

Abstract
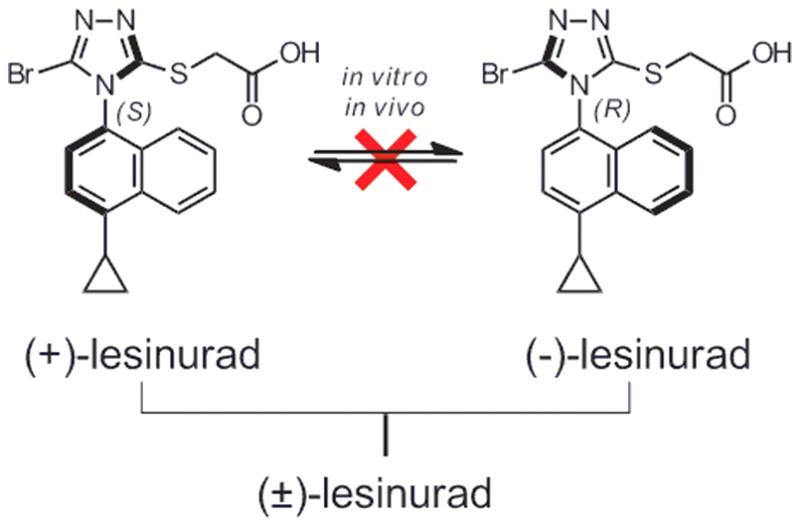
(+)- and (−)-Lesinurad were isolated as atropisomers from racemic lesinurad for the first time. No interconversion was observed between the two atropisomers under various conditions tested. The two atropisomers showed significant differences in hURAT1 highly expressed HEK293 cell-based inhibition assays, monkey pharmacokinetic studies, and in vitro human recombinant CYP2C9 stability studies. It was speculated that (+)-lesinurad might offer a better hyperuricemia/gout therapy than (−)-lesinurad or the racemate.
Keywords: Atropisomer, lesinurad, gout, hyperuricemia
Gout is a crystal-deposition arthritis caused by supersaturation and precipitation of monosodium urate (MSU) in tissues or joints, which affects over 20 million people worldwide.1−5 Hyperuricemia is the biological fundamental of gout.6 It is a purine related metabolic disease defined as serum uric acid (sUA) level >6.8 mg/dL.7 The standard medical treatment of hyperuricemia is urate-lowing therapy (ULT) with a goal to lower sUA level below 6 mg/dL.4,5,8 Several ULTs are available, including xanthine-oxidase inhibitors (XOIs, e.g., allopurinol and febuxostat) to reduce sUA formation, and uric acid reabsorption inhibitors (URIs, e.g., benzbromarone and probenecid) to increase the excretion of uric acid. In recent years, selectively blocking hURAT1 (human uric acid transporter 1, SLC22A12) to reduce reabsorption of uric acid at kidney tubules gained attention for the treatment of hyperuricemia and gout.9−12 Among them, Zurampic (lesinurad, Scheme 1), a first-in-class selective hURAT1 inhibitor, was approved by the FDA as an oral therapy for hyperuricemia and gout in late 2015. The approved and recommended dose of Zurampic is 200 mg once daily in combination with allopurinol or febuxostat.13,14 The 400 mg dose of lesinurad, albeit demonstrating good therapeutic efficacy when applied on its own in phase III clinical studies, was not approved due to several unacceptable side effects, including renal-related adverse events and serum creatinine elevations.15
Scheme 1. Discovery of Atropisomers of Lesinurad.

The chemical structure of lesinurad was first reported in 2006 and was composed of a naphthalene ring linked with a triazole.16,17 Based on the structure, the triazole ring should rotate freely along the C–N bond; however, the thio-acetic acid moiety and bromine atom might hinder the free rotation due to steric bulk. This gave us a hint that the less noticed axial chirality possibly existed in lesinurad. A quick chiral supercritical fluid chromatography (SFC) analysis of lesinurad confirmed our suspicion, and two identical atropisomeric peaks were observed. Considering that atropisomers might have different pharmacological and pharmacokinetic properties, we were intrigued to further study the two atropisomers of lesinurad in details.
Herein, we report the discovery, characterization, and assessment of (+)-lesinurad and (−)-lesinurad (Scheme 1) on their stabilities, physicochemical properties, and in vitro and in vivo ADME/PKs. To the best of our knowledge, the existence of atropisomers in lesinurad was not previously described.
Atropisomers are conformational isomers, which epimerize or racemize via rotation along the bond axis.18 Atropisomeric compounds represent an interesting but not fully studied family despite its importance to drug discovery and development.19,20 A recent review from LaPlante and co-workers proposed that atropisomers could be classified into three groups based on rotational energy barriers and racemization rates (t1/2) (Class 1, rapid equilibration with t1/2 less than minutes; Class 2, moderate rate of equilibration with t1/2 from hours to days; Class 3, very slow equilibration with t1/2 in years).18,21 Since atropisomers in Class 3 are as stable as classical chiral compounds, they could be isolated and treated as single enantiomers.
In order to find out whether the two atropisomers of lesinurad could easily interconvert under various conditions, chiral SFC separation was tried on either lesinurad itself or lesinurad-ester 1 as shown in Scheme 1, and (+)/(−)-lesinurad were obtained in good yields. The X-ray crystal structures of both compounds 1A and 1B were obtained (Figure 1) allowing confirmation of the absolute configurations of both (+)/(−)-lesinurad.
Figure 1.

X-ray structures of compound 1A and 1B.
The stability studies of (+)/(−)-lesinurad in solid form, in solvent, and in plasma were performed. It was found that these two atropisomers were very stable in their solid states, and no significant purity reduction or racemization was observed after storage at ambient temperature for over 80 days. (+)/(−)-Lesinurad was also found to be very stable in solutions (ethanol/37 °C and DMSO/80 °C for 4 days), and no interconversion was observed in these two solvents under testing conditions, even after heating in DMSO at 120 °C for 24 h. Additionally, stabilities of (+)/(−)-lesinurad in plasma were also measured, and both isomers remained unchanged after incubation in human plasma/37 °C or SD-rat plasma/37 °C up to 2 h.
Lesinurad was reported to inhibit hURAT1 transporter with IC50 value of 7.3 μM in hURAT1 highly expressed HEK293 cell lines.15 The same assay was applied to evaluate the inhibition activities of the two atropisomers. After multiple tests (n = >7), the IC50 values for (+)/(−)/(±)-lesinurad inhibition of [14C]-uric acid uptake by hURAT1 were determined to be 4.4, 15.1, and 9.6 μM, respectively (Table 1). (+)-Lesinurad showed about 3-fold boost in potency as compared to (−)-lesinurad and (±)-lesinurad. The two atropisomers together with lesinurad were also evaluated for permeability, in vitro microsome stability, cytochrome P450 enzyme isoform, and hERG inhibitions. The two atropisomers showed comparable physiochemical and biological properties in these in vitro tests (Table 1).
Table 1. Physiochemical and Biological Properties Comparison of (+)/(−)-Lesinurad with (±)-Lesinurad.
| physiochemical and biological properties | (±)-lesinurad | (−)-lesinurad | (+)-lesinurad | |
|---|---|---|---|---|
| hURAT1 IC50 (μM) | 9.6 ± 1.7a | 15.1 ± 3.1b | 4.4 ± 1.0b | |
| CYP IC50 (μM) | 1A2 | 17.3 | 39.3 | 21.9 |
| 2C9 | >100 | 78.7 | 31.2 | |
| 2C19 | 61.7 | 10.6 | 10.8 | |
| 2D6 | 32.6 | 92.1 | >100 | |
| 3A4 | 74.4 | 32.4 | 31 | |
| hERG IC50 (μM) | >30 | >30 | >30 | |
| MMS (mL/min/kg) | human | <9.5 | <9.5 | <9.5 |
| rat | <17.3 | <17.3 | <17.3 | |
| mouse | <38.0 | <38.0 | <38.0 | |
| dog | <13.8 | <13.8 | <13.8 | |
| monkey | <13.0 | <13.0 | 18.7 | |
| MDR1-MDCK Papp (10–6 cm/s) | A–B | 1.34 | 1.29 | 1.26 |
| B–A | 1.98 | 1.84 | 1.62 | |
n = 17.
n = 5.
These two atropisomers were stable in vitro in human plasma and SD-rat plasma, and also seemed quite stable in human in vitro microsome stability studies. In order to find out whether interconversion could happen in vivo and whether (+)/(−)-lesinurad could behave differently in vivo, pharmacokinetic studies of these two atropisomers were carried out in animals. In the first study, rats in two groups were administrated with (+)-lesinurad and (−)-lesinurad, respectively, and plasma samples were analyzed using chiral column detecting (+)-lesinurad and (−)-lesinurad simultaneously (Table 2). Following a single IV or PO dose of (+)-lesinurad, the other isomer (−)-lesinurad was not detected in all plasma samples at different time points. Similarly, (+)-lesinurad was not detected after a single IV or PO dose of (−)-lesinurad neither. This result confirmed that there was no enzyme-catalyzed in vivo interconversion between these two atropisomers in rats.
Table 2. Rat PKa Profile of (+)-Lesinurad and (−)-Lesinurad.
| parameters | (−)-lesinurad | (+)-lesinurad | (−)/(+)-index | |
|---|---|---|---|---|
| IV | doseb (mg/kg) | 2 | 2 | |
| AUC0-inf (μM·h) | 14.4 ± 1.5 | 15.0 ± 1.0 | 1.0 | |
| CLp (mL/min/kg) | 5.8 ± 0.6 | 5.5 ± 0.4 | 1.1 | |
| PO | dosec (mg/kg) | 10 | 10 | |
| AUC0-inf (μM·h) | 46.9 ± 11.5 | 32.7 ± 13.4 | 1.4 | |
Rat PK data is mean ± SD, n = 3.
IV formulation: 1 mg/mL in water, pH = 8, nearly clear solution with few particles before filter.
PO formulation: 2 mg/mL in water, pH = 8, nearly clear solution with few particles.
By closer examination of the PK data, it was found that the two atropisomers showed similar apparent clearance and plasma exposure in the IV dosing groups. However, (−)-lesinurad afforded about 1.2-fold higher plasma exposure than (+)-lesinurad upon PO dosing. This observation indicated some potential in vivo difference between the two isomers, although the result could also arise from experimental deviations.
To make a more informative comparison, a second study was carried out to measure the plasma concentration of (+)-lesinurad and (−)-lesinurad following a single IV dose of racemic (±)-lesinurad at 2 mg/kg or oral administration at 10 mg/kg (Table 3). Interestingly, the concentration of (−)-lesinurad was always 10–20% higher than that of (+)-lesinurad in all plasma samples collected at different time points. As a result, ratios of exposure levels between (−)- and (+)-lesinurad were 1.1 and 1.2 in IV and PO groups, respectively. The in vivo differentiation could be rationalized by slight differences in the absorption and metabolism of the two isomers, which were not distinguishable in in vitro experiments.
Table 3. Rat PKa Profile of (+)/(−)-Lesinurad after a Single Dosing of (±)-Lesinurad.
| parameters | (−)-lesinurad | (+)-lesinurad | (−)/(+)-index | |
|---|---|---|---|---|
| IV | doseb | 2 mg/kg of (±)-lesinurad | ||
| AUC0-inf (μM·h) | 9.5 ± 2.5 | 8.9 ± 1.6 | 1.1 | |
| CLp (mL/min/kg) | 9.0 ± 2.1 | 9.4 ± 1.5 | 1.0 | |
| PO | dosec | 10 mg/kg of (±)-lesinurad | ||
| AUC0-inf (μM·h) | 41.9 ± 10.2 | 35.6 ± 8.0 | 1.2 | |
Rat PK data is mean ± SD, n = 3.
IV formulation: 1 mg/mL in water, pH = 8–9, clear solution.
PO formulation: 2 mg/mL in water, pH = 8–9, clear solution.
With this interesting finding, we further examined the pharmacokinetic behavior of (+)-lesinurad and (−)-lesinurad in large animals. Following a single IV and PO dose of racemic lesinurad to male Cynomolgus monkeys, the two isomers were analyzed separately. In the third PK study, the urine samples collected during different time periods were also analyzed since around 20% of (±)-lesinurad was reported to be excreted unchanged to urine and thus gave efficacy.15 Surprisingly, concentrations of (−)-lesinurad in both plasma and urine at all time points/periods were found to be significantly higher than those of (+)-lesinurad in both IV and PO dosing groups (Table 4). The plasma concentration–time curves of (+)-lesinurad and (−)-lesinurad are shown in Figure 2. In the IV dosing group, the apparent clearance of (−)-lesinurad was lower than that of (+)-lesinurad resulting in a 1.4-fold higher plasma exposure for (−)-lesinurad. The mean urine concentration of (−)-lesinurad was also 1.9-fold higher than that of (+)-lesinurad. In the PO dosing group, the ratios of (−)-lesinurad vs (+)-lesinurad for plasma exposure and mean urine concentration were 3.5- and 4.0-fold. Taken together, these results strongly suggested that these two atropisomers were absorbed, metabolized, and excreted differently in monkeys.
Table 4. Monkey PKa Profile of (+)/(−)-Lesinurad after a Single Dosing of (±)-Lesinurad.
| parameters | (−)-lesinurad | (+)-lesinurad | (−)/(+)-index | |
|---|---|---|---|---|
| IV | doseb | 2 mg/kg of (±)-lesinurad | ||
| AUC0-inf (μM·h) | 14.5 ± 3.3 | 10.0 ± 2.8 | 1.4 | |
| CLp (mL/min/kg) | 5.9 ± 1.5 | 8.8 ± 2.7 | 0.7 | |
| C0–24h,urine (μM) | 5.4 ± 0.9 | 2.8 ± 1.4 | 1.9 | |
| PO | dosec | 10 mg/kg of (±)-lesinurad | ||
| AUC0-inf (μM·h) | 99.4 ± 29.7 | 28.2 ± 19.2 | 3.5 | |
| C0–24h,urine (μM) | 153.3 ± 93.5 | 38.0 ± 20.7 | 4.0 | |
Monkey PK data is mean ± SD, n = 3.
IV formulation: 2.00 mg/mL in DMSO/PEG400/H2O(5/40/55), clear solution.
PO formulation: 2 mg/mL in DMSO/0.5%MC(5/95), suspension.
Figure 2.
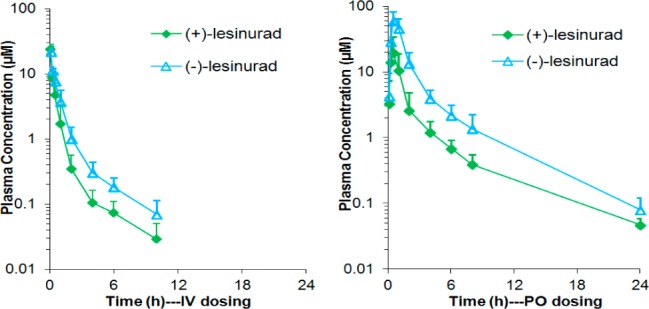
Plasma Concentration–Time Curve of (+)/(−)-Lesinurad in Monkey PK Study.
Considering the potential drug–drug interactions between the two isomers in absorption, metabolism, and excretion, we carried out a fourth pharmacokinetic study dosing them separately in monkeys. As it turned out, the ratios of (−)-lesinurad vs (+)-lesinurad for plasma exposure and mean urine concentration were 1.4- and 1.0-fold for IV dosing group and 2.2- and 2.5-fold for PO dosing group as shown in Table 5. Although smaller ratios were observed in the PO group compared to the codosing study, (−)-lesinurad still demonstrated significantly higher plasma exposure and urine concentration than (+)-lesinurad in monkeys.
Table 5. Monkey PKa Profile of (+)-Lesinurad and (−)-Lesinurad.
| parameters | (−)-lesinurad | (+)-lesinurad | (−)/(+)-index | |
|---|---|---|---|---|
| IV | doseb (mg/kg) | 2 | 2 | |
| AUC0-inf (μM·h) | 26.7 ± 8.8 | 19.2 ± 7.3 | 1.4 | |
| CLp (mL/min/kg) | 3.3 ± 1.0 | 4.7 ± 1.5 | 0.7 | |
| C0–24h,urine (μM) | 16.3 ± 7.4 | 15.7 ± 3.3 | 1.0 | |
| PO | dosec (mg/kg) | 10 | 10 | |
| AUC0-inf (μM·h) | 83.9 ± 12.1 | 37.3 ± 6.9 | 2.2 | |
| C0–24h,urine (μM) | 120.1 ± 82.7 | 47.8 ± 20.3 | 2.5 | |
Monkey PK data is mean ± SD, n = 3.
IV formulation: 2.00 mg/mL in DMSO/PEG400/H2O(5/40/55), clear solution.
PO formulation: 2 mg/mL in DMSO/0.5%MC(5/95), suspension.
Based on the animal pharmacokinetic data, (−)-lesinurad showed much higher plasma/urine exposures than (+)-lesinurad, especially in monkeys. It was reasonable to suspect that the two atropisomers behave differently in human. According to the NDA reports of lesinurad, human liver cytochrome P450 isozyme CYP2C9 played a major role in the formation of oxidative metabolites while other CYPs contributed minimally for the metabolism of lesinurad in vitro.15 In order to predict which atropisomer would be more stable in human, we evaluated the stabilities of (+)-lesinurad and (−)-lesinurad in pooled human recombinant CYP2C9. Interestingly, it was found that (−)-lesinurad was metabolized faster by CYP2C9 with t1/2 around 90.3 min (Table 6), while (+)-lesinurad showed prolonged t1/2 over 145 min. From the CYP2C9 stability data, it was anticipated that (+)-lesinurad might be metabolically more stable in human than (−)-lesinurad and might give higher exposure in plasma and urine. This would be different from what we had observed in animals. As described early in this letter, (+)-lesinurad was also identified to be about 3-fold more potent than (−)-lesinurad. Therefore, it was suspected that (+)-lesinurad might demonstrate better clinical efficacy than (−)-lesinurad and (±)-lesinurad. Moreover, from a Met-ID of the two isomers, three mono-oxidative metabolites (P+16; m/z 420) were detected for (−)-lesinurad, while only one mono-oxidative metabolite (P+16; m/z 420) was detected for (+)-lesinurad. It has been reported that the clinical side effects observed in lesinurad might be attributed to the oxidative metabolites.15 Taken together, (+)-lesinurad might demonstrate a larger therapeutic index than (−)-lesinurad or the racemate.
Table 6. Microsome Stability of (+)/(−)-Lesinurad in Recombinant Human CYP2C9.
| compound | t1/2a | CLintb | stability 1c (%) | stability 2d (%) |
|---|---|---|---|---|
| (−)-lesinurad | 90.3 | 0.04 | 61.8 | 110.5 |
| (+)-lesinurad | >145.0 | <0.02 | 74.9 | 108.9 |
| diclofenac | 12.3 | 5.6 | 3.3 | 112.2 |
Value was reported in min.
Value was reported in μL/min/pmol.
Value was reported as the % remaining at T = 60 min in the presence of cofactor NADPH.
Value was reported as the % remaining at T = 60 min in the absence of cofactor NADPH.
Next, with the effort to develop a readily accessible way to prepare (+)-lesinurad and (−)-lesinurad without costly SFC separation, a chiral synthetic route was developed as described in Scheme 2. Acylation of (R)-1-phenylethanol 2 with 2-bromoacetyl chloride 3 provided (R)-1-phenylethyl-2-bromoacetate 4. S-alkylation of thio-trizole 5 gave intermediate 6. Further bromination yielded key intermediate 7, and 7A and 7B could be separated by column. Subsequent hydrolysis of 7A and 7B gave (+)-lesinurad and (−)-lesinurad in high yields. Interestingly, we did not observe the existence of stable atropisomers in compound 6 under various SFC conditions, which indicated the observed axial chirality of (+)-lesinurad and (−)-lesinurad was maintained by the introduction of steric bulky bromine atom.
Scheme 2. Chiral Synthesis of (+)/(−)-Lesinurad.

In conclusion, existence of stable atropisomers was discovered in lesinurad. (+)-/(−)-Lesinurad were isolated and fully characterized. No interconversion was observed between the two isomers under heat, in solutions, and in in vivo pharmacokinetic studies. Although the two atropisomers showed comparable data in most of the physiochemical properties, (+)-lesinurad showed significant higher in vitro inhibition potency against hURAT1 than (−)-lesinurad. Furthermore, the two atropisomers showed significantly different pharmacokinetic profiles in Cynomolgus monkeys. In vitro human recombinant CYP2C9 stability study indicated that (+)-lesinurad was more stable than (−)-lesinurad. Considering the significant difference between the two isomers, it was speculated that the (+)-lesinurad might offer a better hyperuricemia/gout therapy than (−)-lesinurad or the racemate.
Acknowledgments
We acknowledge Dr. Yikai Wang for the helpful discussions and review of this manuscript.
Glossary
ABBREVIATIONS
- hURAT1
human uric acid transport 1
- PK
pharmacokinetics
- CYP
Cytochrome P450
- hERG
human ether-a-go-go related gene
- MMS
microsomes metabolic stability
- Met-ID
metabolism identification
- IV
intravenous
- PO
per oral
- AUC
area under curve
- CLp
plasma clearance
- CLint
intrinsic clearance
- SD rat
Sprague–Dawley rat
- NADPH
β-nicotinamide adenine dinucleotide phosphate
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00465.
Full experimental details, in vitro and in vivo assay (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Liu B.; Wang T.; Zhao H. N.; Yue W. W.; Yu H. P.; Liu C. X.; Yin J.; Jia R. Y.; Nie H. W. The prevalence of hyperuricemia in China: a meta-analysis. BMC Public Health 2011, 11, 832. 10.1186/1471-2458-11-832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y.; Pandya B. J.; Choi H. K. Prevalence of gout and hyperuricemia in the US general population: the National Health and Nutrition Examination Survey 2007–2008. Arthritis Rheum. 2011, 63, 3136–3141. 10.1002/art.30520. [DOI] [PubMed] [Google Scholar]
- DeOliveira E. P.; Burini R. C. High plasma uric acid concentration: causes and consequences. Diabetol. Metab. Syndr. 2012, 4, 12. 10.1186/1758-5996-4-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W.; Doherty M.; Pascual E. EULAR evidence based recommendations for gout. Part I: Diagnosis. Report of a task force of the Standing Committee for International Clinical Studies Including Therapeutics (ESCISIT). Ann. Rheum. Dis. 2006, 65, 1301–1311. 10.1136/ard.2006.055251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W.; Doherty M.; Bardin T. EULAR evidence based recommendations for gout. Part II: Management. Report of a task force of the EULAR Standing Committee for International Clinical Studies Including Therapeutics (ESCISIT). Ann. Rheum. Dis. 2006, 65, 1312–1324. 10.1136/ard.2006.055269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crittenden D. B.; Pillinger M. H. New Therapies for Gout. Annu. Rev. Med. 2013, 64, 325–337. 10.1146/annurev-med-080911-105830. [DOI] [PubMed] [Google Scholar]
- Shahid H.; Singh J. A. Investigational drugs for hyperuricemia. Expert Opin. Invest. Drugs 2015, 24, 1013–1030. 10.1517/13543784.2015.1051617. [DOI] [PubMed] [Google Scholar]
- Shoji A.; Yamanaka H.; Kamatani N. A retrospective study of the relationship between serum urate level and recurrent attacks of gouty arthritis: evidence for reduction of recurrent gouty arthritis with ntihyperuricemic therapy. Arthritis Rheum. 2004, 51, 321–325. 10.1002/art.20405. [DOI] [PubMed] [Google Scholar]
- Diaz-Torne C.; Perez-Herrero N.; Perez-Ruiz F. New medications in development for the treatment of hyperuricemia of gout. Curr. Opin. Rheumatol. 2015, 27, 164–169. 10.1097/BOR.0000000000000146. [DOI] [PubMed] [Google Scholar]
- Matsuo H.; Nakayama A.; Sakiyama M. ABCG2 dysfunction causes hyperuricemia due to both renal urate underexcretion and renal urate overload. Sci. Rep. 2014, 4, 3755–3799. 10.1038/srep03755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn S. O.; Ohtomo S.; Kiyokawa J.; Nakagawa T.; Yamane M.; Lee K. J.; Kim K. H.; Kim B. H.; Tanaka J.; Kawabe Y.; Horiba N. Stronger uricosuric effects of the novel selective URAT1 inhibitor UR-1102 lowered plasma urate in tufted capuchin monkeys to a greater extent than benzbromarone. J. Pharmacol. Exp. Ther. 2016, 357, 157–166. 10.1124/jpet.115.231647. [DOI] [PubMed] [Google Scholar]
- Hiratochi M.; Tatani K.; Shimizu K.; Kuramochi Y.; Kikuchi N.; Kamada N.; Itoh F.; Isaji M. Hypouricemic effects of novel concentrative nucleoside transporter 2 inhibitors through suppressing intestinal absorption of purine nucleosides. Eur. J. Pharmacol. 2012, 690, 183–191. 10.1016/j.ejphar.2012.06.015. [DOI] [PubMed] [Google Scholar]
- Yeh L.; Shen Z.; Kerr B. RDEA594: a potent URAT1 inhibitor without affecting other important renal transporters, OAT1 and OAT 3. Ann. Rheum Dis. 2009, 68, 320. [Google Scholar]
- Fleischmann R.; Kerr B.; Yeh L. T.; Suster M.; Shen Z. C.; Polvent E.; Hingorani V.; Quart B.; Manhard K.; Miner J. N.; Baumgartner S. Pharmacodynamic, pharmacokinetic and tolerability evaluation of concomitant administration of lesinurad and febuxostat in gout patients with hyperuricaemia. Rheumatology 2014, 53, 2167–2174. 10.1093/rheumatology/ket487. [DOI] [PubMed] [Google Scholar]
- Center for Drug Evaluation and Research. Clinical Pharmacology and Biopharmaceutics Review(s). http://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/207988Orig1s000ClinPharmR.pdf (accessed Jan 19, 2016).
- Pema K. M. Lesinurad sodium. Solute carrier family 22 member 12 (URAT1) inhibitor, uricosuric, treatment of gout. Drugs Future 2011, 36, 875–880. 10.1358/dof.2011.036.12.1665561. [DOI] [Google Scholar]
- Girardet J. L.; Koh Y. H.; Delarosa M.; Gunic E.; Hong Z.; Lang S.; Kim H. W.. WO 2006/026356.
- LaPlante S. R.; Fader L. D.; Fandrick K. R.; Fandrick D. R.; Hucke O.; Kemper R.; Miller S. P. F.; Edwards P. J. Assessing Atropisomer Axial Chirality in Drug Discovery and Development. J. Med. Chem. 2011, 54, 7005–7022. 10.1021/jm200584g. [DOI] [PubMed] [Google Scholar]
- Bringmann G.; Mortimer A. J. P.; Keller P. A.; Gresser M. J.; Garner J.; Breuning M. Atroposelective Synthesis of Axially Chiral Biaryl Compounds. Angew. Chem., Int. Ed. 2005, 44, 5384–5427. 10.1002/anie.200462661. [DOI] [PubMed] [Google Scholar]
- Maryanoff B. E.; Greco M. N.. Stereochemical Lability in Drug Molecules: Cases Where Chirality May Not Be Critical for Drug Development. In Comprehensive Chirality; Carriera E. M., Yamamto H., Eds.; Elsevier, Amsterdam, 2012; Vol. 1, pp 105–119. [Google Scholar]
- LaPlante S. R.; Edwards P. J.; Fader L. D.; Jakalian A.; Hucke O. Revealing atropisomer axial chirality in drug discovery. ChemMedChem 2011, 6, 505–513. 10.1002/cmdc.201000485. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.