

Abstract
Human African trypanosomiasis (HAT), Chagas disease, and leishmaniasis present a significant burden across the developing world. Existing therapeutics for these protozoal neglected tropical diseases suffer from severe side effects and toxicity. Previously, NEU-1045 (3) was identified as a promising lead with cross-pathogen activity, though it possessed poor physicochemical properties. We have designed a library of analogues with improved calculated physicochemical properties built on the quinoline scaffold of 3 incorporating small, polar aminoheterocycles in place of the 4-(3-fluorobenzyloxy)aniline substituent. We report the biological activity of these inhibitors against Trypanosoma brucei (HAT), T. cruzi (Chagas disease), and Leishmania major (cutaneous leishmaniasis) and describe the identification of N-(5-chloropyrimidin-2-yl)-6-(4-(morpholinosulfonyl)phenyl)quinolin-4-amine (13t) as a promising inhibitor of L. major proliferation and 6-(4-(morpholinosulfonyl)phenyl)-N-(pyrimidin-4-yl)quinolin-4-amine (13j), a potent inhibitor of T. brucei proliferation with improved drug-like properties.
Keywords: Antiparasitic agents, Trypanosoma brucei, Trypanosoma cruzi, Leishmania major, Plasmodium falciparum, Chagas disease, leishmaniasis, human African trypanosomiasis
Taken together, human African trypanosomiasis (HAT), Chagas disease, and leishmaniasis are responsible for 4.4 million disability adjusted life years (DALY) and 70,000 deaths annually.1 Caused by the protozoan parasites Trypanosoma brucei, T. cruzi, and Leishmania spp., respectively, these diseases are spread through insect vectors across Latin America, Africa, and parts of southern Asia.2−6 Current therapeutics suffer from severe side effects, complex and prolonged dosing regimens, high costs, and emerging resistance necessitating the need for new treatments.7−12
Our group has reported the discovery of the antitrypanosomal activity of a library of compounds derived from a known tyrosine kinase inhibitor, lapatinib (1, Figure 1).13−16 Owing to the high degree of homology in kinetoplastid protein kinases (PKs) in T. brucei, T. cruzi, and L. major,17 we expected these compounds to be active against other trypanosomatid parasites. Thus, we tested new compounds against cultures of T. brucei, T. cruzi, and L. major. We also included Plasmodium falciparum, the causative agent of malaria, in this work. We identified two promising compounds as new leads for leishmaniasis, NEU-554 (2) and NEU-1045 (3, Figure 1), which were evolved from the highly potent inhibitor of T. brucei growth, NEU-617 (4).14 Physicochemical analysis of this collection of compounds revealed they generally showed poor drug-like properties (Figure 1 and Table 1), and subsequent efforts have been focused on the development of new analogues aimed at improving these properties and overall lead quality (as described by lipophilic ligand efficiency (LLE))18 while maintaining or improving in vitro activity against the parasites. Consistent with our practice, we tested compounds against multiple kinetoplastid parasites in parallel. The results of these efforts are reported herein.
Figure 1.
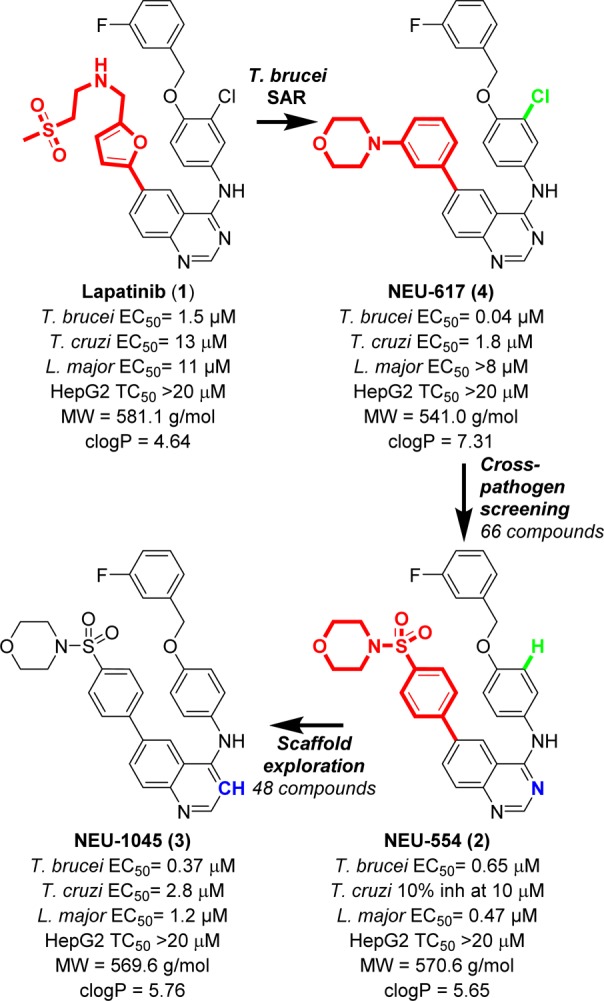
SAR development of NEU-1045 from lapatinib.
Table 1. Druglike Properties of 3.
| 3 | goal | |
|---|---|---|
| molecular weight | 569.6 | ≤500 |
| clogP | 5.76 | ≤5 |
| protein binding (% free) | <0.03 | >10 |
| aqueous solubility (μM) | 2 | >50 |
| human liver microsomes median CLint (μL/min/mg) | 98 | <47 |
| male rat hepatocytes median CLint (μL/min/106 cells) | 19 | <27 |
The broad spectrum antitrypanosomal activity of 3 attracted our attention as a modestly potent lead for HAT, Chagas disease, and leishmaniasis. The primary shortcoming of 3 is in its poor predicated physicochemical properties, driven by a high molecular weight (569.6 g/mol) and clogP (5.76). Much of the MW and clogP of 3 may be attributed to the large, lipophilic 4-(3-fluorobenzyloxy)aniline “head group”. We had previously observed that truncation of the similar 4-benzyloxy-3-chloroaniline group in our quinazoline series led only to a 3-fold loss in activity (cf. NEU-369, 5 versus NEU-555, 6, Figure 2).14 Replacement of the 3-fluorobenzyl group with a methyl led to significant reductions in heavy atom count (ΔHAC= −7) and lipophilicity (ΔclogP = −1.87), giving rise to a similar ligand efficiency (LE = pEC50/heavy atom count, 0.14 → 0.15) and improved LLE (pEC50 – clogP, −0.43 → 0.93).18−21
Figure 2.

Truncation of lipophilic groups with partial retention of potency. All L. major data are in the intracellular amastigote life stage.
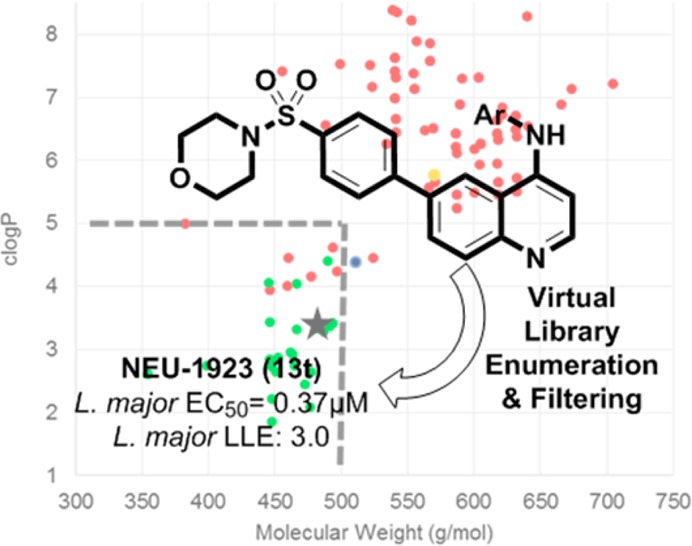
While the SAR of compounds built on the quinazoline scaffold were well explored13 little attention had to this point been given to compounds built on the quinoline scaffold, despite their widespread prevalence as a privileged scaffold in medicinal chemistry.22−24 When tested in a cross-parasite screening campaign, 3 inhibited T. brucei, T. cruzi, and L. major proliferation with good potency (T. brucei EC50 = 0.37 μM; T. cruzi EC50 = 2.8 μM; L. major EC50 = 1.2 μM). We believed that reducing the molecular size and lipophilicity would lead to an improvement in the overall ADME profile of the series. As such, we hypothesized that smaller, more polar analogues built upon the 3 scaffold could lead to compounds that maintained antiparasite activity and were more soluble in water.
In order to accomplish our goal, we enumerated a virtual library of analogues by using JChem Reactor (ChemAxon, Inc.) and a set of 246 heterocyclic amines that are commercially available in preweighed quantities. Using Vortex (Dotmatics, Inc.) we shaped the library on the basis of clogP (<5) and molecular weight (<500). From this filtered virtual library of 132 compounds, we selected 23 analogues for synthesis via inspection, eliminating compounds with multiple reactive moieties, or those with anticipated difficulties in chemical stability or purification. Also selected for synthesis was the quinoline analogue of 6, bearing the truncated 3-chloro-4-methoxyaniline group that was featured in the active truncated analogues of 1 and 4. Calculated physicochemical properties of these compounds in comparison with the properties of compounds previously described are plotted in Figure 3.13,14
Figure 3.
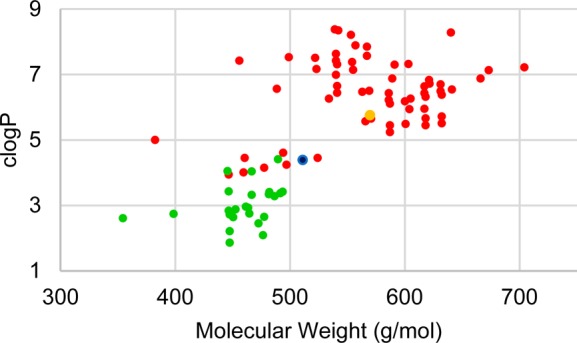
Physicochemical properties analysis of lapatinib analogues selected after in silico enumeration and filtering. Related compounds that were previously disclosed are shown in red,13,14 and the new shaped virtual library is shown in green. Compound 3 is shown in orange, and 6 is shown in blue.
The synthesis of our selected library commenced with generation of the sulfonamide 8 from 4-bromophenylsulfonyl chloride, 7 (Scheme 1). Halogen-lithium exchange of 8 followed by quenching with triisopropyl borate, and subsequent hydrolysis yielded boronic acid 9.
Scheme 1. NEU-1045 Analogue Western Half Synthesis.

Reagents and conditions: (a) morpholine, THF, rt, o.n.; (b) n-BuLi, THF, −78 °C, 1 h; B(Oi-Pr)3 rt, o.n.; HCl rt.
Suzuki-Miyaura coupling of 9 and 1014 produced 6-arylquinolinone 11 (Scheme 2). Deoxychlorination of 11 generated 12 in nearly quantitative yield. Conversion to the corresponding 4-aminoquinolines 13a–z could be achieved under acidic or basic conditions or via Buchwald–Hartwig amination.
Scheme 2. NEU-1045 Analogue Synthesis.

Reagents and conditions: (a) 9, Et3N, Pd(PPh3)2Cl2, 1:1 EtOH/H2O, ↓↑, 2 h; (b) POCl3, ↓↑, 2 h; (c) TsOH, ArNH2, DMSO, 80 °C, 24 h; (d) ArNH2, Cs2CO3, Pd2(dba)3, xantphos, N2, 1,4-dioxane, ↓↑, 24 h; (e) ArNH2, KOt-Bu, Pd2(dba)3, xantphos, N2, 1,4-dioxane, ↓↑, 24 h; (f) ArNH2, NaH, 2:1 1,4-dioxane/DMF, 0 → 100 °C, 1 h.
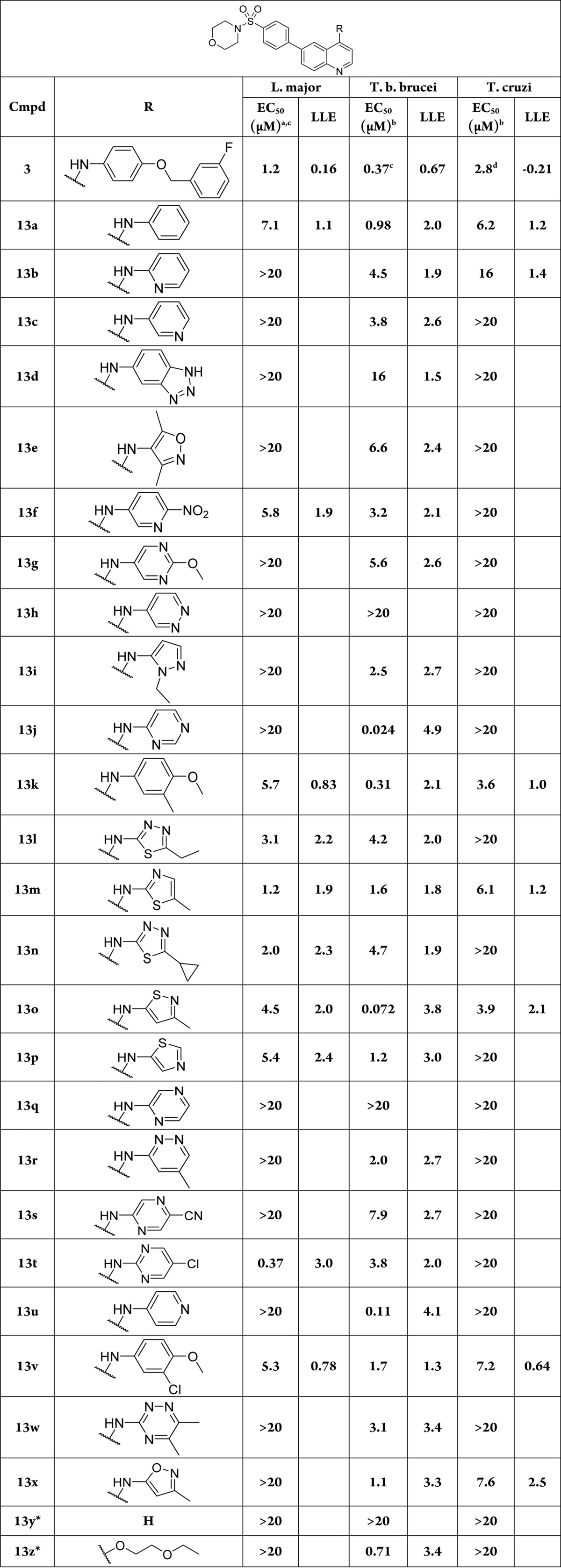
Compounds were tested in an in vitro assay of RAW 264.7 macrophages infected with intracellular amastigotes of L. major, 3T3 fibroblasts infected with intracellular amastigotes of T. cruzi, and bloodstream form of T. brucei. The data are summarized in Table 2. All compounds synthesized were also screened against P. falciparum, the causative agent of malaria (Table S1, Supporting Information). Compound toxicity to mammalian cells was assessed against HepG2 cells as well as NIH 3T3 cells (Table S1).
Table 2. Anti-Kinetoplastid Activity of Analogues of Compound 3.
All r2 values >0.75.
All SEM values within 25%.
n = 1.
n = 2.
Isolated as a side product.
All analogues possessing a thiazole, isothiazole, or thiadiazole (13l–p) consistently maintained anti-leishmanial activity in the single digit micromolar range (1.2–5.4 μM). Compound 13v, incorporating the truncated aniline of 6, and the isostere 13k also displayed activity in this range. The incorporation of nitrogen atom(s) into six-membered arenes was poorly tolerated against L. major amastigotes, with the exception of 13f (EC50 = 5.8 μM) and 13t (EC50= 0.37 μM).
Compound 13t is of particular interest as the only compound in this library to achieve submicromolar activity against L. major amastigotes. Compound 13y, devoid of an aminoheterocycle was inactive against all parasites tested, implicating these groups are essential structural motifs for antiparasitic activity. All compounds in this set were found to possess low host cell toxicity against HepG2 (TC50 > 20 μM) except for 13k (TC50 = 9.0 μM).
Highlighted by 13t, a dramatic improvement in LLE was achieved in this series (13t LLE = 3.02, compared to 3, LLE = 0.16) driven by the reduced clogP (13t = 3.41, 3 = 5.76; Table S2, Supporting Information). Despite the improved lipophilicity profile, aqueous solubility (1 μM) and PPB (99.6%) remained poor. Taken together, these properties limited these compounds’ progression to further studies against Leishmania.
Noting that the targeted potency range for anti-T. cruzi lead compounds is <10 μM,25 we were pleased to observe that approximately half of the compounds tested showed some activity against T. cruzi, with six in the single-digit micromolar range. The most potent were 13k (EC50 = 3.6 μM) and 13o (EC50 = 3.9 μM). Importantly, we observed all compounds were nontoxic to NIH 3T3 host cells (TC50 > 20 μM) with the exception of 13k (TC50 = 15 μM).
Sixteen analogues showed activity in the 1–10 μM range against T. brucei, and an additional six were <1 μM, including the highly active 13o (EC50 = 72 nM) and 13j (EC50 = 24 nM). We were intrigued by the activity of 13j against T. brucei. Compared to 3, 13j has a lower molecular weight (447.5 vs 569.6 g/mol) and clogP (2.72 vs 5.76; Table S2, Supporting Information), reflected in the improved LE (0.24) and excellent LLE (4.9). This compound showed an acceptable logD7.4 (3.3) and improvement in human plasma protein binding (95.6%), though the thermodynamic aqueous solubility remained poor (1.4 μM).
We note that, despite the similarity between the kinomes of the three kinetoplastid parasites, we see distinct differences in selectivity across species. There are some potential explanations for this. First, though homologous across pathogens, certain targets may not have similar essentiality. Second, there are potential confounding factors involved by virtue of the nature of the parasite (intra- versus extracellular), and there could be potentiating effects from host cell targets. We do note, however, that we do not know the target(s) of action for these compounds; this is of interest for future work.
Given the attractive potency and drug-like properties of 13j, we performed pharmacokinetic experiments in mice. Compound 13j was dosed by intraperitoneal injection to 18 female BALB/c mice at 10 mg/kg. Blood and brain samples were collected from groups of three at 0.08, 0.25, 1, 4, 8, and 24 h and analyzed by LC–MS/MS. The results are visualized in Figure S1 and tabulated in Tables S3–S6 in the Supporting Information. We observed plasma exposure >120-fold of the EC50 for 4 h, and the maximum brain to plasma ratio was 0.01 at 0.25 h after dosing 13j. In conjunction with the low levels of 13j in the brain, we noted the rapid clearance in human liver microsomes (Clint = 207.5 μL/min/mg) and rat hepatocytes (Clint = 31.5 μL/min/106 cells) suggested 13j would show poor efficacy in in vivo models of infection. We continue to work toward compounds with improved PK properties and will report these results in due course.
In summary, through a strategy of generation and filtering of a virtual library of analogues of 3 on the basis of computed physicochemical properties, we have identified potent inhibitors of L. major, T. cruzi, and T. brucei proliferation. These compounds possess improved drug-like properties and efficiency metrics as a result, in part, of the reduction in molecular weight and lipophilicity designed in the virtual library. Further work aimed at improving the in vivo ADME properties and requisite properties for the different protozoan parasites will be undertaken. In addition, attention must be paid to the nonparallel SAR that we have observed between parasites. As a result, a single, multipathogen-targeting agent would seem unlikely, and pathogen-specific optimization is needed at this point.
Acknowledgments
ChemAxon is gratefully acknowledged for the free academic license provided for their software suite. Dr. Lori Ferrins is gratefully acknowledged for her assistance in preparing this manuscript. We are grateful to AstraZeneca for the determination of in vitro ADME properties in support of this work.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00011.
Chemical synthesis and characterization of new compounds reported, antimalarial and physicochemical property data tables (annotated with NEU registry numbers), and biological methods (PDF)
Funding from the National Institutes of Health (R01AI124046 and R56AI099476 to M.P.P. and K.M.-W.) and Northeastern University are gratefully acknowledged.
The authors declare no competing financial interest.
Supplementary Material
References
- Moran M.; Guzman J.; Ropars A.-L.; McDonald A.; Jameson N.; Omune B.; Ryan S.; Wu L. Neglected Disease Research and Development: How Much Are We Really Spending?. PLOS Med. 2009, 6, e1000030. 10.1371/journal.pmed.1000030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization. Investing to overcome the global impact of neglected tropical diseases: Third WHO report on neglected tropical diseases; 2015.
- Simarro P. P.; Cecchi G.; Franco J. R.; Paone M.; Diarra A.; Ruiz-Postigo J. A.; Fevre E. M.; Mattioli R. C.; Jannin J. G. Estimating and mapping the population at risk of sleeping sickness. PLoS Neglected Trop. Dis. 2012, 6, e1859. 10.1371/journal.pntd.0001859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvar J.; Velez I. D.; Bern C.; Herrero M.; Desjeux P.; Cano J.; Jannin J.; den Boer M.; Team W. H. O. L. C. Leishmaniasis worldwide and global estimates of its incidence. PLoS One 2012, 7, e35671. 10.1371/journal.pone.0035671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Souza A.; Ishikawa E.; Braga R.; Silveira F.; Lainson R.; Shaw J. Psychodopygus complexus, a new vector of Leishmania braziliensis to humans in Para State, Brazil. Trans. R. Soc. Trop. Med. Hyg. 1996, 90, 112–113. 10.1016/S0035-9203(96)90103-0. [DOI] [PubMed] [Google Scholar]
- Andrade L. O.; Andrews N. W. The Trypanosoma cruzi-host-cell interplay: location, invasion, retention. Nat. Rev. Microbiol. 2005, 3, 819–823. 10.1038/nrmicro1249. [DOI] [PubMed] [Google Scholar]
- Chulay J. D.; Spencer H. C.; Mugambi M. Electrocardiographic changes during treatment of leishmaniasis with pentavalent antimony (sodium stibogluconate). Am. J. Trop. Med. Hyg. 1985, 34, 702–709. [DOI] [PubMed] [Google Scholar]
- Delgado J.; Macias J.; Pineda J. A.; Corzo J. E.; Gonzalez-Moreno M. P.; de la Rosa R.; Sanchez-Quijano A.; Leal M.; Lissen E. High frequency of serious side effects from meglumine antimoniate given without an upper limit dose for the treatment of visceral leishmaniasis in human immunodeficiency virus type-1-infected patients. Am. J. Trop. Med. Hyg. 1999, 61, 766–769. [DOI] [PubMed] [Google Scholar]
- Herwaldt B. L.; Berman J. D. Recommendations for treating leishmaniasis with sodium stibogluconate (Pentostam) and review of pertinent clinical studies. Am. J. Trop. Med. Hyg. 1992, 46, 296–306. [DOI] [PubMed] [Google Scholar]
- Chappuis F.; Sundar S.; Hailu A.; Ghalib H.; Rijal S.; Peeling R. W.; Alvar J.; Boelaert M. Visceral leishmaniasis: what are the needs for diagnosis, treatment and control?. Nat. Rev. Microbiol. 2007, 5, 873–882. 10.1038/nrmicro1748. [DOI] [PubMed] [Google Scholar]
- Maltezou H. C. Drug resistance in visceral leishmaniasis. J. Biomed. Biotechnol. 2010, 2010, 617521. 10.1155/2010/617521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messori A.; Fadda V.; Maratea D.; Trippoli S.; Marinai C. Nephrotoxicity of different formulations of amphotericin B: summarizing evidence by network meta-analysis. Clin. Infect. Dis. 2013, 57, 1783–1784. 10.1093/cid/cit588. [DOI] [PubMed] [Google Scholar]
- Patel G.; Karver C. E.; Behera R.; Guyett P. J.; Sullenberger C.; Edwards P.; Roncal N. E.; Mensa-Wilmot K.; Pollastri M. P. Kinase scaffold repurposing for neglected disease drug discovery: discovery of an efficacious, lapatinib-derived lead compound for trypanosomiasis. J. Med. Chem. 2013, 56, 3820–3832. 10.1021/jm400349k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devine W.; Woodring J. L.; Swaminathan U.; Amata E.; Patel G.; Erath J.; Roncal N. E.; Lee P. J.; Leed S. E.; Rodriguez A.; Mensa-Wilmot K.; Sciotti R. J.; Pollastri M. P. Protozoan Parasite Growth Inhibitors Discovered by Cross-Screening Yield Potent Scaffolds for Lead Discovery. J. Med. Chem. 2015, 58, 5522–5537. 10.1021/acs.jmedchem.5b00515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katiyar S.; Kufareva I.; Behera R.; Thomas S. M.; Ogata Y.; Pollastri M.; Abagyan R.; Mensa-Wilmot K. Lapatinib-Binding Protein Kinases in the African Trypanosome: Identification of Cellular Targets for Kinase-Directed Chemical Scaffolds. PLoS One 2013, 8, e56150. 10.1371/journal.pone.0056150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackey K. E. Lessons from the Drug Discovery of Lapatinib, a Dual ErbB1/2 Tyrosine Kinase Inhibitor. Curr. Top. Med. Chem. 2006, 6, 435–460. 10.2174/156802606776743156. [DOI] [PubMed] [Google Scholar]
- Parsons M.; Worthey E. A.; Ward P. N.; Mottram J. C. Comparative analysis of the kinomes of three pathogenic trypanosomatids: Leishmania major, Trypanosoma brucei and Trypanosoma cruzi. BMC Genomics 2005, 6, 1–19. 10.1186/1471-2164-6-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shultz M. D.; Cheung A. K.; Kirby C. A.; Firestone B.; Fan J.; Chen C. H.; Chen Z.; Chin D. N.; Dipietro L.; Fazal A.; Feng Y.; Fortin P. D.; Gould T.; Lagu B.; Lei H.; Lenoir F.; Majumdar D.; Ochala E.; Palermo M. G.; Pham L.; Pu M.; Smith T.; Stams T.; Tomlinson R. C.; Toure B. B.; Visser M.; Wang R. M.; Waters N. J.; Shao W. Identification of NVP-TNKS656: The Use of Structure-Efficiency Relationships To Generate a Highly Potent, Selective, and Orally Active Tankyrase Inhibitor. J. Med. Chem. 2013, 56, 6495–6511. 10.1021/jm400807n. [DOI] [PubMed] [Google Scholar]
- Hopkins A. L.; Groom C. R.; Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discovery Today 2004, 9, 430–431. 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- Kuntz I. D.; Chen K.; Sharp K. A.; Kollman P. A. The maximal affinity of ligands. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 9997–10002. 10.1073/pnas.96.18.9997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeson P. D.; Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discovery 2007, 6, 881–890. 10.1038/nrd2445. [DOI] [PubMed] [Google Scholar]
- Zhao H.; Dietrich J. Privileged scaffolds in lead generation. Expert Opin. Drug Discovery 2015, 10, 781–790. 10.1517/17460441.2015.1041496. [DOI] [PubMed] [Google Scholar]
- Welsch M. E.; Snyder S. A.; Stockwell B. R. Privileged scaffolds for library design and drug discovery. Curr. Opin. Chem. Biol. 2010, 14, 347–361. 10.1016/j.cbpa.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bongarzone S.; Bolognesi M. L. The concept of privileged structures in rational drug design: focus on acridine and quinoline scaffolds in neurodegenerative and protozoan diseases. Expert Opin. Drug Discovery 2011, 6, 251–268. 10.1517/17460441.2011.550914. [DOI] [PubMed] [Google Scholar]
- Katsuno K.; Burrows J. N.; Duncan K.; van Huijsduijnen R. H.; Kaneko T.; Kita K.; Mowbray C. E.; Schmatz D.; Warner P.; Slingsby B. T. Hit and lead criteria in drug discovery for infectious diseases of the developing world. Nat. Rev. Drug Discovery 2015, 14, 751–758. 10.1038/nrd4683. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.