

Abstract
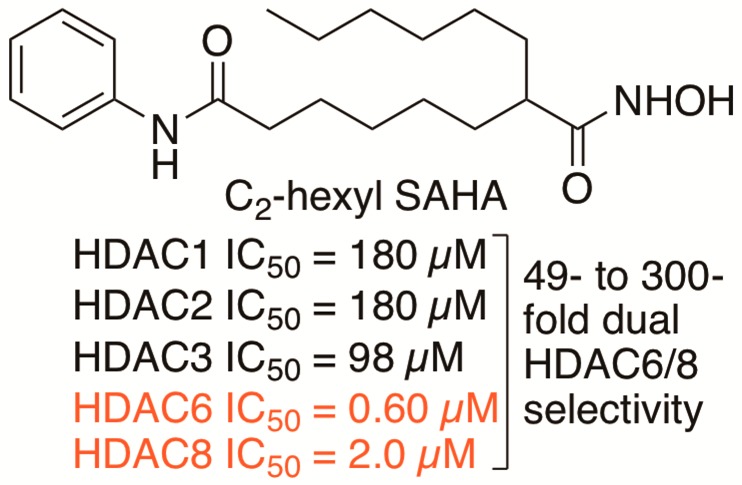
Histone deacetylase (HDAC) proteins are epigenetic regulators that deacetylate protein substrates, leading to subsequent changes in cell function. HDAC proteins are implicated in cancers, and several HDAC inhibitors have been approved by the FDA as anticancer drugs, including SAHA (suberoylanilide hydroxamic acid; Vorinostat and Zolinza). Unfortunately, SAHA inhibits most HDAC isoforms, which limits its use as a pharmacological tool and may lead to side effects in the clinic. In this work SAHA analogues substituted at the C2 position were synthesized and screened for HDAC isoform selectivity in vitro and in cells. The most potent and selective compound, C2-n-hexyl SAHA, displayed submicromolar potency with 49- to 300-fold selectivity for HDAC6 and HDAC8 compared to HDAC1, -2, and -3. Docking studies provided a structural rationale for selectivity. Modification of the nonselective inhibitor SAHA generated HDAC6/HDAC8 dual selective inhibitors, which can be useful lead compounds toward developing pharmacological tools and more effective anticancer drugs.
Keywords: Histone deacetylase, HDAC inhibitor, HDAC6 HDAC8 selective inhibitor, HDAC isoform selectivity, Vorinostat
Histone deacetylase (HDAC) proteins play an important role in the epigenetic regulation of transcription. HDAC proteins deacetylate acetyllysine residues on nucleosomal histone proteins, which influences gene expression.1 The HDAC family contains 18 proteins, which are grouped into four classes according to their size, cellular localization, and phylogenetic analysis.2 Classes I, II, and IV are metal dependent, while class III are NAD+ dependent.2 The metal-dependent HDACs are the focus in this work.
Through the deacetylation of nucleosomal histones, HDAC proteins regulate both the expression and activity of cancer-related proteins that are involved in transcription, tumor suppression, and cell signaling.3,4 Through the deacetylation of nonhistone substrates, HDAC proteins affect protein stability, localization, and intracellular interactions, including protein–protein interactions and protein–DNA interactions.4,5 Due to their fundamental role in regulating gene expression and protein activity, overexpression of HDAC proteins is linked to cancer formation.5 For example, HDAC1 was overexpressed in prostate6 and breast,7 while HDAC8 has been implicated in neuroblastoma and T-cell lymphoma and acute myeloid leukemia.8 Among the class II proteins, HDAC6 was overexpressed in oral squamous cell carcinoma.9 In addition, HDAC6 is implicated in several nonepigenetic cancer-related intracellular functions.10 Both HDAC6 and HDAC8 were found to be highly expressed and implicated in the invasion and progression of breast cancer cells.11
With a role in cancer, HDAC proteins have emerged as important targets for cancer treatment, with a wide variety of HDAC inhibitor drugs available.12−15 The effect of HDAC inhibitors on both histone and nonhistone proteins can lead to cell signaling dysregulation, transcription and expression changes, and protein degradation. Through these effects on tumor cells, HDAC inhibitors can reduce proliferation, migration, and angiogenesis, enhance differentiation and immunogenicity, and promote apoptosis.16 In fact, several HDAC inhibitors are used for treatment of T-cell lymphoma, including SAHA (suberoylanilide hydroxamic acid, Vorinostat, Figure 1) and Belinostat (Figure S41).12−14 Panobinostat (Figure S41) was recently approved for treatment of multiple myeloma.15 Unfortunately, these FDA approved drugs are relatively nonselective and inhibit most of the 11 metal-dependent HDAC isoforms.17 Nonspecific inhibition may account for several mild to severe side effects associated with treatment, including dehydration, thrombocytopenia, anorexia, and cardiac arrhythmia.17,18 In addition, the nonselectivity of these drugs limits their use as biological tools to probe HDAC function in cancer biology.
Figure 1.

Chemical structures of the FDA approved drug SAHA (suberoylanilide hydroxamic acid; Vorinostat and Zolinza) and the C2-modified SAHA analogues reported here.
Several isoform selective inhibitors have been developed. MS-275 (Figure S41) is in clinical trials and is class I selective, with 4- to 400-fold selectivity for HDAC1, 2, and 3 over the other isoforms.17 RGFP966 (Figure S41) showed more than 188-fold selectivity for HDAC3 over the other isoforms.19 Tubastatin (Figure S41) is HDAC6 selective with 87-fold or 1000-fold selectivity for HDAC6 over HDAC1, 2, and 3 according to different reports.20,21 Selective inhibitors can be used as biological tools to elucidate the function of each isoform in the development of cancer. In addition, modification of nonselective inhibitors currently used in clinic can possibly improve their selectivity and reduce their clinical side effects.
Toward development of selective HDAC inhibitors, we previously created SAHA analogues containing substituents in the linker region between the hydroxamic acid and the anilide ends (Figure 1). A C3-modified SAHA analogue showed modest preference for HDAC3, while C6-modified SAHA analogues displayed selectivity for HDAC1 and -6 compared to HDAC3.22−24 In addition, modifying the amine of the hydroxamic acid reduced potency, but enhanced preference for HDAC1.25 C2-modified SAHA analogues (Figure 1) were also generated and showed μM potency with HeLa cell lysates (Table S1), but no selectivity assessment was performed.22 We report here a selectivity assessment of C2-modified SAHA analogues both in vitro and in cells. Modification at the C2 position led to reduced potency but enhanced selectivity compared to SAHA, with preference for HDAC6 and -8 over HDAC1, -2, and -3. HDAC6/8 dual inhibitors can be used as biological tools to study breast cancer metastasis.11,26 In addition, the SAHA analogues reported here are useful lead compounds for further development of pharmacological agents and anticancer drug targeting HDAC6 and -8. More generally, these studies confirm that modification of the SAHA linker region can enhance isoform selectivity.
Synthesis of C2-Modified SAHA Analogues
The syntheses of seven C2-modified SAHA analogues (1a–g) were previously described.22 Two new derivatives, C2-n-pentyl SAHA (1h) and C2-n-hexyl SAHA (1i) are reported here. Synthesis began with ring opening of ε-caprolactone 2 with aniline to give anilide alcohol 3. Activation of the hydroxyl as a mesylate with subsequent substitution with dimethyl malonate gave intermediate diester 5. Substitution with 1-bromopentane or 1-bromohexane generated the substituted diesters 6h–i. Decarboxylation and subsequent saponification, followed by coupling with N-benzyl protected hydroxylamine afforded protected hydroxamic acids 7h–i. Finally, hydrogenolysis gave the desired C2-n-pentyl (1h) and C2-n-hexyl SAHA (1i) analogues (Scheme 1).
Scheme 1. Synthesis of C2-n-pentyl and C2-n-hexyl SAHA Analogues (1h–i).

In Vitro Screening of C2-Modified SAHA Analogues
Prior work showed that C2-modified SAHA analogues displayed weak μM potency with the HDAC activity from HeLa cell lysates.22 However, no selectivity assessment was performed. In this work we used the recently developed ELISA-based HDAC activity assay to screen the analogues against mammalian-derived HDAC1, HDAC2, HDAC3, and HDAC6.21 As an initial test of selectivity, the potency of each C2-modified SAHA derivative was tested with HDAC1, -2, -3, and -6 at single concentrations of either 5 or 10 μM. All analogues (1a–i) displayed some selectivity for HDAC6 compared to HDAC1, HDAC2, and HDAC3 (Figure 2). Among them, the C2-benzyl (1g), C2-n-pentyl (1h), and C2-n-hexyl (1i) analogues showed the greatest difference in inhibitory activity comparing HDAC6 to HDAC1, HDAC2, and HDAC3. In contrast, the C2-methyl SAHA analogue was least selective, with similar activity against all four isoforms (Figure 2).
Figure 2.

Isoform selectivity screening of C2-modified SAHA analogues (1a–i) against HDAC1, -2, -3, and -6 using the ELISA-based HDAC activity assay.21 All analogues were tested at 5 μM concentration, except for 1d, which was tested at 10 μM. SAHA was tested at 1 μM.21 Mean percent deacetylase activities from a minimum of three independent trials with standard errors were plotted (Table S2). The substituent below each compound number corresponds to the R group in C2-modified SAHA (Figure 1).
To further assess selectivity, IC50 values for the most selective compounds in the initial screen, compounds 1g–i, were determined with HDAC1, -2, -3, -6, and -8 (Table 1). As controls, the IC50 values of both SAHA and tubastatin were included. As expected, SAHA displayed similar IC50 values with HDAC1–6, but 5-fold reduced activity with HDAC8, which is consistent with prior reports.17,27 Tubastatin showed at least 87-fold selectivity for HDAC6 over class I HDAC1, -2, and -3, but only 10-fold selectivity versus HDAC8.21 Interestingly, the C2-modified SAHA analogues showed selectivity for HDAC6 and HDAC8, with IC50 values in the submicromolar to micromolar range (0.6–2.0 μM, Table 1). The C2-benzyl 1g and C2-n-pentyl 1h analogues displayed 33- to 92-fold selectivity for HDAC6 and HDAC8 over the Class I isoforms (Tables 1, S5, and S6 and Figures S44 and S45). The most selective compound C2-n-hexyl SAHA 1i displayed 49- to 300-fold selectivity for HDAC6 and HDAC8 compared to the class I isoforms (Tables 1 and S7 and Figure S46).
Table 1. IC50 Values for SAHA, Tubastatin, SAHA Analogues 1g–1i against HDAC1, -2, -3, -6, and -8a.
| IC50 values (μM) |
|||||
|---|---|---|---|---|---|
| compd | HDAC1 | HDAC2 | HDAC3 | HDAC6 | HDAC8 |
| SAHA21 | 0.033 ± 0.001 | 0.096 ± 0.01 | 0.020 ± 0.001 | 0.033 ± 0.003 | 0.54 ± 0.01 |
| Tubastatin21 | 2.7 ± 0.2 | 3.9 ± 0.4 | 2.9 ± 0.5 | 0.031 ± 0.004 | 0.33 ± 0.01 |
| 1g (benzyl) | 84 ± 6 | 110 ± 10 | 91 ± 4 | 1.5 ± 0.2 | 1.2 ± 0.1 |
| 1h (pentyl) | 48 ± 2 | 58 ± 2 | 43 ± 2 | 0.85 ± 0.05 | 1.3 ± 0.1 |
| 1i (hexyl) | 180 ± 20 | 180 ± 30 | 98 ± 10 | 0.60 ± 0.05 | 2.0 ± 0.1 |
| (S)-1i (hexyl) | 330 ± 30 | 580 ± 30 | 530 ± 50 | ND | 3.1 ± 0.1 |
| (R)-1i (hexyl) | ND | ND | ND | ND | 0.71 ± 0.01 |
Mean IC50 value and standard error of at least two independent trials are shown (Figures S42–S48 and Tables S3–S9). ND = not determined.
It is notable that the selectivity of C2-n-hexyl SAHA 1i for HDAC6 (>163-fold) is elevated compared to tubastatin (>87-fold), while it showed 20-fold less potency than tubastatin (0.60 vs 0.031 μM IC50 values). The conclusion is that C2-substituents impart selectivity by discriminating against HDAC1, HDAC2, and HDAC3.
In-Cell Selectivity Testing
To assess the HDAC6 selectivities of the analogues in a biologically relevant context, C2-benzyl (1g), C2-n-pentyl (1h), and C2-n-hexyl (1i) SAHA were tested for their abilities to increase the acetylation levels of HDAC substrates. Acetylated-α-tubulin (AcTub) was monitored as a known substrate of HDAC6, whereas acetylated-histone H3 (AcH3) was observed as a substrate for HDAC1, -2, and -3. U937 myeloid leukemia cells were used in these cellular HDAC6 selectivity study. HDAC6 is overexpressed in several acute myeloid leukemia (AML) cell lines, suggesting that HDAC6 is a promising target for development of antileukemic drugs.28 SAHA or the analogues were incubated with U937 cells before lysis and Western blot analysis of protein acetylation. As expected, SAHA increased the acetylation levels of both α-tubulin and histone H3 (Figure 3a, lane 2), consistent with its broad inhibition. In contrast, the HDAC6 selective inhibitor tubastatin affected only α-tubulin acetylation (Figure 3a, lane 3). Similar to tubastatin, the three analogues 1h–i increased acetylation levels of α-tubulin to a greater level than histone H3 (Figure 3a, lanes 4–6). Quantification confirmed that 1h–i significantly increased acetylation of α-tubulin compared to DMSO, but not acetyl histone H3 levels (Figure 3b and Table S10). In addition, the C2-n-hexyl analogue 1i promoted a dose-dependent increase in acetylation of α-tubulin, but not histone H3 (Figure 3c, lanes 2–7), compared to the DMSO control (Figure 3c, lane 1). The HDAC6-dependent acetylation of tubulin observed in cells is consistent with the HDAC6 selectivity observed in vitro (Table 1 and Figure 2).
Figure 3.

Cell-based selectivity testing of the SAHA analogues. U937 cells were treated with (a) DMSO (1%), SAHA (2 μM), tubastatin (2 μM), C2-benzyl SAHA (1g, 30 μM), C2-n-pentyl SAHA (1h, 30 μM), C2-n-hexyl SAHA (1i, 30 μM), or (c) increasing concentrations of C2-n-hexyl SAHA analogue (1i, 10–60 μM) before lysis, SDS-PAGE separation, and Western blot analysis of acetyl-histone H3 (AcH3) and acetyl-α-tubulin (AcTub). GAPDH was a load control. Repetitive trials are shown in Figures S49 and S50. (b) Fold increase in AcH3 or AcTub after quantification of band intensities from part a, with mean fold increase from four independent trials and standard error (Table S10).
Inhibitor Cytotoxicity
To test the anticytotoxic properties of the HDAC6-selective inhibitors, analogues 1g–i were tested in cell-based cytotoxicity assays using leukemia cell lines.28 First, the analogues were tested with the Jurkat cell line at 1 and 10 μM concentrations using an MTT assay (Figure 4, Table S11). SAHA was also tested as a control. All compounds showed reduced cytotoxicity compared to the SAHA (Figure 4). Of the analogues, C2-n-hexyl SAHA (1i) showed the greatest cytotoxic effect, with only 47% viability at 10 μM concentration.
Figure 4.

Cytotoxicity screening of 1g, 1h, 1i, and SAHA at 1 and 10 μM concentrations using an MTT assay with Jurkat cells. Mean percent viability from at least three independent trials with standard error were plotted (Table S11).
To further assess cytotoxicity, both SAHA and the most potent analogue 1i were tested to determine EC50 values against three leukemia cancer cell lines: Jurkat, AML MOLM-13, and U937 cells. SAHA showed potent cytotoxicity, with EC50 values of 0.72, 1.2, and 0.88 μM with Jurkat, AML MOLM-13, and U937 cell lines, respectively (Table 2). The observed EC50 values are consistent with previous reports.29−31 The high potency of SAHA may be due to its nonselectivity, as well as the high inhibitory activity against class 1 HDAC1, -2, and -3. The C2-n-hexyl SAHA analogue 1i showed roughly 10-fold reduced cytotoxicity compared to SAHA, with EC50 values of 11.8, 10.5, and 13.8 μM with Jurkat, AML MOLM-13, and U937 cell lines, respectively (Table 2). The reduced cytotoxicity is consistent with the 18-fold reduction in potency against HDAC6 compared to SAHA (Table 1). In addition, the selectivity for HDAC6 and -8 over HDAC1, -2, and -3 might also contribute to the lower cytotoxicity.
Table 2. EC50 Values for SAHA and C2-n-hexyl (1i) SAHA Analogue against Jurkat, AML MOLM-13, and U937 Cells Using MTT Assaya.
| cellular
EC50 values (μM) |
|||
|---|---|---|---|
| compd | Jurkat | AML MOLM-13 | U937 |
| SAHA | 0.72 ± 0.13 | 1.2 ± 0.06 | 0.88 ± 0.13 |
| 1i (hexyl) | 11.8 ± 2.2 | 10.5 ± 3.1 | 13.8 ± 1.7 |
Mean EC50 value and standard error of at least three independent trials are shown (Figures S51–S52 and Tables S12–S13).
Synthesis and Screening of (R)- and (S)-C2-n-hexyl SAHA (1i)
C2-n-hexyl SAHA (1i) contains a stereocenter at the 2 position, and the compounds tested to this point were racemic mixtures. To test the selectivity of each enantiomer, an enantioselective synthesis of C2-n-hexyl SAHA (1i) was employed using Evans chiral auxiliary 8 and octanoyl chloride (Schemes 2 and S1). Allyl bromide was added to the resulting amide 9 to generate chiral compound (R)-10 from auxiliary (R)-8 (Scheme 2) or (S)-10 from auxiliary (S)-8 (Scheme S1). After olefin metathesis with Grubbs’ second generation catalyst32 and removal of the auxiliary, the olefin was reduced to generate (S)-11 and (R)-11. Finally, coupling with hydroxylamine generated the two enantiomers of C2-n-hexyl SAHA, (S)-1i or (R)-1i, in 95 and 92% ee, respectively.
Scheme 2. Synthesis of (S)-C2-n-hexyl SAHA, (S)-1i.

With the two C2-n-hexyl SAHA enantiomers in hand, IC50 values were determined (Table 1). As expected, both enantiomers displayed low micromolar to submicromolar potency with HDAC8 (3.1 ± 0.1 or 0.71 ± 0.01 μM), similar to racemic 1i (2.0 ± 0.1). The data suggested that (R)-1i is more potent than (S)-1i, although only by 4-fold. The (S)-1i enantiomer was further tested for selectivity against HDAC1, -2, and -3. (S)-1i displayed 106- to 187-fold selectivity for HDAC8, which is greater than that observed with racemic 1i (49- to 300-fold). In total, studies with the enantiomers of C2-n-hexyl SAHA indicated that both are low micromolar to submicromolar potency HDAC8 inhibitors, with the expected HDAC8 selectivity compared to HDAC1, -2, and -3.
Docking Studies
To rationalize the HDAC6 selectivity of the C2-n-hexyl SAHA (1i) analogue, we performed docking analysis using the AutoDock 4.2 program.33 Both enantiomers of the analogue were docked into the recently published HDAC6 crystal structure (PDB: 5EEM),34 and both displayed similar binding interactions (Figure 5 and Figure S53), consistent with the similar IC50 values observed experimentally. For example, the hydroxamic acid was positioned in bonding distance (1.9–2.9 Å) of three active site residues (H573, H574, and Y745) and the catalytic zinc atom in HDAC6 active site (Figures 5a and S53A). For comparison, docking of the parent SAHA compound with HDAC6 showed similar distances between the hydroxamic acid and the active site (1.6–2.4 Å, Figure S54A). To explore the HDAC6 selectivity, compound 1i was also docked into the HDAC2 crystal structure (PDB: 3MAX).35 In contrast to the bonding distances observed with HDAC6, elongated distances between the hydroxamic acid group and H145 (5.7–5.9 Å), H146 (3.8 Å), and Y308 (3.0–5.5 Å) were observed (Figures 5b and S53B). Metal binding was also weakened with longer bond distances (3.5–4.7 Å, Figures 5b and S53B). One possibility accounting for the weak binding with HDAC2 is that the bulky C2-n-hexyl substituent cannot favorably fit into the relatively narrow catalytic active channel of HDAC2.20 Consistent with this possibility, superimposition of the docked poses of compound 1i and SAHA with HDAC2 showed that the C2-n-hexyl substituent is positioned toward the solvent exposed surface of the active site, which consequently places the hydroxamic acid distant from the metal (Figure S55C,D). In contrast, the relatively wide catalytic pocket in HDAC6 allowed compound 1i and SAHA to similarly position the hydroxamic acid within bonding distances of the catalytic metal and nearby residues (Figure S55A,B).
Figure 5.
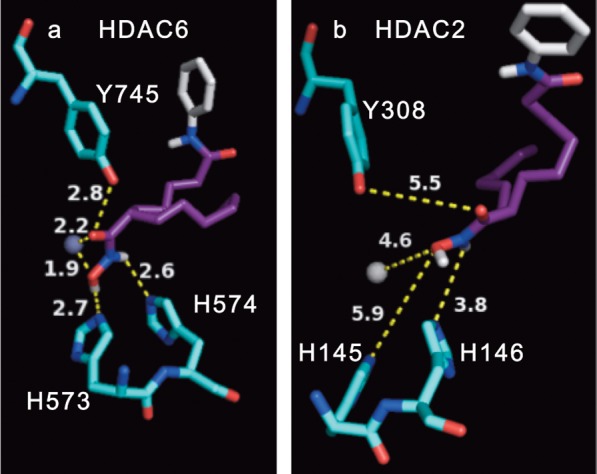
Docked pose of (S)-C2-hexyl SAHA ((S)-1i) in the (a) HDAC6 (PDB: 5EEM)34 or (b) HDAC2 (PDB: 3MAX)35 crystal structures (b) using Autodock 4.2.33 Binding distances between the hydroxamic acid atoms and active site residues (numbered in figure) or the metal are displayed in Angstroms. The (R) enantiomer is shown in Figure S53. The atomic radius of the metal (Zn2+) was set at 0.5 Å for clarity. Atom color-coding: (S)-C2-n-hexyl SAHA (C = purple/white; O = red; N = blue; H = white); amino acids (C = deep teal; O = red, N = blue); Zn2+ metal ion (gray sphere).
Because all HDAC isoforms show high conservation among their active site residues,36,37 previous studies suggested that the shape of the active sites might explain the HDAC6 selectivity of reported compounds.38 In particular, HDAC6 maintains a wider active site entrance compared to the class I isoforms.20 In previous work, HDAC6-selective inhibitors were generated by replacing the solvent-exposed anilide group of SAHA with bulky aryl groups.20,39,40 In addition, compounds with an aryl, cyclic, or unsaturated group attached in the linker region directly adjacent to the hydroxamic acid demonstrated HDAC6 selectivity.40 For example, tubastatin is a highly HDAC6-selective inhibitor that displays a series of bulky aryl groups near the hydroxamic acid (Figure S41).20 More closely related to this work, valpropylhydroxamic acid (Figure S41) positions an alkyl group adjacent to a hydroxamic acid and showed HDAC6/8 dual selectivity, with 16 and 39 μM IC50 against HDAC6 and -8, respectively, but only 9–17-fold reduced potency with HDAC1, -2, and -3.41 The data with valpropylhydroxamic acid are consistent with our data showing that the linker can influence potency and selectivity. Our docking analysis and these prior studies suggest that the selectivity of the C2-modified SAHA analogues is due to the bulky substituent adjacent to the hydroxamic acid that takes advantage of the wider active site entrance of HDAC6 and HDAC8.20,42
In conclusion, we report the synthesis and screening of several SAHA analogues substituted at the C2 position. C2-modified SAHA analogues displayed selectivity for HDAC6 and HDAC8 over HDAC1, -2, and -3. The highest selectivity observed was with C2-n-hexyl SAHA analogue 1i, which displayed 49- to 300-fold selectivity for HDAC6 and -8 over HDAC1, -2, and -3. Importantly, the selectivity of C2-n-hexyl SAHA is elevated compared to the widely used HDAC6-selective inhibitor, tubastatin. Cell-based selectivity testing of analogues 1g–i reproduced the selectivity observed in vitro. The dual HDAC6/8 selective C2-modified SAHA analogues reported in this work can be useful as lead compounds to develop pharmacological tools and anticancer drugs targeting HDAC6 and HDAC8. More generally, these studies with SAHA analogues suggest that modifying known drugs can significantly improve their properties.
Acknowledgments
We thank the Lumigen Instrument Center at Wayne State University for NMR and MS instrumentation, Dr. Y. Ge for the AML MOLM-13 and U937 cell lines, Dr. K. Honn for use of the Alpha Innotech FluorChem imaging system, Dr. M. Allen for use of the chiral HPLC column, Dr. Y. Danylyuk for HRMS analysis, and Dr. D. Nalawansha for helpful comments.
Glossary
ABBREVIATIONS
- HDAC
histone deacetylase
- SAHA
suberoylanilide hydroxamic acid
- NAD
nicotinamide adenine dinucleotide
- FDA
Food and Drug Administration
- ELISA
enzyme-linked immunosorbent assay
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- AML
acute myloid leukemia
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- CDI
carbonyldiimidazole
- TEA
triethylamine
- EDCI
N-(3-(dimethylamino)propyl)-N′-ethylcarbodiimide hydrochloride
- NaHMDS
sodium bis(trimethylsilyl)amide
- DCM
dichloromethane
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00124.
Experimental procedure, procedure for biological screening, docking procedure, compound characterization, IC50 curves and tables, and docking figures (PDF)
Author Present Address
† (A.V.B) Ash Stevens, Inc., 18655 Krause Street, Riverview, Michigan 48193, United States.
Author Contributions
A.T.N performed the enantioselective syntheses of (S)-1i and (R)-1i, purified and tested compounds 1g–i, and performed all in cells selectivity, cytotoxicity, and docking studies. G.P. tested compounds 1a–f. A.V.B. synthesized analogues (1a–I). M.K.H.P. conceived of the project and assisted in experimental design and interpretation. All authors contributed to the writing of the manuscript.
We thank National Institutes of Health (GM067657 and GM121061) and Wayne State University for funding. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The authors declare no competing financial interest.
Supplementary Material
References
- Walsh C. T.; Garneau-Tsodikova S.; Gatto G. J. Protein posttranslational modifications: the chemistry of proteome diversifications. Angew. Chem., Int. Ed. 2005, 44 (45), 7342–7372. 10.1002/anie.200501023. [DOI] [PubMed] [Google Scholar]
- Gregoretti I.; Lee Y.-M.; Goodson H. V. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J. Mol. Biol. 2004, 338 (1), 17–31. 10.1016/j.jmb.2004.02.006. [DOI] [PubMed] [Google Scholar]
- Glozak M. A.; Seto E. Histone deacetylases and cancer. Oncogene 2007, 26 (37), 5420–32. 10.1038/sj.onc.1210610. [DOI] [PubMed] [Google Scholar]
- Chuang C.; Pan J.; Hawke D. H.; Lin S.-H.; Yu-Lee L.-y. NudC deacetylation regulates mitotic progression. PLoS One 2013, 8 (9), e73841. 10.1371/journal.pone.0073841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newbold A.; Salmon J. M.; Martin B. P.; Stanley K.; Johnstone R. W. The role of p21waf1/cip1 and p27Kip1 in HDACi-mediated tumor cell death and cell cycle arrest in the E[mu]-myc model of B-cell lymphoma. Oncogene 2014, 33 (47), 5415–5423. 10.1038/onc.2013.482. [DOI] [PubMed] [Google Scholar]
- Halkidou K.; Gaughan L.; Cook S.; Leung H. Y.; Neal D. E.; Robson C. N. Upregulation and nuclear recruitment of HDAC1s in hormone refractory prostate cancer. Prostate 2004, 59 (2), 177–189. 10.1002/pros.20022. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Yamashita H.; Toyama T.; Sugiura H.; Ando Y.; Mita K.; Hamaguchi M.; Hara Y.; Kobayashi S.; Iwase H. Quantitation of HDAC1 mRNA expression in invasive carcinoma of the breast. Breast Cancer Res. Treat. 2005, 94 (1), 11–6. 10.1007/s10549-005-6001-1. [DOI] [PubMed] [Google Scholar]
- Oehme I.; Deubzer H. E.; Wegener D.; Pickert D.; Linke J.-P.; Hero B.; Kopp-Schneider A.; Westermann F.; Ulrich S. M.; von Deimling A.; Fischer M.; Witt O. Histone deacetylase 8 in neuroblastoma tumorigenesis. Clin. Cancer Res. 2009, 15 (1), 91–99. 10.1158/1078-0432.CCR-08-0684. [DOI] [PubMed] [Google Scholar]
- Sakuma T.; Uzawa K.; Onda T.; Shiiba M.; Yokoe H.; Shibahara T.; Tanzawa H. Aberrant expression of histone deacetylase 6 in oral squamous cell carcinoma. Int. J. Oncol. 2006, 29 (1), 117–24. 10.3892/ijo.29.1.117. [DOI] [PubMed] [Google Scholar]
- Aldana-Masangkay G. I.; Sakamoto K. M. The role of HDAC6 in cancer. J. Biomed. Biotechnol. 2011, 2011, 875824. 10.1155/2011/875824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S. Y.; Jun J. A.; Jeong K. J.; Heo H. J.; Sohn J. S.; Lee H. Y.; Park C. G.; Kang J. Histone deacetylases 1, 6 and 8 are critical for invasion in breast cancer. Oncol. Rep. 2011, 25 (6), 1677–81. 10.3892/or.2011.1236. [DOI] [PubMed] [Google Scholar]
- Warrell R. P. Jr.; He L. Z.; Richon V.; Calleja E.; Pandolfi P. P. Therapeutic targeting of transcription in acute promyelocytic leukemia by use of an inhibitor of histone deacetylase. J. Natl. Cancer Inst. 1998, 90 (21), 1621–1625. 10.1093/jnci/90.21.1621. [DOI] [PubMed] [Google Scholar]
- Grant S.; Easley C.; Kirkpatrick P. Vorinostat. Nat. Rev. Drug Discovery 2007, 6 (1), 21–22. 10.1038/nrd2227. [DOI] [PubMed] [Google Scholar]
- Plumb J. A.; Finn P. W.; Williams R. J.; Bandara M. J.; Romero M. R.; Watkins C. J.; La Thangue N. B.; Brown R. Pharmacodynamic response and inhibition of growth of human tumor xenografts by the novel histone deacetylase inhibitor PXD101. Mol. Cancer Ther. 2003, 2 (8), 721–728. [PubMed] [Google Scholar]
- Laubach J. P.; Moreau P.; San-Miguel J. F.; Richardson P. G. Panobinostat for the treatment of multiple myeloma. Clin. Cancer Res. 2015, 21 (21), 4767–4773. 10.1158/1078-0432.CCR-15-0530. [DOI] [PubMed] [Google Scholar]
- West A. C.; Johnstone R. W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Invest. 2014, 124 (1), 30–39. 10.1172/JCI69738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan N.; Jeffers M.; Kumar S.; Hackett C.; Boldog F.; Khramtsov N.; Qian X.; Mills E.; Berghs S. C.; Carey N.; Finn P. W.; Collins L. S.; Tumber A.; Ritchie J. W.; Jensen P. B.; Lichenstein H. S.; Sehested M. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem. J. 2008, 409 (2), 581–589. 10.1042/BJ20070779. [DOI] [PubMed] [Google Scholar]
- Kelly W. K.; O’Connor O. A.; Krug L. M.; Chiao J. H.; Heaney M.; Curley T.; MacGregore-Cortelli B.; Tong W.; Secrist J. P.; Schwartz L.; Richardson S.; Chu E.; Olgac S.; Marks P. A.; Scher H.; Richon V. M. Phase I study of an oral histone deacetylase inhibitor, suberoylanilide hydroxamic acid, in patients with advanced cancer. J. Clin. Oncol. 2005, 23 (17), 3923–3931. 10.1200/JCO.2005.14.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malvaez M.; McQuown S. C.; Rogge G. A.; Astarabadi M.; Jacques V.; Carreiro S.; Rusche J. R.; Wood M. A. HDAC3-selective inhibitor enhances extinction of cocaine-seeking behavior in a persistent manner. Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (7), 2647–2652. 10.1073/pnas.1213364110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler K. V.; Kalin J.; Brochier C.; Vistoli G.; Langley B.; Kozikowski A. P. Rational design and simple chemistry yield a superior, neuroprotective HDAC6 inhibitor, tubastatin A. J. Am. Chem. Soc. 2010, 132 (31), 10842–10846. 10.1021/ja102758v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padige G.; Negmeldin A. T.; Pflum M. K. H. Development of an ELISA-based HDAC activity assay for characterization of isoform-selective inhibitors. J. Biomol. Screening 2015, 20 (10), 1277–1285. 10.1177/1087057115598118. [DOI] [PubMed] [Google Scholar]
- Bieliauskas A.; Weerasinghe S.; Pflum M. H. Structural requirements of HDAC inhibitors: SAHA analogs functionalized adjacent to the hydroxamic acid. Bioorg. Med. Chem. Lett. 2007, 17 (8), 2216–2219. 10.1016/j.bmcl.2007.01.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S. E.; Weerasinghe S. V.; Pflum M. K. The structural requirements of histone deacetylase inhibitors: Suberoylanilide hydroxamic acid analogs modified at the C3 position display isoform selectivity. Bioorg. Med. Chem. Lett. 2011, 21 (20), 6139–6142. 10.1016/j.bmcl.2011.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S. E.; Pflum M. K. H. The structural requirements of histone deacetylase inhibitors: Suberoylanilide hydroxamic acid analogs modified at the C6 position. Bioorg. Med. Chem. Lett. 2012, 22 (23), 7084–7086. 10.1016/j.bmcl.2012.09.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieliauskas A. V.; Weerasinghe S. V. W.; Negmeldin A. T.; Pflum M. K. H. Structural requirements of histone deacetylase inhibitors: SAHA analogs modified on the hydroxamic acid. Arch. Pharm. (Weinheim, Ger.) 2016, 349, 373. 10.1002/ardp.201500472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson D. E.; Wagner F. F.; Kaya T.; Gale J. P.; Aidoud N.; Davoine E. L.; Lazzaro F.; Weiwer M.; Zhang Y. L.; Holson E. B. Discovery of the first histone deacetylase 6/8 dual inhibitors. J. Med. Chem. 2013, 56 (11), 4816–4820. 10.1021/jm400390r. [DOI] [PubMed] [Google Scholar]
- Bradner J. E.; West N.; Grachan M. L.; Greenberg E. F.; Haggarty S. J.; Warnow T.; Mazitschek R. Chemical phylogenetics of histone deacetylases. Nat. Chem. Biol. 2010, 6 (3), 238–243. 10.1038/nchembio.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackanson B.; Rimmele L.; Benkißer M.; Abdelkarim M.; Fliegauf M.; Jung M.; Lübbert M. HDAC6 as a target for antileukemic drugs in acute myeloid leukemia. Leuk. Res. 2012, 36 (8), 1055–1062. 10.1016/j.leukres.2012.02.026. [DOI] [PubMed] [Google Scholar]
- Vickers C. J.; Olsen C. A.; Leman L. J.; Ghadiri M. R. Discovery of HDAC inhibitors that lack an active site Zn2+-binding functional group. ACS Med. Chem. Lett. 2012, 3 (6), 505–508. 10.1021/ml300081u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodji Q. H.; Patil V.; Kornacki J. R.; Mrksich M.; Oyelere A. K. Synthesis and structure–activity relationship of 3-hydroxypyridine-2-thione-based histone deacetylase inhibitors. J. Med. Chem. 2013, 56 (24), 9969–9981. 10.1021/jm401225q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Inks E. S.; Li X.; Hou J.; Chou C. J.; Zhang J.; Jiang Y.; Zhang Y.; Xu W. Discovery of the first N-hydroxycinnamamide-based histone deacetylase 1/3 dual inhibitors with potent oral antitumor activity. J. Med. Chem. 2014, 57 (8), 3324–3341. 10.1021/jm401877m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee A. K.; Choi T.-L.; Sanders D. P.; Grubbs R. H. A General Model for Selectivity in Olefin Cross Metathesis. J. Am. Chem. Soc. 2003, 125 (37), 11360–11370. 10.1021/ja0214882. [DOI] [PubMed] [Google Scholar]
- Morris G. M.; Huey R.; Lindstrom W.; Sanner M. F.; Belew R. K.; Goodsell D. S.; Olson A. J. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30 (16), 2785–2791. 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hai Y.; Christianson D. W. Histone deacetylase 6 structure and molecular basis of catalysis and inhibition. Nat. Chem. Biol. 2016, 12 (9), 741–747. 10.1038/nchembio.2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bressi J. C.; Jennings A. J.; Skene R.; Wu Y.; Melkus R.; Jong R. D.; O’Connell S.; Grimshaw C. E.; Navre M.; Gangloff A. R. Exploration of the HDAC2 foot pocket: synthesis and SAR of substituted N-(2-aminophenyl)benzamides. Bioorg. Med. Chem. Lett. 2010, 20 (10), 3142–3145. 10.1016/j.bmcl.2010.03.091. [DOI] [PubMed] [Google Scholar]
- Weerasinghe S. V. W.; Estiu G.; Wiest O.; Pflum M. K. H. Residues in the 11Å channel of histone deacetylase 1 promote catalytic activity: Implications for designing isoform-selective histone deacetylase inhibitors. J. Med. Chem. 2008, 51 (18), 5542–5551. 10.1021/jm800081j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wambua M. K.; Nalawansha D. A.; Negmeldin A. T.; Pflum M. K. Mutagenesis studies of the 14 Å internal cavity of histone deacetylase 1: Insights towards the acetate escape hypothesis and selective inhibitor design. J. Med. Chem. 2014, 57 (3), 642–650. 10.1021/jm401837e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estiu G.; Greenberg E.; Harrison C. B.; Kwiatkowski N. P.; Mazitschek R.; Bradner J. E.; Wiest O. Structural origin of selectivity in class II-selective histone deacetylase inhibitors. J. Med. Chem. 2008, 51 (10), 2898–2906. 10.1021/jm7015254. [DOI] [PubMed] [Google Scholar]
- Kozikowski A. P.; Chen Y.; Gaysin A. M.; Savoy D. N.; Billadeau D. D.; Kim K. H. Chemistry, biology, and QSAR studies of substituted biaryl hydroxamates and mercaptoacetamides as HDAC inhibitors-nanomolar-potency inhibitors of pancreatic cancer cell growth. ChemMedChem 2008, 3 (3), 487–501. 10.1002/cmdc.200700314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner F. F.; Olson D. E.; Gale J. P.; Kaya T.; Weiwer M.; Aidoud N.; Thomas M.; Davoine E. L.; Lemercier B. C.; Zhang Y. L.; Holson E. B. Potent and selective inhibition of histone deacetylase 6 (HDAC6) does not require a surface-binding motif. J. Med. Chem. 2013, 56 (4), 1772–1776. 10.1021/jm301355j. [DOI] [PubMed] [Google Scholar]
- Fass D. M.; Shah R.; Ghosh B.; Hennig K.; Norton S.; Zhao W.-N.; Reis S. A.; Klein P. S.; Mazitschek R.; Maglathlin R. L.; Lewis T. A.; Haggarty S. J. Short-chain HDAC inhibitors differentially affect vertebrate development and neuronal chromatin. ACS Med. Chem. Lett. 2011, 2 (1), 39–42. 10.1021/ml1001954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KrennHrubec K.; Marshall B. L.; Hedglin M.; Verdin E.; Ulrich S. M. Design and evaluation of ’Linkerless’ hydroxamic acids as selective HDAC8 inhibitors. Bioorg. Med. Chem. Lett. 2007, 17 (10), 2874–2878. 10.1016/j.bmcl.2007.02.064. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.