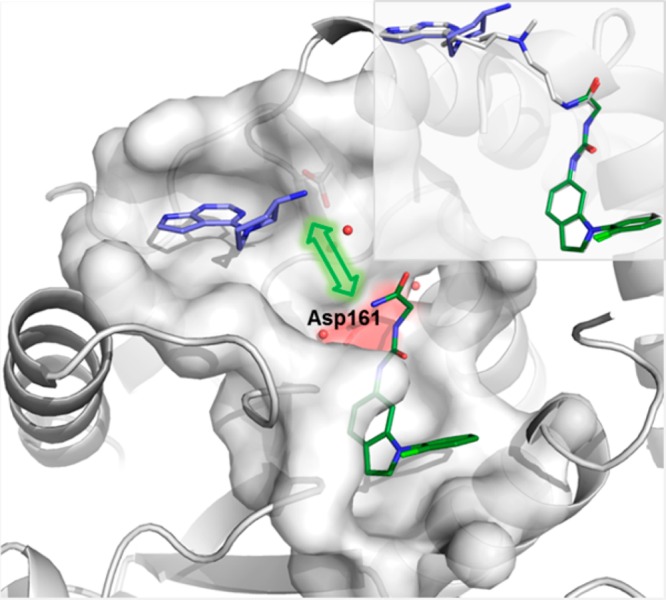
Model of ternary complex of 2 and 3 bound to Dot1L based on the individual X-ray cocrystal structures of 2 and 3. Surface representation of Dot1L (gray) and stick models of ligands 2 (blue) and 3 (green). Green arrow represents linking vector between 2 and 3. Inset: Overlay of 2 and 3 with a model of 6 (gray).
Both analogues, with the three-carbon and the four-carbon linker, were synthesized. To our delight, compounds 4 and 5 turned out to be highly potent Dot1L inhibitors (IC50 = 4 nM for both compounds) (Figure 7). Of note, and assuming that IC50 = 2Ki for both compounds, the binding energy (ΔG = −RTlnKi) of 4 and 5 is approximately the sum of the binding energy of fragments 2 and 3 (additive linking with linking coefficient E = 3.7 M–1).23,26
Figure 7.

N-Linking of 2 and 3 by a C3 or C4 linker to give 4 and 5.
Linked compound 4 was amenable for further potency optimization. Methylation of the secondary amine of 4 led to the dramatically more potent analogue 6 (IC50 = 0.15 nM) (Figure 8). We speculate that the additional methyl group stabilizes the almost eclipsed syn conformation of 4 (central torsion 20°) to a regular gauche conformation (central torsion 58°),27 although shielding of the hydrogen bond may further contribute to the more than 20-fold reduced IC50. Of note, the N-methyl derivative of fragment 2 is roughly equipotent to 2 (IC50 = 141 μM), formally resulting in superadditive linking with fragment 3 to compound 6 (E = 0.14 M–1). The high linking efficiency is remarkable, considering that the binding mode of the initial fragment 2 (and its N-Me analogue) is likely to be slightly altered (Figure 6 inset).
Figure 8.

Optimization of indole 4 to benzothiophene 7.
Furthermore, a series of bicyclic 5,6-, 6,5-, and 6,6-systems was explored as indole replacements. It turned out that benzothiophene is one of the most suitable cores, as exemplified by compound 7, a highly active Dot1L inhibitor (IC50 < 0.1 nM) (Figure 8).
Analogue 7 is characterized by a Ki in the very low picomolar range and a very long on-target residence time (τ > 5 h, the detection limit of our internal SPR assay) as assessed by surface plasmon resonance experiments (Table 1). Importantly, 7 performs equal or better than EPZ-5676 in a head-to-head comparison in our cellular assays. It potently suppresses H3K79 dimethylation (IC50 = 3 nM), the direct product of the Dot1L-catalyzed reaction, as well as the activity of the HoxA9 promoter (IC50 = 17 nM) in HeLa and Molm-13 cells, respectively (Table 1). Furthermore, 7 very effectively inhibits proliferation of the human MLL-rearranged leukemia cell line MV4-11 carrying the oncogenic MLL-AF4 fusion (IC50 = 5 nM) (Table 1). Noteworthy, 7 displays a favorable selectivity profile against a panel of 22 PKMTs and PRMTs with no inhibitory activity up to 50 μM (Supporting Information).
Table 1. Biochemical, Biophysical, and Cellular Characterization of Lead Compound 7.
| 7 | EPZ-5676 | |
|---|---|---|
| IC50 (Dot1L SPA)a | <0.1 | <0.1 |
| Ki (Dot1L SPA)a | 0.002 | 0.012 |
| τ (Dot1L SPR)+ | >5 | >5 |
| IC50 (HeLa, H3K79me2 ELISA)a | 3 | 7 |
| IC50 (Molm-13, HoxA9 RGA)a | 17 | 52 |
| IC50 (MV4-11, prolif.)a | 5 | 15 |
[nM], +[h], all data are the results of at least two assay runs with the mean value reported. The coefficient of variation was less than 60% in all cases. Biochemical IC50 values were determined at KM for SAM. Ki values were determined by applying the Morrison tight binding model (Supporting Information).
The X-ray cocrystal structure of 7 bound to Dot1L confirmed the expected binding mode (Figure 9). The urea glycine amide core overlaid perfectly with 3 in the ternary complex, the only marked difference being a flip of the o-Cl aryl due to an interaction of the pyridine nitrogen with Ser140. The amino piperidine was slightly pushed down relative to 2 in the ternary complex, so that the charged amine replaces the crystal water from the ternary complex and assumes a gauche conformation.
Figure 9.

X-ray cocrystal structure of Dot1L with 7 (pdb code 5mw4). Surface representation of Dot1L (gray) and stick model of ligand 7 (blue). Amino acid side chains engaging in key interactions with the ligand illustrated as sticks and polar contacts highlighted as dotted red lines. Inset: Overlay of the purine region of 7 (blue) and 2 (gray, crystal water in red) from the ternary structure.
The synthetic route to linked analogues is modular and allows for parallel SAR interrogation of the adenosine pocket binder, the induced pocket binder, and the linker. As an example, the synthesis of 7 is shown in Scheme 1. Adenosine pocket binder 2 is readily prepared in two steps from commercially available (R)-tert-butyl piperidin-3-ylcarbamate 8. The linker building block 11, accessed from Cbz-protected glycine 10 by amide bond formation with 3,3-diethoxypropan-1-amine, is fused to 2 by reductive amination. Methylation of 12, followed by Cbz-deprotection and CDI-mediated urea formation with amino-benzothiophene 15 completes the synthesis of 7 in a convergent manner. Please note that 15 is prepared by Suzuki coupling from 14, the synthesis for which has been described earlier.18
Scheme 1. Synthetic Route to Compound 7.

Reagents and conditions: (a) 4-chloro-7H-pyrrolo[2,3-d]pyrimidine, Hünig’s base, BuOH, 52 h, 80 °C, quant.; (b) HCl(aq), dioxane, 4 h, rt, then H+ exchange resin, MeOH and elution with NH3, MeOH, 95%; (c) TPTU, Hünig’s base, DMA, 7 min, 0 °C, then 3,3-diethoxypropan-1-amine, 45 min, rt, 67%; (d) 2, NaOAc, MeOH/CH2Cl2, 30 min, rt, then NaBH3CN, 16 h, rt, 58%; (e) 37% formaldehyde(aq), NaBH3CN, AcOH, MeOH, 45 min, rt, 80%; (f) 10% Pd–C, H2, MeOH/THF, 7.5 h, rt, 84%; (g) 2-methyl-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine, PdCl2(Amphos), Na2CO3(aq), EtOH/toluene, 20 min, 60 °C, 36%; (h) CDI, MeCN/DMF, 1 h, rt, then 15, NEt3, 1 h, 45 °C, 33%.
In summary, we have discovered by customized screening an attractive fragment 2 mimicking the adenosine pocket interactions of SAM bound to Dot1L. Fragment linking of 2 and 3, an induced pocket binder identified in earlier studies, delivered a highly potent, selective and structurally novel Dot1L inhibitor 7, lacking any nucleoside-like features. Future research is directed at the improvement of ADME properties, with a focus on the reduction of hydrogen bond donors and number of rotatable bonds.
PyMol was used for structural visualization and figure preparation.28
Acknowledgments
We are grateful to our medicinal chemistry colleagues Christophe Mura, Rainer Tschan, Meral Ilhan, Michael Hediger, and Stephan Kläusler for their scientific contributions. We thank Paul J. Westwood, Kerstin Pollehn, Dirk Erdmann, Claudio Thoma, and Andreas Theuer for developing and supporting project-related biophysical, biochemical, and cellular assays. The authors would like to acknowledge Elke Koch, Céline Be, Aurelie Winterhalter, Julia Klopp, Aude Izaac, and Patrick Graff for their contributions to the protein structural work. The crystallographic experiments were performed on the X10SA beamline at the Swiss Light Source, Paul Scherrer Institut, Villigen, Switzerland supported by the team of Expose GmbH.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00519.
Synthetic procedures, compound characterization, and assay protocols (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Nguyen A. T.; Taranova O.; He J.; Zhang Y. Dot1L, the H3K79 Methyltransferase, Is Required for MLL-AF9-Mediated Leukemogenesis. Blood 2011, 117, 6912–6922. 10.1182/blood-2011-02-334359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S. Y.; Granowicz E. M.; Maillard I.; Thomas D.; Hess J. L. Requirement for Dot1L in Murine Postnatal Hematopoiesis and Leukemogenesis by MLL Translocation. Blood 2011, 117, 4759–4768. 10.1182/blood-2010-12-327668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang M.-J.; Wu H.; Achille N. J.; Reisenauer M. R.; Chou C.-W.; Zeleznik-Le N. J.; Hemenway C. S.; Zhang W. Histone H3 Lysine 79 Methyltransferase Dot1 Is Required for Immortalization by MLL Oncogenes. Cancer Res. 2010, 70, 10234–10242. 10.1158/0008-5472.CAN-10-3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernt K. M.; Zhu N.; Sinha A. U.; Vempati S.; Faber J.; Krivtsov A. V.; Feng Z.; Punt N.; Daigle A.; Bullinger L.; Pollock R. M.; Richon V. M.; Kung A. L.; Armstrong S. A. MLL-Rearranged Leukemia Is Dependent on Aberrant H3K79 Methylation by Dot1L. Cancer Cell 2011, 20, 66–78. 10.1016/j.ccr.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande A. J.; Chen L.; Fazio M.; Sinha A. U.; Bernt K. M.; Banka D.; Dias S.; Chang J.; Olhava E. J.; Daigle S. R.; Richon V. M.; Pollock R. M.; Armstrong S. A. Leukemic Transformation by the MLL-AF6 Fusion Oncogene Requires the H3K79 Methyltransferase Dot1L. Blood 2013, 121, 2533–2541. 10.1182/blood-2012-11-465120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L.; Deshpande A. J.; Banka D.; Bernt K. M.; Dias S.; Buske C.; Olhava E. J.; Daigle S. R.; Richon V. M.; Pollock R. M.; Armstrong S. A. Abrogation of MLL-AF10 and CALM-AF10-Mediated Transformation through Genetic Inactivation or Pharmacological Inhibition of the H3K79 Methyltransferase Dot1L. Leukemia 2013, 27, 813–822. 10.1038/leu.2012.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivtsov A. V.; Feng Z.; Lemieux M. E.; Faber J.; Vempati S.; Sinha A. U.; Xia X.; Jesneck J.; Bracken A. P.; Silverman L. B.; Kutok J. L.; Kung A. L.; Armstrong S. A. H3K79 Methylation Profiles Define Murine and Human MLL-AF4 Leukemias. Cancer Cell 2008, 14, 355–368. 10.1016/j.ccr.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kühn M. W. M.; Hadler M. J.; Daigle S. R.; Koche R. P.; Krivtsov A. V.; Olhava E. J.; Caligiuri M. A.; Huang G.; Bradner J. E.; Pollock R. M.; Armstrong S. A. MLL Partial Tandem Duplication Leukemia Cells Are Sensitive to Small Molecule Dot1L Inhibition. Haematologica 2015, 100, e190–e193. 10.3324/haematol.2014.115337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande A. J.; Deshpande A.; Sinha A. U.; Chen L.; Chang J.; Cihan A.; Fazio M.; Chen C.; Zhu N.; Koche R.; Dzhekieva L.; Ibáñez G.; Dias S.; Banka D.; Krivtsov A.; Luo M.; Roeder R. G.; Bradner J. E.; Bernt K. M.; Armstrong S. A. AF10 Regulates Progressive H3K79 Methylation and HOX Gene Expression in Diverse AML Subtypes. Cancer Cell 2014, 26, 896–908. 10.1016/j.ccell.2014.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkaria S. M.; Christopher M. J.; Klco J. M.; Ley T. J. Primary Acute Myeloid Leukemia Cells with IDH1 or IDH2Mutations Respond to a Dot1L Inhibitor in Vitro. Leukemia 2014, 28, 2403–2406. 10.1038/leu.2014.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Methods for the Detection and Treatment of Leukemias That Are Responsive to Dot1L Inhibition. WO2015/017863.
- Daigle S. R.; Olhava E. J.; Therkelsen C. A.; Basavapathruni A.; Jin L.; Boriack-Sjodin P. A.; Allain C. J.; Klaus C. R.; Raimondi A.; Scott M. P.; Waters N. J.; Chesworth R.; Moyer M. P.; Copeland R. A.; Richon V. M.; Pollock R. M. Potent Inhibition of Dot1L as Treatment of MLL-Fusion Leukemia. Blood 2013, 122, 1017–1025. 10.1182/blood-2013-04-497644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen C.; Jo S. Y.; Liao C.; Hess J. L.; Nikolovska-Coleska Z. Targeting Recruitment of Disruptor of Telomeric Silencing 1-like (Dot1L): Characterizing the Interactions between Dot1L and Mixed Lineage Leukemia (MLL) Fusion Proteins. J. Biol. Chem. 2013, 288, 30585–30596. 10.1074/jbc.M113.457135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W.; Chory E. J.; Wernimont A. K.; Tempel W.; Scopton A.; Federation A.; Marineau J. J.; Qi J.; Barsyte-Lovejoy D.; Yi J.; Marcellus R.; Iacob R. E.; Engen J. R.; Griffin C.; Aman A.; Wienholds E.; Li F.; Pineda J.; Estiu G.; Shatseva T.; Hajian T.; Al-Awar R.; Dick J. E.; Vedadi M.; Brown P. J.; Arrowsmith C. H.; Bradner J. E.; Schapira M. Catalytic Site Remodelling of the Dot1L Methyltransferase by Selective Inhibitors. Nat. Commun. 2012, 3, 1288–1298. 10.1038/ncomms2304. [DOI] [PubMed] [Google Scholar]
- Yao Y.; Chen P.; Diao J.; Cheng G.; Deng L.; Anglin J. L.; Prasad B. V. V; Song Y. Selective Inhibitors of Histone Methyltransferase Dot1L: Design, Synthesis, and Crystallographic Studies. J. Am. Chem. Soc. 2011, 133, 16746–16749. 10.1021/ja206312b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anglin J. L.; Deng L.; Yao Y.; Cai G.; Liu Z.; Jiang H.; Cheng G.; Chen P.; Dong S.; Song Y. Synthesis and Structure-Activity Relationship Investigation of Adenosine-Containing Inhibitors of Histone Methyltransferase Dot1L. J. Med. Chem. 2012, 55, 8066–8074. 10.1021/jm300917h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anglin J. L.; Song Y. A. Medicinal Chemistry Perspective for Targeting Histone H3 Lysine-79 Methyltransferase Dot1L. J. Med. Chem. 2013, 56, 8972–8983. 10.1021/jm4007752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.; Zhu H.; Stauffer F.; Caravatti G.; Vollmer S.; Machauer R.; Holzer P.; Möbitz H.; Scheufler C.; Klumpp M.; Tiedt R.; Beyer K. S.; Calkins K.; Guthy D.; Kiffe M.; Zhang J.; Gaul C. The Discovery of Novel Dot1L Inhibitors through a Structure-Based Fragmentation Approach. ACS Med. Chem. Lett. 2016, 7, 735–740. 10.1021/acsmedchemlett.6b00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheufler C.; Be C.; Fernandez C.; Möbitz H.; Ragot C.; Beyer K.; Gaul C.; Stauffer F. Optimization Towards Potent Dot1L Inhibitors of a Fragment-Based Screen Hit Containing a 2,6-Dichlorophenyl Hydrophobic Anchor and Interacting in an Induced Binding Pocket. ACS Med. Chem. Lett. 2016, 7, 730–734. 10.1021/acsmedchemlett.6b00168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichihara O.; Barker J.; Law R. J.; Whittaker M. Compound Design by Fragment-Linking. Mol. Inf. 2011, 30, 298–306. 10.1002/minf.201000174. [DOI] [PubMed] [Google Scholar]
- Howard N.; Abell C.; Blakemore W.; Chessari G.; Congreve M.; Howard S.; Jhoti H.; Murray C. W.; Seavers L. C. A.; van Montfort R. L. M. Application of Fragment Screening and Fragment Linking to the Discovery of Novel Thrombin Inhibitors. J. Med. Chem. 2006, 49, 1346–1355. 10.1021/jm050850v. [DOI] [PubMed] [Google Scholar]
- Petros A. M.; Dinges J.; Augeri D. J.; Baumeister S. A.; Betebenner D. A.; Bures M. G.; Elmore S. W.; Hajduk P. J.; Joseph M. K.; Landis S. K.; Nettesheim D. G.; Rosenberg S. H.; Shen W.; Thomas S.; Wang X.; Zanze I.; Zhang H.; Fesik S. W. Discovery of a Potent Inhibitor of the Antiapoptotic Protein Bcl-XL from NMR and Parallel Synthesis. J. Med. Chem. 2006, 49, 656–663. 10.1021/jm0507532. [DOI] [PubMed] [Google Scholar]
- Borsi V.; Calderone V.; Fragai M.; Luchinat C.; Sarti N. Entropic Contribution to the Linking Coefficient in Fragment Based Drug Design: A Case Study. J. Med. Chem. 2010, 53, 4285–4289. 10.1021/jm901723z. [DOI] [PubMed] [Google Scholar]
- Hebeisen P.; Kuhn B.; Kohler P.; Gubler M.; Huber W.; Kitas E.; Schott B.; Benz J.; Joseph C.; Ruf A. Allosteric FBPase Inhibitors Gain 10(5) Times in Potency When Simultaneously Binding Two Neighboring AMP Sites. Bioorg. Med. Chem. Lett. 2008, 18, 4708–4712. 10.1016/j.bmcl.2008.06.103. [DOI] [PubMed] [Google Scholar]
- Dimeric IAP Inhibitors. WO2012/080271.
- Shuker S. B.; Hajduk P. J.; Meadows R. P.; Fesik S. W. Discovering High-Affinity Ligands for Proteins: SAR by NMR. Science 1996, 274, 1531–1534. 10.1126/science.274.5292.1531. [DOI] [PubMed] [Google Scholar]
- Observation from unpublished X-ray cocrystal structures of 4 and 6 bound to Dot1L.
- The PyMOL Molecular Graphics System, version 1.5.0.4; Schrödinger, LLC. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.