

Abstract
Dysregulation of glycogen synthase kinase-3β (GSK-3β) is implicated in the pathogenesis of neurodegenerative and psychiatric disorders. Thus, development of GSK-3β radiotracers for positron emission tomography (PET) imaging is of paramount importance, because such a noninvasive imaging technique would allow better understanding of the link between the activity of GSK-3β and central nervous system disorders in living organisms, and it would enable early detection of the enzyme’s aberrant activity. Herein, we report the synthesis and biological evaluation of a series of fluorine-substituted maleimide derivatives that are high-affinity GSK-3β inhibitors. Radiosynthesis of a potential GSK-3β tracer [18F]10a is achieved. Preliminary in vivo PET imaging studies in rodents show moderate brain uptake, although no saturable binding was observed in the brain. Further refinement of the lead scaffold to develop potent [18F]-labeled GSK-3 radiotracers for PET imaging of the central nervous system is warranted.
Keywords: Glycogen synthase kinase-3, PET imaging, radiofluorination, maleimide
Glycogen synthase kinase-3 (GSK-3) is a serine-threonine protein kinase family consisting of two highly homologous isoforms (GSK-3α and GSK-3β). This enzyme is ubiquitously expressed in a variety of tissues and is particularly abundant in the central nervous system (CNS).1 GSK-3 is associated with a number of cellular functions and physiological events, such as cell proliferation, stem cell renewal, neuropathological events, and apoptosis.2−5 A growing body of literature has linked dysregulation of GSK-3β activity to numerous severe pathologies, such as diabetes,6 neurodegenerative conditions, such as Alzheimer’s disease (AD)7,8 and Parkinson disease,9 psychiatric disorders, such as bipolar disorder and major depression,10−12 pain,13,14 cancer,15−17 and cardiac hypertrophy.18,19 Accordingly, GSK-3β has emerged as an important target for development of new drugs for the treatment of these diseases.
Positron emission tomography (PET) is a noninvasive in vivo imaging technique using radiotracers to characterize, visualize, and quantify physiological processes at the molecular level.20 In addition, due to the high penetrating power of γ photons (511 keV) generated in the annihilation process, PET is especially suitable for brain imaging, which renders it a powerful tool to study GSK-3β dysregulation in neurological disorders. Thus, development of a selective GSK-3β PET tracer may enable early detection of GSK-3β aberrant activity and accelerate GSK-3β therapeutic drug discovery. To date, several PET radiotracers for GSK-3 were reported, but only a few of them were successfully used in PET imaging (Figure 1). [11C]Methoxy-AR-A014418 (IC50 = 104 nM)21,22 and [11C]PyrATP-1 (IC50 = 4.9 nM)23 were radiolabeled and evaluated in rodents. Unfortunately, both of them lacked sufficient brain penetration and thus were unsuitable for neuroimaging purposes.21,23 In 2015, Zhang et al. reported two PET radiotracers, [11C]1 (IC50 = 66 nM) and [11C]3 (IC50 = 42 nM), that could pass the blood-brain-barrier (BBB).24 In the same year, Li showed that [11C]SB-216763 (IC50 = 34 nM) was able to cross the BBB and enter the CNS in both rodents and nonhuman primates.25 Most recently, [11C]PF-04082367 (IC50 = 2.1 nM) was reported to be a selective GSK-3 PET tracer with reasonable brain permeability and specificity in a nonhuman primate brain.26
Figure 1.
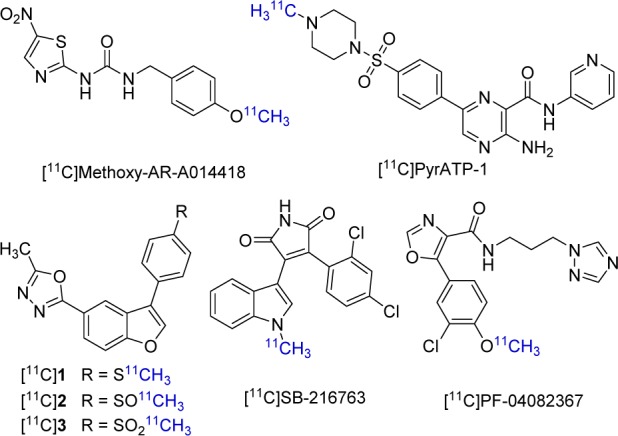
Structures of published GSK-3 PET radiotracers.
Despite the recent promising attempts to study GSK-3β in the CNS, further efforts to quantify GSK-3β activity in vivo using brain-penetrating radiotracers are needed. A major drawback of the current PET radiotracers is the use of a short-lived carbon-11 (t1/2 = 20.4 min), which limits their applications to PET imaging centers equipped with an in-house cyclotron. In contrast, the longer half-life of fluorine-18 (t1/2 = 109.7 min) compared with that of carbon-11 offers the potential advantage of facilitating widespread use and distribution of the tracer as well as longer imaging protocols to be carried out. In addition, 18F radioisotope emits positrons with a short positron range, which offers a better spatial resolution and image quality. Thus, development of [18F]-labeled brain-penetrating GSK-3β PET tracers is highly desired.27−29 Herein, we report our efforts in development of a [18F]-labeled maleimide-based radiotracer for GSK-3β PET imaging.
We selected substituted maleimides as a platform for development of high-affinity GSK-3β tracers because this class of compounds were used as scaffolds for ATP competitive GSK-3 inhibitors and showed an exceptional potency and selectivity profile for GSK-3.30−40 It was recently demonstrated that radiolabeled maleimide [11C]SB-216763 (Figure 1) is able to cross the BBB,25 which provides evidence that maleimides show sufficient brain uptake to potentially study GSK-3β activity in the CNS. Additionally, the maleimide scaffold is highly modular and allows fast assembly of a wide range of molecules through condensation of methylindole-3-glyoxalates (6a–k, 11a–c) and (hetero)arylacetamide fragments (9, 13a–g) (Scheme 1 and Scheme 2) in a few synthetic steps. We focused our efforts on the synthesis of maleimide derivatives with different aryl/heteroaryl substituents at the C3′ and C4′ positions (Figure 2). We also incorporated a fluoroethyl group in an array of compounds synthesized with a fluorine atom at a position to which [18F]fluoride could be incorporated in the PET radiotracer.
Scheme 1. General Synthesis of Compounds 10a–k.

Reagents and conditions: (a) (i) (COCl)2, Et2O, 0 °C to rt, 1 h; (ii) NaOMe, MeOH, −78 °C to rt, 2 h; (b) NaH, DMF, R1X, 0 °C to rt, overnight; (c) (i) MeOH, H2SO4, reflux, 5 h; (ii) NH3(aq), rt, overnight; (d) K2CO3, DMF, NaI, 1-bromo-2-fluoroethane, 60 °C, 20 h; (e) t-BuOK, THF, 0 °C, 4 h.
Scheme 2. General Synthesis of Compounds 14a–i.

Reagents and conditions: (a) NaH, DMF, 1-bromo-2-fluoroethane, 0 °C to rt, overnight; (b) (i) SOCl2, CH2Cl2, rt to reflux, 2 h; (ii) NH3(aq), rt; overnight; (c) t-BuOK, THF, 0 °C, 4 h.
Figure 2.
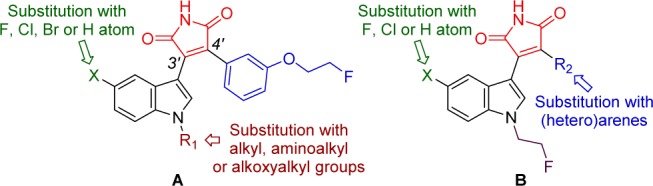
Synthetic modifications of maleimide scaffold.
Target compounds (10a–k) were synthesized according to literature procedures as depicted in Scheme 1.35,41,42 Commercially available indole derivatives 4a–d were first treated with oxalyl chloride and then with sodium methoxide to afford methylindole-3-glyoxalates 5a–d. N-Alkylation of 5a–d with alkyl halides in the presence of sodium hydride afforded 6a–k. Preparation of the coupling partner, 2-(3-(2-fluoroethoxy)phenyl)acetamide 9 began with an acid catalyzed esterification of 2-(3-hydroxyphenyl)acetic acid 7 followed by amidation of the resulting ester with aqueous ammonia to give acetamide 8. O-Alkylation of 8 with 1-bromo-2-fluoroethane in the presence of potassium carbonate and sodium iodide afforded 9. With 6a–k and 9 in hand, we performed a condensation reaction using potassium tert-butoxide in tetrahydrofuran at 0 °C to provide the desired fluorinated maleimides 10a–k in 35–69% yield. It is noteworthy that maintaining reaction temperature at 0 °C and dropwise addition of the potassium tert-butoxide solution (in THF) are critical in minimizing formation of an alkene side product (HF elimination) and obtaining the desired product in good yields.
The synthetic route to inhibitors containing different aromatic and heteroaromatic moieties at the C4′ position of the maleimide (14a–i) is illustrated in Scheme 2. N-Fluoroethylindole derivatives 11a–c were prepared from 5a–c and 1-bromo-2-fluoroethane in the presence of sodium hydride in DMF. The amide coupling partners 13a–g were synthesized from the corresponding carboxylic acids 12a–g in a two-step procedure: 12a–g were treated with thionyl chloride to produce acyl chloride intermediates, which were subsequently treated with ammonia to afford acetamides 13a–g. Condensation of N-fluoroethylindole derivatives 11a–c with substituted acetamides 13a–g afforded 14a–i in 10–65% yield.
With fluorine-substituted maleimides in hand, we first investigated the relationship between indole rings with different substitution patterns and the inhibitory activity of the maleimides (10a–k), while keeping the fluoroethyl substituent fixed on the aromatic ring (Figure 2A). As indicated in Table 1a, most of the compounds tested showed potent GSK-3β inhibitory activity.43 The maleimide derivative with an ethyl group on the indole nitrogen and a fluorine atom at the C5-position of the indole ring (10a) was found to be the most potent GSK-3β inhibitor among the whole series, with an IC50 value of 1.70 nM, which is of equivalent potency to the known and highly selective GSK-3β inhibitor CHIR-99021 (IC50 = 1.50 nM). We observed that both shortening (10b, IC50 = 45.6 nM) and lengthening of the alkyl chain (10c, IC50 = 63.9 nM; 10d, IC50 = 144 nM, 10e, IC50 > 10,000 nM) diminished the GSK-3β inhibition. Chemical modifications were carried out to decrease the lipophilicity of the maleimides, which is commonly accomplished by introduction of an oxygen- or nitrogen-containing functional group. The aminoethyl (10f, IC50 = 169 nM) and 2-morpholinylethyl (10g, IC50 = 50.0 nM) analogues were 99- and 29-fold less potent than the ethyl analogue 10a, respectively. Substitution of ethyl by a 2-methoxyethyl group at the indole nitrogen (10h, IC50 = 5.60 nM) resulted in an approximately 3-fold decrease in potency. Comparison of the substituents at the 5-position of indole showed that 5-halogen analogues (10b, IC50 = 45.6 nM; 10j, IC50 = 27.0 nM; and 10k, IC50 = 127 nM) were about 1–6-fold higher in potency than the unsubstituted indole analogue 10i (IC50 = 164 nM). The observation that the presence of a chlorine or fluorine atom at the 5-position of the indole improves inhibition of GSK-3β is consistent with the previously reported data.34
Table 1. In Vitro Evaluation of GSK-3β Inhibition by Maleimides 10a–k and 14a–ia.
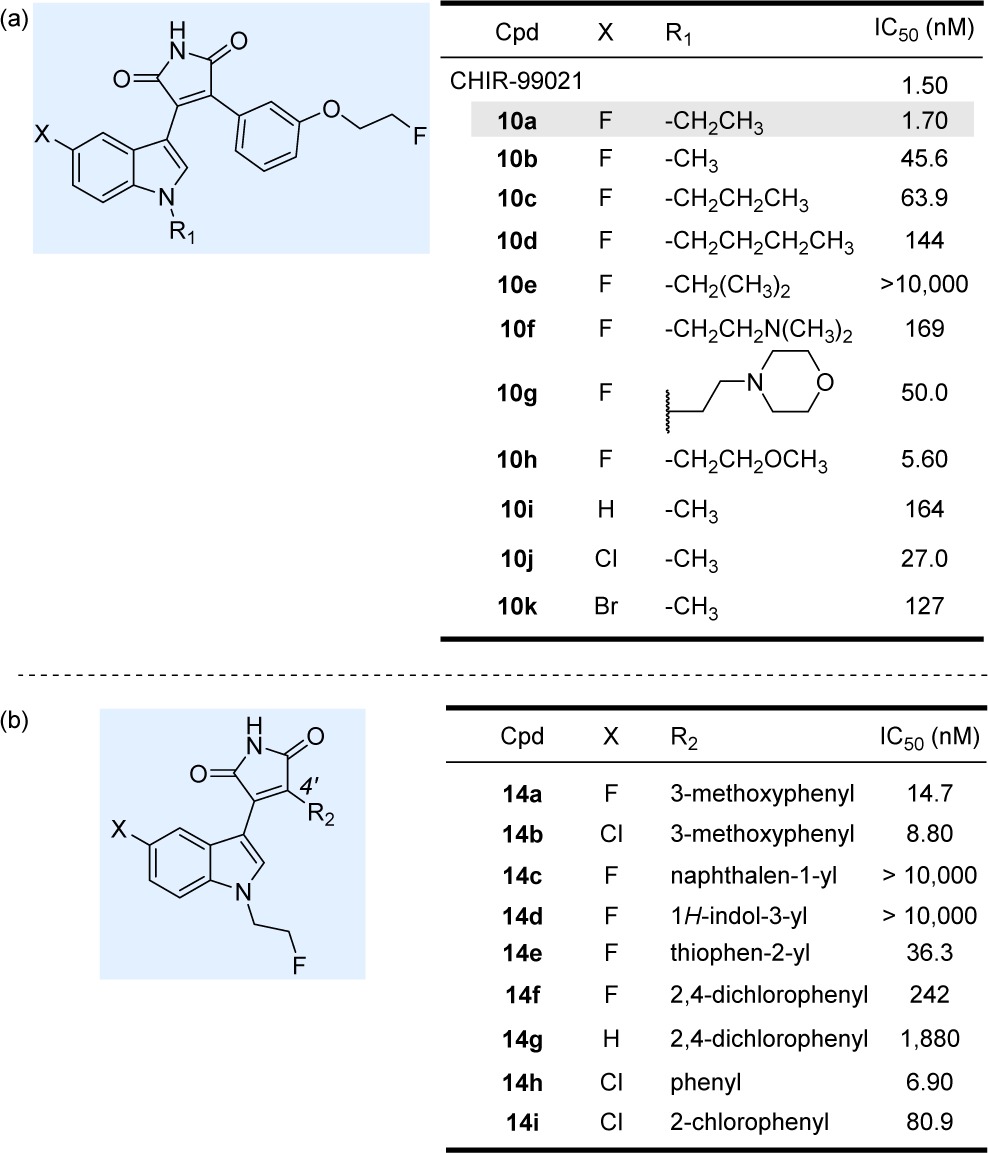
These IC50 values were experimentally determined under our identical assay conditions as used for the GSK-3 tracer [11C]PF-04082367.44 Kinase reactions were performed with 9 μM GSP-2, 15 μM ATP, and 10 nM GSK-3β in reaction buffer (50 mM Tris pH 7.5, 5 mM MgCl2, 0.01% Brij-35, 3 mM DTT) in the presence of 0–30 μM of the maleimides. For comparison purposes, the IC50 values of the known GSK-3β inhibitor CHIR-9902145 in the same assay are shown.
In the next set of SAR modifications, the fluoroethyl group was fixed at indole’s nitrogen, while the substituent at the C4′ position of the maleimide scaffold was varied (Table 1b). Previous studies showed that incorporation of a 3-methoxyphenyl group to the structure of maleimide analogues resulted in potent GSK-3β inhibitors (IC50 < 10 nM).34 Accordingly, 3-methoxyphenyl maleimide analogues 14a and 14b were very potent inhibitors, with IC50 values of 14.7 and 8.80 nM, respectively. Replacement of the 3-methoxyphenyl substituent with either a 1-naphthalenyl (14c) or a 1H-indol-3-yl (14d) moiety was found to be deleterious for the inhibitory activity. Substitution of 3-methoxyphenyl with 2-thiophenyl (14e) resulted in an approximately 2-fold decrease in potency. 2,4-Dichlorophenyl analogues (14f and 14g) significantly diminished GSK-3β inhibition. Replacement of 3-methoxyphenyl with a phenyl substituent (14h, IC50 = 6.90 nM) rendered an inhibitor with the highest potency in the series. An inhibitor with a 2-chlorophenyl group at the C4′ position (14i, IC50 = 81.0 nM) was almost 10-fold less potent than the 3-methoxyphenyl analogue 14b.
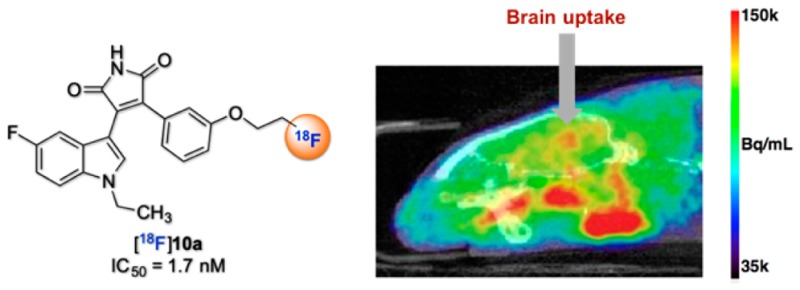
Based on the in vitro pharmacological results, compound 10a (IC50 = 1.70 nM) was selected as the potential PET radioligand for in vivo imaging of GSK-3β. The required precursor 15 was prepared (Scheme S1, SI), and fully automated radiosynthesis of [18F]10a was performed on a GE Tracerlab FXFN synthesis module. Treatment of precursor 15 with [18F]K/K222 complex at 90 °C for 10 min afforded the desired [18F]10a in 36% radiochemical yield (non-decay-corrected) with excellent radiochemical purity (>99%) and high specific activity (>148 GBq/μmol) at the end of the synthesis (Figure 3a). These results demonstrate the feasibility of synthesizing our radiotracer using a fully automated module, which thereby facilitated our preclinical studies with [18F]10 and should be readily translatable to structurally related [18F]-analogs.
Figure 3.
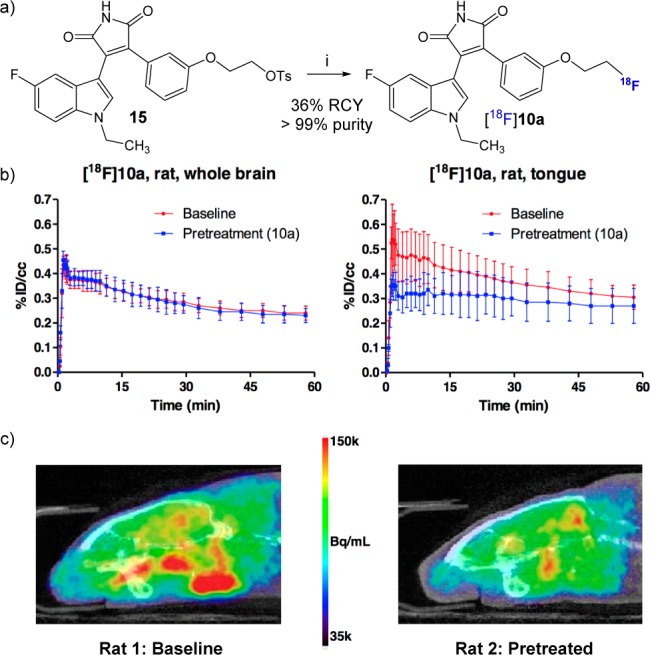
(a) Radiosynthesis of [18F]10a: (i) [18F]KF, K222 complex, MeCN, 90 °C, 10 min. Ts = toluenesulfonyl, RCY = radiochemical yield. (b) Time-course of [18F]10a activity of the brain and tongue of the rat without (red) and with (blue) pretreatment of nonradioactive tracer 10a. (c) Brain PET-CT scans of rat with [18F]10a. Images were converted to units of Bq/mL, and a color scale running from 35k-150k Bq/mL was used. Rat 2 was pretreated with nonradioactive tracer 10a.
Preliminary PET imaging was conducted in Sprague–Dawley rats at baseline (n = 2) and after pretreatment with nonradioactive 10a (1 mg/kg, iv, n = 2) to determine brain uptake and binding saturability (Figure 3). Radiotracer uptake in whole-brain was reasonable, reached a peak >0.4%ID/cc at 1–2 min postinjection, and cleared slowly over 60 min. Interestingly, greater uptake (>0.5%ID/cc) was observed at baseline in certain loci outside of the brain, including harderian glands and tissue near the jaws. Pretreatment did not measurably affect whole-brain radiotracer uptake, but did diminish uptake in other VOIs to ∼0.4%ID/cc. Although [18F]10a did not display saturable binding in vivo in the rodent brain, our efforts reveal the first report with biological evaluation and imaging studies attempting to develop a [18F]-labeled GSK-3β PET tracer for the central nervous system.46
In conclusion, we synthesized and evaluated a series of maleimide-based GSK-3β inhibitors containing a fluoroethyl group as candidates for in vivo PET imaging. Through structural modifications of the maleimide scaffold at the C3′ and C4′ positions, we identified a number of highly potent GSK-3β inhibitors with nanomolar IC50 values. Radiosynthesis of tracer [18F]10a on an automated module was achieved with excellent radiochemical purity and high specific activity. Thus, using conventional chemical transformations to quickly and reliably access the [18F]-labeled maleimide-based radiotracers is one of the advantages of the reported chemistry. In vivo PET-CT imaging studies in rodents showed that [18F]10a enters the rodent brain. Further refinement of this scaffold to improve the potency of the lead compounds is underway in our laboratories in an attempt to discover optimized [18F]-labeled GSK-3 inhibitors for PET imaging of the central nervous system.
Glossary
Abbreviations
- GSK-3
glycogen synthase kinase-3
- CNS
central nervous system
- AD
Alzheimer’s disease
- PET
positron emission tomography
- CT
computed tomography
- SAR
structure–activity relationship
- NMR
nuclear magnetic resonance
- RCY
radiochemical yield
- ATP
adenosine triphosphate
- DTT
dithiothreitol
- K222
4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo[8.8.8]hexacosane
- VOI
volume of interest
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00405.
Full experimental details and copies of NMR spectra for all compounds synthesized. Procedures for in vitro evaluation of GSK-3β inhibitors, radiochemical syntheses, quality control, and in vivo PET-CT imaging in rodents (PDF)
Author Present Address
∇ Kongzhen Hu’s current address is Department of PET Center, Nanfang Hospital, Guangzhou, China.
This work was partially supported by National Institute of General Medical Sciences (R35GM119652 to M.-Y.N.) and National Institute on Aging of the National Institute of Health and (R01AG054473 to N.V.), start-up funds from Stony Brook University (M.-Y.N.), and National Natural Science Foundation of China (Grant no. 21403208 to Z.Y.). K.N.L. received a graduate fellowship from an NIH Chemical-Biology training grant (T32GM092714). S.J.H. received support from the Tau Consortium. S.H.L is a recipient of NIH career development award from the National Institute on Drug Abuse (DA038000). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The authors declare no competing financial interest.
Dedication
This paper is dedicated to Prof. Barry M. Trost on the occasion of his 75th birthday.
Supplementary Material
References
- Woodgett J. R. Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J. 1990, 9, 2431–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P.; Frame S. The renaissance of GSK3. Nat. Rev. Mol. Cell Biol. 2001, 2, 769–76. 10.1038/35096075. [DOI] [PubMed] [Google Scholar]
- Kaidanovich-Beilin O.; Woodgett J. R. GSK-3: Functional Insights from Cell Biology and Animal Models. Front. Mol. Neurosci. 2011, 4, 40. 10.3389/fnmol.2011.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P.; Goedert M. GSK3 inhibitors: development and therapeutic potential. Nat. Rev. Drug Discovery 2004, 3, 479–87. 10.1038/nrd1415. [DOI] [PubMed] [Google Scholar]
- Jope R. S.; Johnson G. V. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem. Sci. 2004, 29, 95–102. 10.1016/j.tibs.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Eldar-Finkelman H.; Schreyer S. A.; Shinohara M. M.; LeBoeuf R. C.; Krebs E. G. Increased glycogen synthase kinase-3 activity in diabetes- and obesity-prone C57BL/6J mice. Diabetes 1999, 48, 1662–6. 10.2337/diabetes.48.8.1662. [DOI] [PubMed] [Google Scholar]
- Hooper C.; Killick R.; Lovestone S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008, 104, 1433–9. 10.1111/j.1471-4159.2007.05194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avila J.; Wandosell F.; Hernandez F. Role of glycogen synthase kinase-3 in Alzheimer’s disease pathogenesis and glycogen synthase kinase-3 inhibitors. Expert Rev. Neurother. 2010, 10, 703–10. 10.1586/ern.10.40. [DOI] [PubMed] [Google Scholar]
- Morales-Garcia J. A.; Susin C.; Alonso-Gil S.; Perez D. I.; Palomo V.; Perez C.; Conde S.; Santos A.; Gil C.; Martinez A.; Perez-Castillo A. Glycogen Synthase Kinase-3 Inhibitors as Potent Therapeutic Agents for the Treatment of Parkinson Disease. ACS Chem. Neurosci. 2013, 4, 350–360. 10.1021/cn300182g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J. Q.; Lewis M. C.; Ketterman J. K.; Clore E. L.; Riley M.; Richards K. R.; Berry-Scott E.; Liu X.; Wagner F. F.; Holson E. B.; Neve R. L.; Biechele T. L.; Moon R. T.; Scolnick E. M.; Petryshen T. L.; Haggarty S. J. AKT Kinase Activity Is Required for Lithium to Modulate Mood-Related Behaviors in Mice. Neuropsychopharmacology 2011, 36, 1397–1411. 10.1038/npp.2011.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaidanovich-Beilin O.Neurological functions of the masterswitch protein kinase–GSK-3; Frontiers E-books: 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valvezan A. J.; Klein P. S.. GSK-3 and Wnt signaling in neurogenesis and bipolar disorder. Front. Mol. Neurosci. 2012, 5. 10.3389/fnmol.2012.00001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins D. F.; Rosa A. O.; Gadotti V. M.; Mazzardo-Martins L.; Nascimento F. P.; Egea J.; López M. G.; Santos A. R. The antinociceptive effects of AR-A014418, a selective inhibitor of glycogen synthase kinase-3 beta, in mice. J. Pain 2011, 12, 315–322. 10.1016/j.jpain.2010.06.007. [DOI] [PubMed] [Google Scholar]
- Maixner D. W.; Weng H. R. The Role of Glycogen Synthase Kinase 3 Beta in Neuroinflammation and Pain. J. Pharm. Pharmacol. 2013, 1, 001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J. Glycogen synthase kinase 3beta (GSK3beta) in tumorigenesis and cancer chemotherapy. Cancer Lett. 2009, 273, 194–200. 10.1016/j.canlet.2008.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez A.; Castro A.; Dorronsoro I.; Alonso M. Glycogen synthase kinase 3 (GSK-3) inhibitors as new promising drugs for diabetes, neurodegeneration, cancer, and inflammation. Med. Res. Rev. 2002, 22, 373–384. 10.1002/med.10011. [DOI] [PubMed] [Google Scholar]
- Eldar-Finkelman H. Glycogen synthase kinase 3: an emerging therapeutic target. Trends Mol. Med. 2002, 8, 126–32. 10.1016/S1471-4914(01)02266-3. [DOI] [PubMed] [Google Scholar]
- Haq S.; Choukroun G.; Kang Z. B.; Ranu H.; Matsui T.; Rosenzweig A.; Molkentin J. D.; Alessandrini A.; Woodgett J.; Hajjar R.; Michael A.; Force T. Glycogen synthase kinase-3 beta is a negative regulator of cardiomyocyte hypertrophy. J. Cell Biol. 2000, 151, 117–129. 10.1083/jcb.151.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorn G. W.; Force T. Protein kinase cascades in the regulation of cardiac hypertrophy. J. Clin. Invest. 2005, 115, 527–537. 10.1172/JCI24178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller P. W.; Long N. J.; Vilar R.; Gee A. D. Synthesis of C-11, F-18, O-15, and N-13 Radiolabels for Positron Emission Tomography. Angew. Chem., Int. Ed. 2008, 47, 8998–9033. 10.1002/anie.200800222. [DOI] [PubMed] [Google Scholar]
- Vasdev N.; Garcia A.; Stableford W. T.; Young A. B.; Meyer J. H.; Houle S.; Wilson A. A. Synthesis and ex vivo evaluation of carbon-11 labelled N-(4-methoxybenzyl)-]N ′-(5-nitro-1,3-thiazol-2-yl)urea ([C-11]AR-A014418): A radiolabelled glycogen synthase kinase-3 beta specific inhibitor for PET studies. Bioorg. Med. Chem. Lett. 2005, 15, 5270–5273. 10.1016/j.bmcl.2005.08.037. [DOI] [PubMed] [Google Scholar]
- Hicks J. W.; Wilson A. A.; Rubie E. A.; Woodgett J. R.; Houle S.; Vasdev N. Towards the preparation of radiolabeled 1-aryl-3-benzyl ureas: Radiosynthesis of [(11)C-carbonyl] AR-A014418 by [(11)C]CO(2) fixation. Bioorg. Med. Chem. Lett. 2012, 22, 2099–101. 10.1016/j.bmcl.2011.12.139. [DOI] [PubMed] [Google Scholar]
- Cole E. L.; Shao X.; Sherman P.; Quesada C.; Fawaz M. V.; Desmond T. J.; Scott P. J. Synthesis and evaluation of [(11)C]PyrATP-1, a novel radiotracer for PET imaging of glycogen synthase kinase-3beta (GSK-3beta). Nucl. Med. Biol. 2014, 41, 507–12. 10.1016/j.nucmedbio.2014.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumata K.; Yui J.; Xie L.; Zhang Y.; Nengaki N.; Fujinaga M.; Yamasaki T.; Shimoda Y.; Zhang M. R. Radiosynthesis and preliminary PET evaluation of glycogen synthase kinase 3beta (GSK-3beta) inhibitors containing [(11)C]methylsulfanyl, [(11)C]methylsulfinyl or [(11)C]methylsulfonyl groups. Bioorg. Med. Chem. Lett. 2015, 25, 3230–3. 10.1016/j.bmcl.2015.05.085. [DOI] [PubMed] [Google Scholar]
- Li L.; Shao X.; Cole E. L.; Ohnmacht S. A.; Ferrari V.; Hong Y. T.; Williamson D. J.; Fryer T. D.; Quesada C. A.; Sherman P.; Riss P. J.; Scott P. J.; Aigbirhio F. I. Synthesis and Initial in Vivo Studies with [(11)C]SB-216763: The First Radiolabeled Brain Penetrative Inhibitor of GSK-3. ACS Med. Chem. Lett. 2015, 6, 548–52. 10.1021/acsmedchemlett.5b00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang S. H.; Chen J. M.; Normandin M. D.; Chang J. S.; Chang G. C.; Taylor C. K.; Trapa P.; Plummer M. S.; Para K. S.; Conn E. L.; Lopresti-Morrow L.; Lanyon L. F.; Cook J. M.; Richter K. E.; Nolan C. E.; Schachter J. B.; Janat F.; Che Y.; Shanmugasundaram V. L.; B A.; Enerson B. E.; Livni E.; Wang L.; Guehl N. J.; Patnaik D.; Wagner F. F.; Perlis R.; Holson E. B.; Haggarty S. J.; El Fakhri G.; Kurumbail R. G.; Vasdev N. Discovery of a Highly Selective Glycogen Synthase Kinase-3 Inhibitor (PF-04802367) That Modulates Tau Phosphorylation in the Brain: Translation for PET Neuroimaging. Angew. Chem., Int. Ed. 2016, 55, 9601–05. 10.1002/anie.201603797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Several [18F]-labeled tracers were reported, but their biological evaluation and imaging studies are still ongoing (also see refs (28 and 29)).Mossine A.; Li L.; Cole E.; Brooks A.; Aigbirhio F.; Scott P. Glycogen Synthase Kinase 3β Inhibitor Scaffolds for PET Radiotracer Development. J. Nucl. Med. 2015, 56, 1094–1094.25977460 [Google Scholar]
- Mossine A.; Brooks A.; Scott P. Synthesis and evaluation of 4-[18F] fluorophenacylbromide (4FPB), a selective and irreversible inhibitor of Glycogen Synthase Kinase 3β (GSK-3β). J. Nucl. Med. 2016, 57, 1099–1099. [Google Scholar]
- Pandey M. K.; DeGrado T. R. Glycogen Synthase Kinase-3 (GSK-3)-Targeted Therapy and Imaging. Theranostics 2016, 6, 571–93. 10.7150/thno.14334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D. G.; Buffet M.; Fenwick A. E.; Haigh D.; Ife R. J.; Saunders M.; Slingsby B. P.; Stacey R.; Ward R. W. 3-anilino-4-arylmaleimides: Potent and selective inhibitors of glycogen synthase kinase-3 (GSK-3). Bioorg. Med. Chem. Lett. 2001, 11, 635–639. 10.1016/S0960-894X(00)00721-6. [DOI] [PubMed] [Google Scholar]
- Bertrand J. A.; Thieffine S.; Vulpetti A.; Cristiani C.; Valsasina B.; Knapp S.; Kalisz H. M.; Flocco M. Structural characterization of the GSK-3 beta active site using selective and non-selective ATP-mimetic inhibitors. J. Mol. Biol. 2003, 333, 393–407. 10.1016/j.jmb.2003.08.031. [DOI] [PubMed] [Google Scholar]
- Coghlan M. P.; Culbert A. A.; Cross D. A. E.; Corcoran S. L.; Yates J. W.; Pearce N. J.; Rausch O. L.; Murphy G. J.; Carter P. S.; Cox L. R.; Mills D.; Brown M. J.; Haigh D.; Ward R. W.; Smith D. G.; Murray K. J.; Reith A. D.; Holder J. C. Selective small molecule inhibitors of glycogen synthase kinase-3 modulate glycogen metabolism and gene transcription. Chem. Biol. 2000, 7, 793–803. 10.1016/S1074-5521(00)00025-9. [DOI] [PubMed] [Google Scholar]
- Kramer T.; Schmidt B.; Lo Monte F. Small-Molecule Inhibitors of GSK-3: Structural Insights and Their Application to Alzheimer’s Disease Models. Int. J. Alzheimer's Dis. 2012, 2012, 381029. 10.1155/2012/381029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong L.; Hirschfeld D.; Tan Y. C.; Heather Hogg J.; Peltz G.; Avnur Z.; Dunten P. Discovery of potent and bioavailable GSK-3beta inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 1693–6. 10.1016/j.bmcl.2010.01.038. [DOI] [PubMed] [Google Scholar]
- Gunosewoyo H.; Midzak A.; Gaisina I. N.; Sabath E. V.; Fedolak A.; Hanania T.; Brunner D.; Papadopoulos V.; Kozikowski A. P. Characterization of maleimide-based glycogen synthase kinase-3 (GSK-3) inhibitors as stimulators of steroidogenesis. J. Med. Chem. 2013, 56, 5115–29. 10.1021/jm400511s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quesada-Romero L.; Mena-Ulecia K.; Tiznado W.; Caballero J. Insights into the interactions between maleimide derivates and GSK3beta combining molecular docking and QSAR. PLoS One 2014, 9, e102212. 10.1371/journal.pone.0102212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaisina I. N.; Gallier F.; Ougolkov A. V.; Kim K. H.; Kurome T.; Guo S.; Holzle D.; Luchini D. N.; Blond S. Y.; Billadeau D. D.; Kozikowski A. P. From a Natural Product Lead to the Identification of Potent and Selective Benzofuran-3-yl-(indol-3-yl)maleimides as Glycogen Synthase Kinase 3 beta Inhibitors That Suppress Proliferation and Survival of Pancreatic Cancer Cells. J. Med. Chem. 2009, 52, 1853–1863. 10.1021/jm801317h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coghlan M. P.; Culbert A. A.; Cross D. A.; Corcoran S. L.; Yates J. W.; Pearce N. J.; Rausch O. L.; Murphy G. J.; Carter P. S.; Cox L. R. Selective small molecule inhibitors of glycogen synthase kinase-3 modulate glycogen metabolism and gene transcription. Chem. Biol. 2000, 7, 793–803. 10.1016/S1074-5521(00)00025-9. [DOI] [PubMed] [Google Scholar]
- Engler T. A.; Henry J. R.; Malhotra S.; Cunningham B.; Furness K.; Brozinick J.; Burkholder T. P.; Clay M. P.; Clayton J.; Diefenbacher C. Substituted 3-imidazo [1, 2-a] pyridin-3-yl-4-(1, 2, 3, 4-tetrahydro-[1, 4] diazepino-[6, 7, 1-hi] indol-7-yl) pyrrole-2, 5-diones as highly selective and potent inhibitors of glycogen synthase kinase-3. J. Med. Chem. 2004, 47, 3934–3937. 10.1021/jm049768a. [DOI] [PubMed] [Google Scholar]
- Kulkarni N. H.; Onyia J. E.; Zeng Q.; Tian X.; Liu M.; Halladay D. L.; Frolik C. A.; Engler T.; Wei T.; Kriauciunas A. Orally Bioavailable GSK-3α/β Dual Inhibitor Increases Markers of Cellular Differentiation In Vitro and Bone Mass In Vivo. J. Bone Miner. Res. 2006, 21, 910–920. 10.1359/jbmr.060316. [DOI] [PubMed] [Google Scholar]
- Faul M. M.; Winneroski L. L.; Krumrich C. A. A New, Efficient Method for the Synthesis of Bisindolylmaleimides. J. Org. Chem. 1998, 63, 6053–6058. 10.1021/jo980513c. [DOI] [PubMed] [Google Scholar]
- Wang M.; Gao M.; Miller K. D.; Sledge G. W.; Hutchins G. D.; Zheng Q. H. The first synthesis of [(11)C]SB-216763, a new potential PET agent for imaging of glycogen synthase kinase-3 (GSK-3). Bioorg. Med. Chem. Lett. 2011, 21, 245–9. 10.1016/j.bmcl.2010.11.026. [DOI] [PubMed] [Google Scholar]
- Estimated Ki values of compounds 10a–k and 14a–i were calculated and reported in the SI.
- Wagner F. F.; Bishop J. A.; Gale J. P.; Shi X.; Walk M.; Ketterman J.; Patnaik D.; Barker D.; Walpita D.; Campbell A. J. Inhibitors of Glycogen Synthase Kinase 3 with Exquisite Kinome-wide Selectivity and their Functional Effects. ACS Chem. Biol. 2016, 11, 1952–63. (See also ref (26)). 10.1021/acschembio.6b00306. [DOI] [PubMed] [Google Scholar]
- Ring D. B.; Johnson K. W.; Henriksen E. J.; Nuss J. M.; Goff D.; Kinnick T. R.; Ma S. T.; Reeder J. W.; Samuels I.; Slabiak T.; Wagman A. S.; Hammond M. E.; Harrison S. D. Selective glycogen synthase kinase 3 inhibitors potentiate insulin activation of glucose transport and utilization in vitro and in vivo. Diabetes 2003, 52, 588–95. 10.2337/diabetes.52.3.588. [DOI] [PubMed] [Google Scholar]
- In vitro metabolic stability of compound 10a in liver microsomes from human (HLM) and rat (RLM) was evaluated. The data show that (i) the half-lives of compound 10a in HLM and RLM are less than 15 min, and (ii) there is no major differentiation between human and rat. Thus, we do not exclude the possibility that the signal is stemmed from a metabolite. See SI for details.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.