

Abstract
Here we report the structure–activity relationship (SAR) investigations of QL-XII-47 (QL47), a compound that possesses broad-spectrum antiviral activity against dengue virus and other RNA viruses. A medicinal chemistry campaign initiated from QL47, a previously reported covalent BTK inhibitor, to derive YKL-04-085, which is devoid of any kinase activity when screened against a panel of 468 kinases and with improved pharmacokinetic properties. Both QL47 and YKL-04-085 are potent inhibitors of viral translation and exhibit cellular antiviral activity at 35-fold lower concentrations relative to inhibition of host-cell proliferation.
Keywords: Dengue virus, broad-spectrum antiviral, QL47, structure−activity relationship
Dengue virus (DENV) is a significant human pathogen for which no effective drugs or vaccines currently exist. DENV is a plus-stranded RNA virus and a member of the Flaviviridae family. The four DENV serotypes (DENV-1, -2, -3, -4) are defined by their immunoreactivity and differ from one another by 25–40% at the amino acid level.1 Due to the large number of people at risk for infection, DENV is the most widespread mosquito-borne viral disease affecting humans today. An estimated 2.5 billion people live in areas at risk for epidemic transmission, and an estimated 390 million DENV infections occur annually, of which 96 million (67–136) manifest clinically.2 Infection with DENV is responsible for disease ranging from dengue fever to the much more severe and life-threatening dengue hemorrhagic fever (DHF) and dengue shock syndrome (DSS) that are characterized by vascular leakage. An estimated 500,000 cases of DHF/DSS occur annually and are associated with 2.5% fatality although fatality rates for DHF/DSS can exceed 20% if untreated.3 Since both the number of DENV infections and the proportion of infections resulting in DHF have escalated in recent years, the absence of anti-DENV therapeutics or an anti-DENV vaccine is a growing public health concern.
Classical antiviral approaches involve development of so-called “direct-acting” antivirals that target essential, virally encoded proteins, which in the case of DENV would include the viral polymerase and protease.4 Despite tremendous success in developing antivirals that target the analogous enzymes of HIV and HCV,5−8 to-date efforts to develop antivirals against the dengue polymerase or protease have not yielded promising clinical candidates. We pursued a complementary approach to identify compounds that target host-factors that are essential for viral replication. The potential advantage of this approach is that viral resistance might emerge more slowly because there is not a direct route to viral resistance arising from mutations that block drug-binding to a viral target. A second advantage is that host-directed antivirals might possess broader spectrum antiviral activity if they target processes commonly utilized by different viruses.9
Previously we have implemented cellular phenotypic screens of DENV infection that resulted in the identification of inhibitors of host signaling and lipid metabolism.10,11 We extended this screening to a large collection of cysteine-reactive, covalent kinase inhibitors that have the potential to target a subset of the approximately 150 kinases that possess a cysteine near the ATP-binding site of the kinase. From this screen,31 we identified a tricyclic quinoline compound, QL-XII-47 (QL47), previously identified as a potent and covalent inhibitor of BTK and other Tec-family kinases.13 Here we describe a medicinal chemistry campaign focused on the goal of understanding the structural features required for antiviral activity of this scaffold relative to inhibition of host-cell kinases. Surprisingly, we discovered compounds such as YKL-04-085, which lack the critical quinoline nitrogen required for hydrogen bonding to the kinase hinge and are devoid of any kinase activity, that retain the potent antiviral activity. We demonstrate that this series of compounds inhibit DENV infection at concentrations that are not cytotoxic and also inhibit the replication of other RNA viruses in cell culture.
To identify new host-targeted inhibitors of DENV, we used two different approaches to screen small molecule libraries. Since a detailed account of the two screens is described elsewhere,31 we present here a qualitative description as background. A first screen was based on the utilization of DENV reporter viral particles (RVP), which are composed of DENV2 E and prM/M proteins and contain a nucleocapsid formed by DENV2 C and a subgenomic replicon of the closely related West Nile virus (WNV), another Flavivirus.14 In a second screen, we employed a previously characterized HEK-293T derived stable cell line encoding a GFP tagged DENV2 replicon, the T-REx-293-DGZ cell line.14 In both screens, the expression levels of the reporter gene encoded by either the RVP-packaged WNV replicon or the DENV2 replicon were measured after treatment with a library of small molecules.
The library of small molecules screened was a collection of acrylamide-modified, ATP-site directed heterocycles designed to target cysteine residues located in or around the kinase ATP-site. This library contains experimentally validated covalent inhibitors of EGFR,15 BTK,16 BMX,17 BLK,16 TEC,16 JAK3,18 FGFR1,2,3,4,19 JNK1,2,3,20 CDK7,20 CDK12,21 and CDK1321 as well as potential inhibitors of a subset of over 150 kinases that possess a potentially reactive cysteine.22 In addition, proteomic studies have demonstrated that these compounds can target other non-kinase targets either due to the presence of a hyper-reactive cysteine or due to the presence of a suitable binding pocket and an adjacent cysteine residue.23
The screen resulted in the identification of a series of small molecules belonging to a class of tricyclic quinoline inhibitors including QL47 (Figure 1A),31 a previously reported inhibitor of BTK, and other TEC-family kinases.13 Among the screening hits, QL47 exhibited the most potent antiviral activity, with IC90 value against DENV2 of 0.343 μM, and a desirable window between potency and cytotoxicity (Figure 1B). In order to determine whether BTK/BMX kinases are the pharmacologically relevant targets for viral replication, we further evaluated other known BTK/BMX kinase inhibitors, CGI-1746,24 Ibrutinib,16 and BMX-IN-1.25 However, none of these inhibitors showed comparable antiviral activity (Figure 2), indicating BTK/BMX are not likely to be the pharmacologically relevant targets of QL47. To further investigate the potent antiviral activity of QL47, we examined the inhibition of other unrelated viruses and observed that QL47 exhibited broad-spectrum antiviral activity against other Flaviviruses, such as West Nile virus (WNV), as well as against Enteroviruses, Vesiculoviruses, Pneumoviruses (Figure 3), among others. The potent inhibition of a variety of viruses by QL47 indicates that the molecular target of QL47 might be a host cell factor commonly utilized in the infectious cycle of these viruses. Despite the risk of side effects, targeting host cell proteins could be used as a complementary approach to direct-targeting of essential viral proteins.
Figure 1.
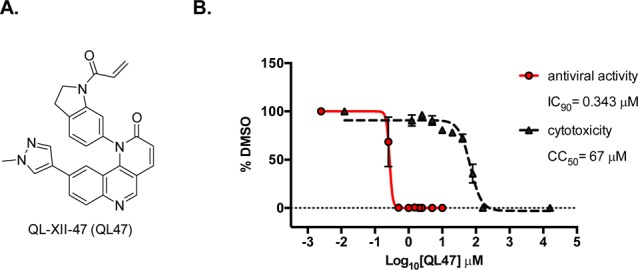
(A) Chemical structure of QL47. (B) Huh7 cells were infected with DENV2 then treated with a range of QL47 concentrations. Cell viability (black) and viral yield (red) were measured 24 hours later. CC50 and IC90 values—respectively, corresponding to the concentrations of QL47 that lead to 50% cytotoxicity and to a 1-log10 unit reduction in the number of infectious progeny virions produced—were determined by non-linear regression. Representative data (mean ± standard deviation of experimental duplicates) out of n ≥ 2 repeats are shown.
Figure 2.
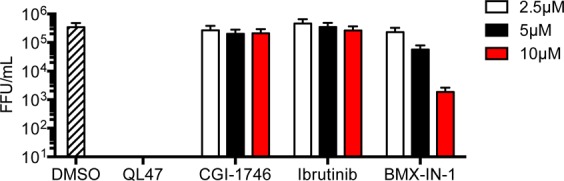
Huh7 cells were infected with DENV2 then treated with each of the indicated small molecules. The antiviral activity of each small molecule against DENV2 was measured by titration of the infectious particles that had been secreted to culture supernatants at 24h post-infection. A representative experiment out of n = 2 repeats is shown. Representative data (mean ± standard deviation of experimental duplicates) out of n = 2 repeats are shown. FFU, focus forming unit.
Figure 3.
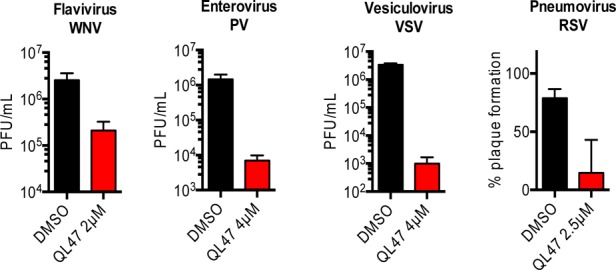
Huh7 cells were infected with West Nile virus (WNV), then treated with 2 μM of QL47 for 24h. Vero cells were infected with poliovirus (PV) or vesicular stomatitis virus (VSV) , then treated with 4 μM of QL47 for 12h and 6h, respectively. The infectious virus released to the supernatants were quantified by PFA. For respiratory syncytial virus (RSV), Vero cells were treated with 2.5 μM of QL47 post-infection. The number of plaque formed at 7 days post-infection were counted and the results were plotted relative to DMSO-treated samples. Representative data (mean ± standard deviation of experimental duplicates) out of n ≥ 2 repeats are shown. PFU, plaque forming unit.
Encouraged by the broad spectrum antiviral activity of QL47, we sought to elucidate the structure–activity relationships required to impart antiviral activity and to improve the poor pharmacokinetic properties of QL47 (Table S1). QL47 was originally developed as a covalent BTK inhibitor using structure-based design starting from the highly potent mTOR inhibitor, Torin2.26,27 Torin2 has been cocrystallized with mTOR, which revealed how this scaffold binds to the ATP-binding cleft (Figure S1).28 These tricyclic quinolines use the quinoline N nitrogen to form an essential hydrogen bond to the kinase hinge segment, while the two side chains extend toward the back and the front of the ATP binding site. The acrylamide arm of QL47 extends toward Cys481 of BTK located at the entrance of the ATP-binding site.13
We first examined whether QL47 acrylamide moiety, which is required for covalent inhibition of BTK, is essential for the antiviral activity. We used QL47R in which the acrylamide moiety was replaced with a nonreactive propyl amide functional group (Figure 4A). QL47R did not inhibit DENV2 at concentrations below 10 μM (Figure 4B), suggesting the acrylamide moiety is essential for antiviral activity. Consistent with the interpretation that QL47 covalently modifies its target, we also found that the full anti-DENV activity was recapitulated when cells were pretreated with QL47 for 6 h followed by extensive washing prior to DENV infection.31
Figure 4.
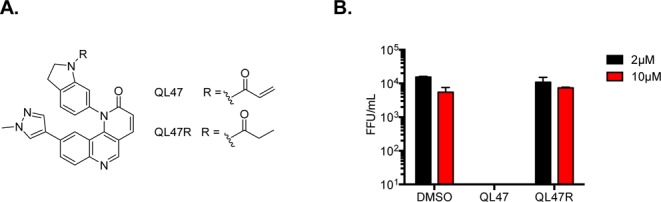
(A) Chemical structures of QL47 and QL47R. (B) Huh7 cells were infected with DENV2 then treated 2 μM with each of the indicated small molecules. The antiviral activity of each small molecule against DENV2 was measured by titration of the infectious particles that had been secreted to culture supernatants at 24h post-infection. Representative data (mean ± standard deviation of experimental duplicates) out of n = 2 repeats are shown.
We next extended the SAR study to other regions of QL47. The quinolinyl analogues of QL47 were prepared based on the reported synthetic routes for QL47,13 Torin1,29 and BEZ235,30 with variations of substituents used in the schemes (Scheme 1A). Starting material S1 was reacted with the appropriate aniline, followed by a series of reduction, oxidation, and ring-closing reactions to give rise to S2. Intermediate S2 was coupled with boronic acids, followed by reduction and acrylation to provide compounds 2–13. To prepare six-membered urea analogues, intermediate S3 underwent a reductive amination reaction, followed by urea formation using triphosgene. A similar sequence of reactions was followed to obtain compound 16 from intermediate S4. Five-membered urea analogues 17–18 were prepared using a similar strategy as has been reported for BEZ23530 from intermediate S5. We have also prepared naphthyl analogues of QL47 that lack a nitrogen that is required for a critical H-bond to the kinase hinge segment (Scheme 1B). Commercially available 7-chlorotetralone S7 was converted to aldehyde S8 via Vilsmeire–Haack-type reaction, followed by dehydrogenation with DBU. Buchwald coupling was performed to provide compound S9, which was subjected to a ring-closing reaction employing triethyl phosphonoacetate in basic conditions to provide common intermediate S10. Suzuki coupling with different aromatic boronic acids, followed by reduction of the nitro group provided S11, which was finally reacted with acryloyl chloride or crotonyl chloride to give rise to the naphthyl analogues 17–22 and YKL-04-085.
Scheme 1. Synthesis of Quinolinyl and Naphthyl Analogues of QL47.

Reagent and conditions: (a) 4-methyl-3-nitroaniline, dioxane, 100 °C; (b) NaBH4, EtOH, 0 °C to rt; (c) MnO2, DCM, rt; (d) triethyl phosphonoacetate, K2CO3, EtOH, 100 °C; (e) R-B(OH)2, Pd(PPh3)2Cl2, tBuXPhos, Na2CO3, dioxane, H2O, 80 °C; (f) SnCl2, EtOAc, 70 °C; (g) acryloyl chloride, THF/NaHCO3 1:1, 0 °C. (h) 2,4-dimethoxybenzylamine, Na(OAc)3BH, AcOH, THF, rt; (i) triphosgene, DIEA, DCM, 0 °C; (j) TFA, DCM, rt; (k) NaH, MeI, THF, 0 °C. (l) i. PBr3, DMF, CHCl3, 70 °C; ii. DDQ, toluene, 110 °C; (m) 4-methyl-3-nitroaniline, Pd2(dba)3, BINAP, Cs2CO3, toluene, 80 °C; (n) i. 4-bromocrotonyl chloride, DIEA, acetonitrile, 0 °C; ii. NHMe2, NMP, 50 °C.
To assess antiviral activity, all compounds were screened in an antiviral assay at concentrations of 2 and 10 μM. The levels of DENV production at 24 h postinfection/treatment were measured using a viral focus-forming assay, and the antiviral potency for each compound was expressed as a log-fold change relative to a DMSO treatment. We demonstrated that QL47 affects steady-state abundance of viral proteins in DENV-infected cells. To evaluate the effect of these drugs on viral protein accumulation, cells were transfected with a nonreplicative DENV2(GVD) replicon, which encodes a luciferase reporter gene.12 The levels of luciferase accumulation after treatment with 2 and 10 μM of all compounds was therefore assessed to evaluate their ability to inhibit the accumulation of DENV viral proteins in vivo. The cytotoxicity of compounds was assessed using a cell viability assay and expressed as the concentration of drug required to kill 50% of the cells (CC50). Finally, as an initial assessment of in cellulo pharmacokinetic properties of the compounds, the half-life of compounds (T1/2) when incubated with mouse liver microsome (MLM) is reported, comparing to QL47, which exhibits a MLM half-life of only 1.2 min.
We initially explored modifications to the methylpyrazole moiety of QL47 because it has previously been determined to be a critical determinant of kinase selectivity. A series of QL47 analogues were prepared including compounds such as 1 (Table 1), but they did not improve the poor mouse microsomal stability exhibited by QL47. We hypothesized that the metabolic instability might derive from the easily oxidized indoline moiety. To address this hypothesis, we turned our attention to the open-ring analogues of QL47 (Table 1, compounds 2–9). Methylpyrazole-substituted analogue 4 showed only modest improvement of microsomal stability but exhibited significantly reduced antiviral activity at 2 μM. Compounds 2 and 3, which bear other monocyclic aromatic moieties, showed antiviral activity, effects on DENV2(GVD) reporter replicon activity (a proxy for translation of the viral RNA), and microsomal stability comparable to 4. We next examined bicyclic aromatic substituents at this position in an effort to regain the antiviral activity exhibited by QL47. Although compounds 5 and 6 showed improved viral inhibition at 2 μM, the stability of the compounds decreased, probably due to the extended conjugated ring systems. Indeed, when nonconjugated ring systems were used (compounds 7–9), the stability was moderately improved. However, these compounds again completely lost viral inhibition even at 10 μM, suggesting a conjugated ring system is required to achieve antiviral activity.
Table 1. Structure–Activity Relationship of QL47 and Quinolinyl Analogues.
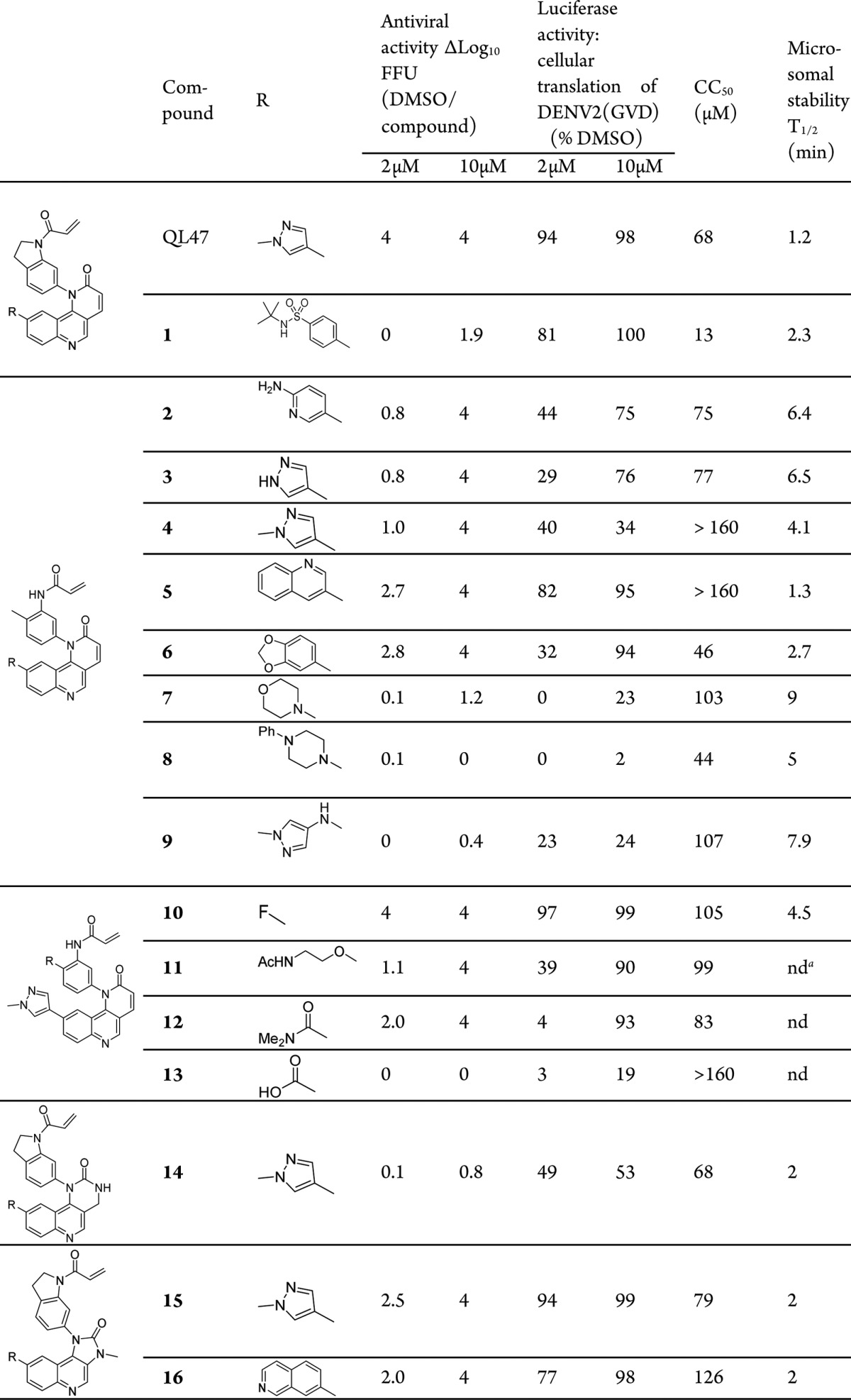
nd = not determined.
Next, we evaluated the effects of the substituents at the position ortho to the acrylamide (Table 1, compounds 10–13). Compound 10 demonstrated comparable potency to QL47, presumably due to the intramolecular hydrogen bonding of the ortho-fluoro group, which forms a pseudo-five-membered ring. Compound 10 also showed potent in cellulo translation inhibition and antiviral activity, but the half-life of the compound was only marginally improved to 4.5 min. The ortho-ether analogue 11 exhibited reduced antiviral activity and translation inhibition. The benzamide analogue 12 showed moderate activity at 2 μM, while the benzoic acid analogue 13 did not inhibit the virus at all, which might be due to the poor cellular permeability.
The unsuccessful effort to improve microsomal stability by varying methylpyrazole and acrylamide moieties prompted us to examine the tricyclic quinolinyl core of QL47 (Table 1, compounds 14–16). Interestingly, compounds containing a six-membered urea moiety, such as 14, only showed minimal antiviral activity at 10 μM, suggesting that this region of the molecule is a critical determinant for antiviral activity. We further evaluated five-membered methylurea analogues 15 and 16. Both displayed increased antiviral activity and inhibition of DENV(GVD) reporter replicon translation, probably due to the restoration of some conjugated features of the tricyclic core. However, none of these compounds exhibited substantially improved metabolic stability.
The SAR indicated that an extended conjugated system from methylpyrazole to acrylamide is required to preserve antiviral activity. Combining the hypothesis that the kinase targets of QL47, BTK/BMX, are not likely responsible for the effects on viral replication, we were interested to assess naphthyl analogues without the critical kinase hinge-interacting nitrogen atom. The antiviral activity, inhibition of reporter replicon translation, and MLM stability for naphthyl analogues are shown in Table 2. As expected, compounds 17–19 exhibited potent viral inhibition at 10 μM, suggesting that the molecular target of these compounds might not be a protein kinase. Among these compounds, 17, with a methylpyrazole substituent, demonstrated potency comparable to QL47, although its MLM half-life was only marginally improved to 4 min. We also tested hydrogenated analogues 20 and 21, which exhibited no antiviral activity, further confirming the necessity of a conjugated ring system to achieve viral inhibition. Intriguingly, when a dimethylamine tail was installed at the acrylamide site, the antiviral activity of compound 22 and YKL-04-085 was greatly improved at 2 μM. More promisingly, the MLM half-life of compound YKL-04-085 was significantly improved to 15.6 min.
Table 2. Structure–Activity Relationship of Naphthyl Analogues.
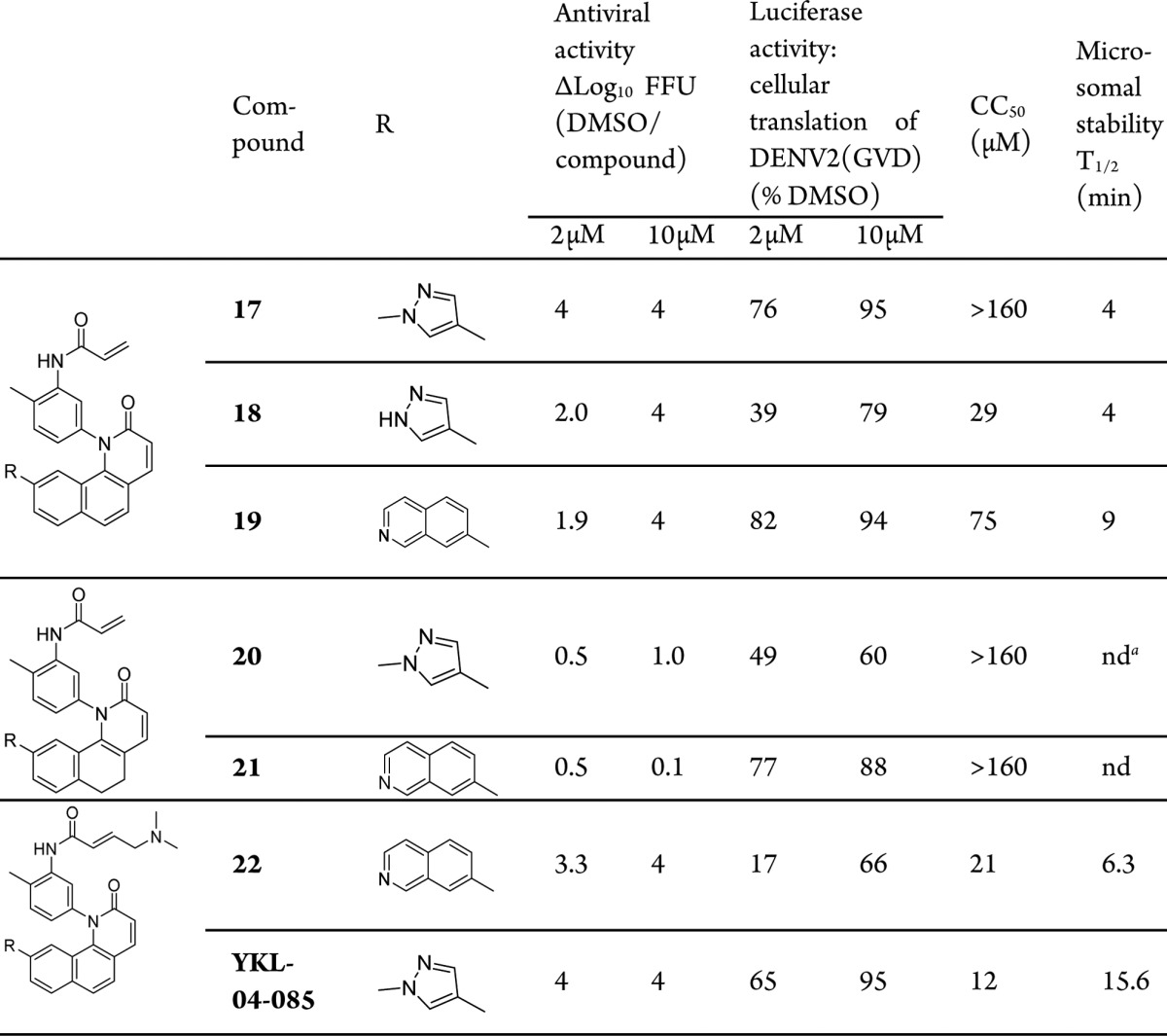
nd = not determined.
To further characterize compound YKL-04-085, it was screened against a panel of 468 kinases and disease relevant mutant variants using an active site-directed competition binding assay at 10 μM drug concentration (Figure 5A). Only a handful of kinases, such as PIMs and DDRs, exhibited more than 80% binding with YKL-04-085. We further determined the IC50 values for these kinases, as well as BTK/BMX (Figure 5B), and none of them showed significant inhibition by YKL-04-085, which confirmed YKL-04-085 is devoid of any kinase activity due to the lack of the critical hinge-contacting nitrogen. The anti-DENV2 IC90 value of YKL-04-085 was determined to be 0.555 μM, and the inhibitor exhibited more than 35-fold window between viral inhibition and cytotoxicity (Figure 5C). In addition, YKL-04-085 demonstrated good inhibition of translation of the DENV2(GVD) reporter replicon in cellulo (Figure 5D). Encouraged by the potent inhibition of DENV2 and improved metabolic stability of YKL-04-085, we then evaluated the mouse PK properties to determine whether it is a suitable candidate for future in vivo efficacy studies (Table 3). Although the compound displayed high clearance via intravenous (IV) route and low bioavailability, intraperitoneal (IP) administration gave acceptable plasma drug exposure. Furthermore, a 40 mg/kg once daily IP dosing regime was both well-tolerated and sufficient to maintain plasma concentrations of YKL-04-085 above the IC90 value measured in cellulo (Figure S2).
Figure 5.
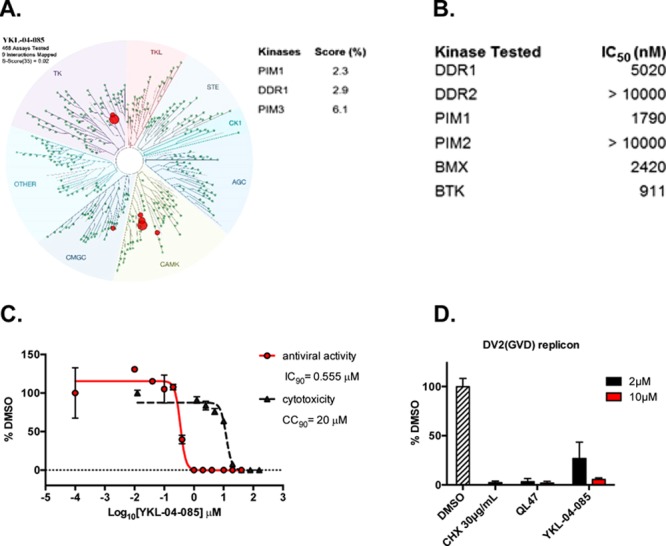
(A) DiscoverX KINOMEscan selectivity profile of YKL-04-085. Compound was screened at 10 μM concentration against 468 kinases and mutants. Scores for primary screen hits were reported as a percent of the DMSO control (% control). The lower the score, the lower the Kd is likely to be. Scores for kinase targets, which showed more than 80% binding, are shown in the table. (B) Enzymatic IC50s of YKL-04-085. IC50s against DDR1/2 were obtained with LanthaScreen binding assays, while IC50s against PIM1/2 and BTK/BMX were obtained with Z′-Lyte activity assays. (C) Huh7 cells were infected with DENV2 and then treated with a range of YKL-04-085 concentrations. Cell viability (black) and viral yield (red) were measured 24 hours later. CC90 and IC90 values—respectively, corresponding to the concentrations of QL47 that lead to 50% cytotoxicity and to a 1-log10 unit reduction in the number of infectious progeny virions produced—were determined by non-linear regression. Representative data (mean ± standard deviation of experimental duplicates) out of n ≥ 2 repeats are shown. (D) Huh7 cells were transfected with the in vitro-transcribed DENV2(GVD) reporter replicon RNA, and then immediately treated with the indicated compounds. Intracellular firefly luciferase activity was quantified at 6h post-transfection as a measure of successful RNA translation. Reporter activity was measured as a function of the luciferase activity obtained with the DMSO treated samples. Representative data (mean ± standard deviation of experimental duplicates) out of n =3 repeats are shown.
Table 3. In Vivo Pharmacokinetic Properties of YKL-04-085.
| route | dose (mg/kg) | T1/2 (h) | Tmax (h) | Cmax (ng/mL) | AUClast (ng·h/mL) | AUCinf (ng·h/mL) | CL (mL/min/kg) | Vss (L/kg) | F (%) |
|---|---|---|---|---|---|---|---|---|---|
| i.v. | 2 | 1.07 | 461.71 | 161.33 | 167.28 | 199.26 | 9.58 | ||
| p.o. | 10 | 1.53 | 1.00 | 40.41 | 95.40 | 117.89 | 12 | ||
| i.p. | 10 | 0.25 | 786.78 | 1112.65 | 1213.28 | ||||
| i.p. | 50 | 0.08 | 4505.75 | 8222.94 | 8554.01 |
In conclusion, we report the identification, characterization, and preliminary structure–activity relationships (SAR) for a novel class of covalent inhibitors of DENV. Preliminary mechanistic studies of QL47 suggest that its antiviral activity is correlated with effects on the steady-state abundance of viral proteins, likely due to an effect on translation of the viral RNA genome. The correlation of anti-DENV activity with effects on translation of the DENV2(GVD) reporter replicon in the SAR studies reported here provide further support that effects on translation of the viral genome are a key contributor to the antiviral activity of these compounds. Nevertheless, there are several outliers that fall outside this correlation, indicating that additional mechanisms may contribute to the overall antiviral activity of these compounds. While the precise molecular target of this series of compounds is currently under investigation, KINOMEscan of QL47 and YKL-04-085 suggests that protein kinases are unlikely to be targets. Lead optimization yielded compound YKL-04-085, which might be an appropriate candidate for further in vivo studies to test efficacy of this class of compounds. Further optimization of the lead compound, target identification, and in vivo efficacy studies are the subject of ongoing research.
Acknowledgments
We gratefully acknowledge Dr. Tinghu Zhang for the synthesis of relevant tool compounds and helpful discussions; and Dr. Michael Cameron at the Scripps Research Institute for conducting in vitro mouse microsome study and in vivo tolerability study. This work was funded by the Linde program in chemical biology.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00008.
Supplemental figures, tables, materials and methods, experimental procedures, and compound characterization (PDF)
Author Contributions
∥ These authors contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Kuno G.; Chang G.-J. J.; Tsuchiya K. R.; Karabatsos N.; Cropp C. B. Phylogeny of the Genus Flavivirus. J. Virol. 1998, 72, 73–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt S.; Gething P. W.; Brady O. J.; Messina J. P.; Farlow A. W.; Moyes C. L.; Drake J. M.; Brownstein J. S.; Hoen A. G.; Sankoh O.; Myers M. F.; George D. B.; Jaenisch T.; Wint G. R. W.; Simmons C. P.; Scott T. W.; Farrar J. J.; Hay S. I. The global distribution and burden of dengue. Nature 2013, 496, 504–507. 10.1038/nature12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization . Dengue and dengue haemorrhagic fever. http://www.who.int/mediacentre/factsheets/fs117/en/.
- Lim S. P.; Wang Q.-Y.; Noble C. G.; Chen Y.-L.; Dong H.; Zou B.; Yokokawa F.; Nilar S.; Smith P.; Beer D.; Lescar J.; Shi P.-Y. Ten years of dengue drug discovery: Progress and prospects. Antiviral Res. 2013, 100, 500–519. 10.1016/j.antiviral.2013.09.013. [DOI] [PubMed] [Google Scholar]
- Kiser J. J.; Flexner C. Direct-Acting Antiviral Agents for Hepatitis C Virus Infection. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 427–449. 10.1146/annurev-pharmtox-011112-140254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber L.; Welzel T. M.; Zeuzem S. New therapeutic strategies in HCV: polymerase inhibitors. Liver Int. 2013, 33, 85–92. 10.1111/liv.12068. [DOI] [PubMed] [Google Scholar]
- Anderson J.; Schiffer C.; Lee S.-K.; Swanstrom R.. Viral Protease Inhibitors. In Antiviral Strategies; Kräusslich H.-G., Bartenschlager R., Eds.; Springer: Berlin Heidelberg, 2009; Vol. 189, pp 85–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeks S. G.; Smith M.; Holodniy M.; Kahn J. O. Hiv-1 protease inhibitors: A review for clinicians. JAMA, J. Am. Med. Assoc. 1997, 277, 145–153. 10.1001/jama.277.2.145. [DOI] [PubMed] [Google Scholar]
- Tan S.-L.; Ganji G.; Paeper B.; Proll S.; Katze M. G. Systems biology and the host response to viral infection. Nat. Biotechnol. 2007, 25, 1383–1389. 10.1038/nbt1207-1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu J. J. H.; Yang P. L. c-Src protein kinase inhibitors block assembly and maturation of dengue virus. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 3520–3525. 10.1073/pnas.0611681104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carocci M.; Hinshaw S. M.; Rodgers M. A.; Villareal V. A.; Burri D. J.; Pilankatta R.; Maharaj N. P.; Gack M. U.; Stavale E. J.; Warfield K. L.; Yang P. L. The Bioactive Lipid 4-Hydroxyphenyl Retinamide Inhibits Flavivirus Replication. Antimicrob. Agents Chemother. 2015, 59, 85–95. 10.1128/AAC.04177-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wispelaere M.; Carocci M.; Liang Y.; Liu Q.; Sun E.; Vetter M. L.; Wang J.; Gray N. S.; Yang P. L. Discovery of host-targeted covalent inhibitors of dengue virus. Antiviral Res. 2017, 139, 171–179. 10.1016/j.antiviral.2016.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H.; Wang W.; Liu F.; Weisberg E. L.; Tian B.; Chen Y.; Li B.; Wang A.; Wang B.; Zhao Z.; McMillin D. W.; Hu C.; Li H.; Wang J.; Liang Y.; Buhrlage S. J.; Liang J.; Liu J.; Yang G.; Brown J. R.; Treon S. P.; Mitsiades C. S.; Griffin J. D.; Liu Q.; Gray N. S. Discovery of a Potent, Covalent BTK Inhibitor for B-Cell Lymphoma. ACS Chem. Biol. 2014, 9, 1086–1091. 10.1021/cb4008524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansarah-Sobrinho C.; Nelson S.; Jost C. A.; Whitehead S. S.; Pierson T. C. Temperature-dependent production of pseudoinfectious dengue reporter virus particles by complementation. Virology 2008, 381, 67–74. 10.1016/j.virol.2008.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W.; Ercan D.; Chen L.; Yun C.-H.; Li D.; Capelletti M.; Cortot A. B.; Chirieac L.; Iacob R. E.; Padera R.; Engen J. R.; Wong K.-K.; Eck M. J.; Gray N. S.; Janne P. A. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 2009, 462, 1070–1074. 10.1038/nature08622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F.; Zhang X.; Weisberg E.; Chen S.; Hur W.; Wu H.; Zhao Z.; Wang W.; Mao M.; Cai C.; Simon N. I.; Sanda T.; Wang J.; Look A. T.; Griffin J. D.; Balk S. P.; Liu Q.; Gray N. S. Discovery of a Selective Irreversible BMX Inhibitor for Prostate Cancer. ACS Chem. Biol. 2013, 8, 1423–1428. 10.1021/cb4000629. [DOI] [PubMed] [Google Scholar]
- Hur W.; Velentza A.; Kim S.; Flatauer L.; Jiang X.; Valente D.; Mason D. E.; Suzuki M.; Larson B.; Zhang J.; Zagorska A.; DiDonato M.; Nagle A.; Warmuth M.; Balk S. P.; Peters E. C.; Gray N. S. Clinical stage EGFR inhibitors irreversibly alkylate Bmx kinase. Bioorg. Med. Chem. Lett. 2008, 18, 5916–5919. 10.1016/j.bmcl.2008.07.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honigberg L. A.; Smith A. M.; Sirisawad M.; Verner E.; Loury D.; Chang B.; Li S.; Pan Z.; Thamm D. H.; Miller R. A.; Buggy J. J. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 13075–13080. 10.1073/pnas.1004594107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W.; Hur W.; McDermott U.; Dutt A.; Xian W.; Ficarro S. B.; Zhang J.; Sharma S. V.; Brugge J.; Meyerson M.; Settleman J.; Gray N. S. A Structure-Guided Approach to Creating Covalent FGFR Inhibitors. Chem. Biol. 2010, 17, 285–295. 10.1016/j.chembiol.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski N.; Zhang T.; Rahl P. B.; Abraham B. J.; Reddy J.; Ficarro S. B.; Dastur A.; Amzallag A.; Ramaswamy S.; Tesar B.; Jenkins C. E.; Hannett N. M.; McMillin D.; Sanda T.; Sim T.; Kim N. D.; Look T.; Mitsiades C. S.; Weng A. P.; Brown J. R.; Benes C. H.; Marto J. A.; Young R. A.; Gray N. S. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 2014, 511, 616–620. 10.1038/nature13393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T.; Kwiatkowski N.; Olson C. M.; Dixon-Clarke S. E.; Abraham B. J.; Greifenberg A. K.; Ficarro S. B.; Elkins J. M.; Liang Y.; Hannett N. M.; Manz T.; Hao M.; Bartkowiak B.; Greenleaf A. L.; Marto J. A.; Geyer M.; Bullock A. N.; Young R. A.; Gray N. S. Covalent targeting of remote cysteine residues to develop CDK12 and CDK13 inhibitors. Nat. Chem. Biol. 2016, 12, 876–884. 10.1038/nchembio.2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q.; Sabnis Y.; Zhao Z.; Zhang T.; Buhrlage; Sara J.; Jones; Lyn H.; Gray; Nathanael S. Developing Irreversible Inhibitors of the Protein Kinase Cysteinome. Chem. Biol. 2013, 20, 146–159. 10.1016/j.chembiol.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanning B. R.; Whitby L. R.; Dix M. M.; Douhan J.; Gilbert A. M.; Hett E. C.; Johnson T. O.; Joslyn C.; Kath J. C.; Niessen S.; Roberts L. R.; Schnute M. E.; Wang C.; Hulce J. J.; Wei B.; Whiteley L. O.; Hayward M. M.; Cravatt B. F. A road map to evaluate the proteome-wide selectivity of covalent kinase inhibitors. Nat. Chem. Biol. 2014, 10, 760–767. 10.1038/nchembio.1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Paolo J. A.; Huang T.; Balazs M.; Barbosa J.; Barck K. H.; Bravo B. J.; Carano R. A. D.; Darrow J.; Davies D. R.; DeForge L. E.; Diehl L.; Ferrando R.; Gallion S. L.; Giannetti A. M.; Gribling P.; Hurez V.; Hymowitz S. G.; Jones R.; Kropf J. E.; Lee W. P.; Maciejewski P. M.; Mitchell S. A.; Rong H.; Staker B. L.; Whitney J. A.; Yeh S.; Young W. B.; Yu C.; Zhang J.; Reif K.; Currie K. S. Specific Btk inhibition suppresses B cell– and myeloid cell–mediated arthritis. Nat. Chem. Biol. 2011, 7, 41–50. 10.1038/nchembio.481. [DOI] [PubMed] [Google Scholar]
- Tan L.; Akahane K.; McNally R.; Reyskens K. M. S. E.; Ficarro S. B.; Liu S.; Herter-Sprie G. S.; Koyama S.; Pattison M. J.; Labella K.; Johannessen L.; Akbay E. A.; Wong K.-K.; Frank D. A.; Marto J. A.; Look T. A.; Arthur J. S. C.; Eck M. J.; Gray N. S. Development of Selective Covalent Janus Kinase 3 Inhibitors. J. Med. Chem. 2015, 58, 6589–6606. 10.1021/acs.jmedchem.5b00710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q.; Wang J.; Kang S. A.; Thoreen C. C.; Hur W.; Ahmed T.; Sabatini D. M.; Gray N. S. Discovery of 9-(6-Aminopyridin-3-yl)-1-(3-(trifluoromethyl)phenyl)benzo[h][1,6]naphthyridin-2(1H)-one (Torin2) as a Potent, Selective, and Orally Available Mammalian Target of Rapamycin (mTOR) Inhibitor for Treatment of Cancer. J. Med. Chem. 2011, 54, 1473–1480. 10.1021/jm101520v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q.; Xu C.; Kirubakaran S.; Zhang X.; Hur W.; Liu Y.; Kwiatkowski N. P.; Wang J.; Westover K. D.; Gao P.; Ercan D.; Niepel M.; Thoreen C. C.; Kang S. A.; Patricelli M. P.; Wang Y.; Tupper T.; Altabef A.; Kawamura H.; Held K. D.; Chou D. M.; Elledge S. J.; Janne P. A.; Wong K.-K.; Sabatini D. M.; Gray N. S. Characterization of Torin2, an ATP-Competitive Inhibitor of mTOR, ATM, and ATR. Cancer Res. 2013, 73, 2574–2586. 10.1158/0008-5472.CAN-12-1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H.; Rudge D. G.; Koos J. D.; Vaidialingam B.; Yang H. J.; Pavletich N. P. mTOR kinase structure, mechanism and regulation. Nature 2013, 497, 217–223. 10.1038/nature12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q.; Chang J. W.; Wang J.; Kang S. A.; Thoreen C. C.; Markhard A.; Hur W.; Zhang J.; Sim T.; Sabatini D. M.; Gray N. S. Discovery of 1-(4-(4-Propionylpiperazin-1-yl)-3-(trifluoromethyl)phenyl)-9-(quinolin-3-yl)benzo[h][1,6]naphthyridin-2(1H)-one as a Highly Potent, Selective Mammalian Target of Rapamycin (mTOR) Inhibitor for the Treatment of Cancer. J. Med. Chem. 2010, 53, 7146–7155. 10.1021/jm101144f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stowasser F.; Baenziger M.; Garad S. D.. Preparation of salts and crystalline forms of 2-methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]-propionitrile and its use as a drug. WO 2008/064093, May 29, 2008.
- de Wispelaere M.; LaCroix A. J.; Yang P. L. The Small Molecules AZD0530 and Dasatinib Inhibit Dengue Virus RNA Replication via Fyn Kinase. J. Virol. 2013, 87, 7367–7381. 10.1128/JVI.00632-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.