

Abstract
The first posttranslational modification step in the biosynthesis of the tryptophan-derived quinone cofactors is the auto-catalytic hydroxylation of a specific Trp residue at the C-7 position on the indole side-chain. Subsequent modifications are catalyzed by modifying enzymes, but the mechanism by which this first step occurs is unknown. LodA possesses a cysteine tryptophylquinone (CTQ) cofactor. Metal analysis, as well as spectroscopic and kinetic studies of the mature and precursor forms of a D512A LodA variant provide evidence that copper is required for the initial hydroxylation of the precursor protein; and that if alternative metals are bound, the modification does not occur and the precursor is unstable. It is shown that the mature native LodA also contains loosely bound copper which affects the visible absorbance spectrum and quenches the fluorescence spectrum that is attributed to the mature CTQ cofactor. When copper is removed the fluorescence appears and when it is added back to the protein the fluorescence is quenched, indicating that copper reversibly binds in the proximity of CTQ. Removal of copper does not diminish the enzymatic activity of LodA. This distinguishes LodA from enzymes with protein-derived tyrosylquinone cofactors in which copper is present near the cofactor and is absolutely required for activity. Mechanisms are proposed for the role of copper in the hydroxylation of the un-activated Trp side-chain. These results demonstrate that the reason that the highly conserved Asp512 is critical for LodA, and possibly all tryptophylquinone enzymes, is not because it is required for catalysis but because it is necessary for CTQ biosynthesis, more specifically to facilitate the initial copper-dependent hydroxylation of a specific Trp residue.
Graphical Abstract

Posttranslational modification of proteins is an essential process that helps determine structural integrity, cellular signaling, and enzymatic activity of proteins. Protein-derived quinone cofactors are interesting products of posttranslational modifications which convert inactive precursor proteins to ones with enzymatic functionality 1. These quinone cofactors (Figure 1) are typically derived from modification of specific Tyr or Trp residues 2. Studies of the biosynthesis of protein-derived quinone cofactors have revealed that these posttranslational modifications occur via a variety of different and interesting mechanisms, some of which were unprecedented at the time of their discovery 1–3.
Figure 1.
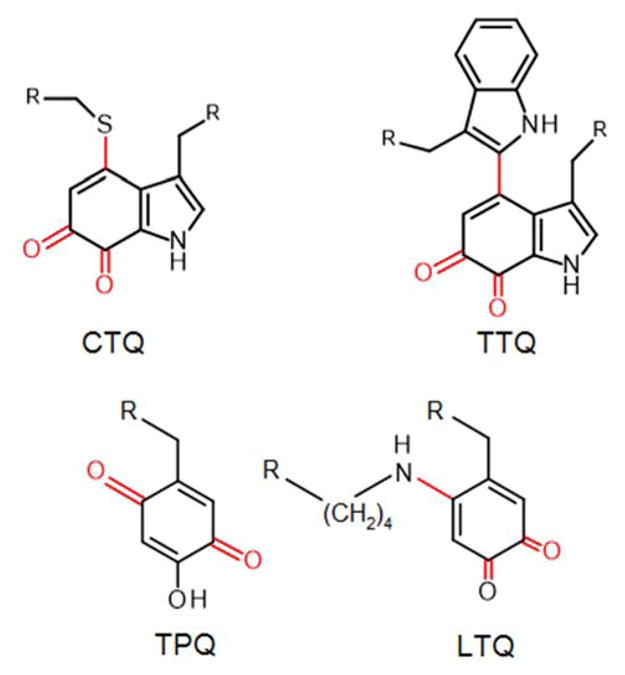
Protein-derived quinone cofactors that are formed by posttranslational modifications. CTQ is cysteine tryptophylquinone. TTQ is tryptophan tryptophylquinone. TPQ is 2,4,5-trihydroxyphenylalanine quinone or topaquinone. LTQ is lysine tyrosylquinone. R is the protein backbone to which the amino acid side chain is attached. The posttranslational modifications to the amino acid residues are shown in red.
The Tyr-derived cofactor, trihydroxyphenylalanine quinone (TPQ), results from the insertion of two oxygen atoms into the phenolic ring of a Tyr residue 4. Another Tyr-derived cofactor, lysine tyrosylquinone (LTQ), requires the insertion of a single oxygen atom into a Tyr ring as well as formation of a covalent cross-link between the Tyr side-chain and that from a Lys residue 5. The complete formation of TPQ and LTQ has been shown to be a series of auto-catalytic events requiring only copper and O2 6, 7. Furthermore, in these enzymes the copper is not only essential for the biogenesis of the cofactor, but remains tightly-bound to the protein and participates in catalytic activity of the enzyme with the mature quinone cofactor 8, 9.
The biogenesis of the Trp-derived cofactors, tryptophan tryptophylquinone (TTQ) and cysteine tryptophylquinone (CTQ), requires that two oxygen atoms be inserted into the indole ring of a specific Trp residue (Figure 1) 10, 11. In addition, a covalent crosslink is formed between this Trp side-chain and that from another Trp residue in TTQ or a Cys residue in CTQ. A common feature in the biosynthesis of TTQ and CTQ in the enzymes that have been characterized thus far is that the first step is an auto-catalytic hydroxylation of the side-chain of a specific Trp residue. However, in contrast to the Tyr-derived cofactors, the subsequent biosynthetic reactions are catalyzed by modifying enzymes. In the case of biosynthesis of TTQ in methylamine dehydrogenase (MADH) the modifying enzyme is a diheme enzyme, MauG 12. In the case of biosynthesis of the CTQ in the L-lysine-ε oxidase (LodA) 13 and glycine oxidase (GoxA) 14, the modifying enzymes are flavoproteins, LodB and GoxB, respectively. Copper has not previously been reported to be present in enzymes with Trp-derived cofactors. While some progress has been made in understanding the roles of the modifying enzymes15, 16, the mechanism of the initial auto-catalytic hydroxylation has remained a mystery.
The mechanism of biosynthesis of TTQ in MADH from Paracoccus denitrificans has been extensively studied 15. When MADH was expressed in the absence of MauG a precursor of MADH (preMADH) was isolated in which residue βTrp57 was monohydroxylated but lacked any further posttranslational modifications 17. MauG was shown to catalyze three two-electron oxidations required for the completion of TTQ biosynthesis from the preMADH substrate in vitro 18–20. Site-directed mutagenesis studies of MADH did reveal that a residue in close proximity to the quinone oxygens of TTQ, βAsp76, is required for the initial hydroxylation 21. Conversion of this residue to Asn resulted in expression of only a trace amount of protein which was shown by mass spectrometry to lack any modifications, including the initial hydroxylation. Unfortunately, it was not possible to isolate sufficient quantities of this unmodified precursor for mechanistic studies.
LodA and GoxA from Marinomonas mediterranea are members of a newly discovered family of enzymes which are oxidases containing CTQ 22. The gene clusters in which lodA and goxA reside each contain only one other gene, lodB and goxB, each of which is predicted to encode a flavoprotein that is required for expression of active LodA and GoxA, respectively 23, 24. When either lodA or goxA was expressed in the absence of lodB or goxB, a precursor protein was again isolated in which the Trp residue which forms CTQ was mono-hydroxylated but lacked any further posttranslational modifications 25.
The active site of LodA contains Asp512 which is in proximity to the quinone portion of CTQ and is structurally conserved with βAsp76 of MADH, the residue that is required for the initial hydroxylation in TTQ biosynthesis in MADH. It was reported that a D512A mutation of LodA also yielded protein which lacked any modifications, including the initial hydroxylation 25. In the current study it has been possible to produce larger yields of D512A LodA than were obtained for the corresponding variant protein in MADH. This has allowed studies to analyze the metal content and to obtain some insight into the mechanism of the auto-catalytic Trp hydroxylation reaction that initiates CTQ biosynthesis in LodA. The data presented in this study indicate that this initial hydroxylation is a copper-dependent process that requires the presence of Asp512, although the copper in the mature protein is relatively weakly bound and is not needed for catalysis.
EXPERIMENTAL PROCEDURES
Protein purification
For recombinant production of WT LodA and D512A LodA the native and variant lodA genes were co-expressed with lodB in E. coli Rosetta cells as described previously23, 26. The generation of the D512A mutation was previously described 25. Cell growth and induction, preparation of cell extracts and initial purification of the His-tagged proteins by affinity chromatography using a Ni-NTA resin were as previously described 26. Active D512A LodA was then further purified by ion exchange chromatography with DEAE cellulose in 50 mM potassium phosphate, pH7.5, and was eluted using 180–270 mM NaCl in the same buffer.
Spectroscopic and kinetic studies
The UV-visible absorbance spectra were recorded using a Shimadzu 1501 Spectrophotometer. Fluorescence spectra were recorded using a Bio Teck Synergy 4. Lysine ε-oxidase activity was determined as described previously using a coupled assay in which the production of the H2O2 product is coupled to the oxidation of Amplex Red by horseradish peroxidase 13, 26.
Binding studies
Specific copper binding was assessed by determining the extent of quenching of fluorescence that was caused by addition of copper to EDTA-treated LodA and D512A LodA. In order to prepare EDTA-treated proteins, samples were incubated in 50 mM potassium phosphate buffer, pH 7.5, containing 10 mM EDTA for 1 hr and then the EDTA was removed by buffer exchange five times with EDTA-free buffer. The EDTA-treated proteins were each then incubated for 30 min with different concentrations of CuSO4. Fluorescence spectra were recorded before and after incubation. The samples were excited at 375 nm and the emission spectra were measured over a range of 420–600 nm. The % quenching of fluorescence for each concentration of added copper was determined and these data were analyzed by Eq 1, where Y is the % fluorescence quenching and S is the [CuSO4].
| (1) |
Metal analysis
Metal analysis was performed using a sector-field ICP-MS (ThermoFisher Element XR) with a Peltier-cooled inlet system (PC3, Elemental Scientific Inc.). Samples were diluted 100 times in ultrapure 1% nitric acid (HNO3) (Fisher Optima) containing 500 ppt indium as an internal standard. Standards were also made in ultrapure 1% HNO3 and checked versus USGS Standard Reference Samples (https://bqs.usgs.gov/srs/). All elements were determined in medium resolution mode (M/ΔM > 4000) to minimize potential isobaric interferences. The analyzed isotopes included 55Mn, 56Fe, 59Co, 60Ni, 63Cu, and 66Zn.
RESULTS
Preparation of different forms of D512A LodA
In the previous characterization of D512 LodA by mass spectrometry which revealed that this protein lacked any posttranslational modifications, the His-tagged protein was purified by affinity chromatography using a Ni-NTA resin 25. In the current study, The D512A protein was further purified by DEAE ion exchange chromatography. This allowed identification of two different forms of D512A LodA. For clarification, the differences in the purification steps that allowed isolation of the different forms are described below. The protein which was initially purified by affinity chromatography will be referred to as total soluble D512A LodA. This preparation did not exhibit enzymatic activity that was significantly above baseline. After DEAE chromatography of the total soluble D512A LodA, only about 5% of the protein was recovered. The recovered protein did exhibit enzymatic activity and will be referred to as active D512A LodA. A possible explanation for the loss of the remaining 95% of the total soluble D512A LodA is that it was the previously characterized unmodified precursor protein which is unstable under the conditions used for ion-exchange chromatography, and lost when trying to purify it further by this technique. In order to test this hypothesis, the same procedure was used with preLodA, the precursor which may be isolated by expression of lodA in the absence of lodB 25. Consistent with this explanation, no preLodA was recovered after subjecting it to the same chromatographic protocol. The inability to observe activity in the total soluble D512A LodA fraction was because 5% of this activity is below the signal to noise levels of the measurements. These results could also explain the inability to observe a CTQ-containing peptide by mass spectrometry in the previous study which analyzed only the total soluble D512A LodA25.
Evidence for bound copper in D512A LodA and WT LodA
The absorbance spectrum of total soluble D512A LodA exhibited a broad featureless visible absorbance in the 300–500 nm range (Figure 2A). Some of this absorbance was retained in the active D512A LodA in the 300–450 nm range but to a much lesser extent (Figure 2AB). This absorbance is reminiscent of features exhibited by metal-binding sites 27–30. To test the hypothesis that a metal is present in active D512A LodA which gives rise to this absorbance, the protein was treated with ethylene diaminetetraacetic acid (EDTA). After EDTA treatment the broad absorbance feature was lost and the spectrum exhibited features more characteristic of CTQ, which were previously masked by the metal-derived absorbance (Figure 2B). A difference spectrum of the protein, before minus after EDTA treatment, shows that the feature that was lost is centered near 315 nm with a broad shoulder extending to higher wavelengths (Figure 2B inset). For comparison, the EDTA treatment protocol was applied to WT LodA. The absorbance spectrum of WT LodA exhibits features in the 350–450 nm range characteristic of the CTQ cofactor (Figure 2C). After treatment with EDTA, a small change was observed in this region of the spectrum (Figure 2D). Examination of the difference spectrum reveals that a similar spectral feature was lost after EDTA treatment; however, it was of much less intensity than that observed for active D512A LodA (Figure 2D inset). This unexpected result suggests that a metal is also present in WT LodA, albeit in smaller amount than in the active D512A LodA.
Figure 2.

Absorbance spectra of different forms of WT LodA and D512A LodA. A. Absorbance spectra of total soluble D512A LodA (blue) and active D512A LodA (black). B. Magnification of the relevant features of the visible absorbance spectrum of active D512A LodA before (black) and after EDTA treatment (red). The inset shows the before minus after difference spectrum. C. Absorbance spectra of WT LodA. D. Magnification of the relevant features of the visible absorbance spectrum of WT LodA before (black) and after EDTA treatment (red). The inset shows the before minus after difference spectrum.
In order to confirm the presence and identity of the putative metal in these proteins, total soluble D512A LodA, active D512A LodA and WT LodA were analyzed using sector-field ICP-MS for the presence of Co, Cu, Fe, Mn, Ni and Zn (Table 1). The sulfur content of each sample was also determined so that molar metal/protein ratio of each metal could be accurately determined by comparison with the know sulfur content of the protein that was determined from the number of Met and Cys residues in the protein. For active D512A LodA, copper has the highest occupancy of any of the metals tested with 2.6 ± 0.5 Cu per protein molecule. Metal analysis of the total soluble D512A LodA, of which only about 5% is active D512A LodA, revealed sub-stoichiometric amounts of multiple metals; among them is Cu but only at 0.24 Cu per protein. This indicates that metals also bind to the inactive unmodified D512A LodA present in the total soluble D512A LodA. However, in contrast to active D512A LodA, there appears to be no specificity for which metal binds.
Table 1.
Metal content of D512A LodA and WT LodAa
| Metal | Active D512A LodA | Total soluble D512A LodA | WT LodA | EDTA-treated WT LodA |
|---|---|---|---|---|
| Co | 0.0016 ± 0.0008 | 0.006 ± 0.006 | 0.0006 ± 0.0004 | 0.0002 ± 0.0001 |
| Cu | 2.7 ± 0.5 | 0.24 ± 0.01 | 1.2 ± 0.04 | 0.006 ± 0.001 |
| Fe | 0.1 ± 0.02 | 0.14 ± 0.10 | 0.09 ± 0.07 | 0.011 ± 0.005 |
| Mn | 0.003 ± 0.003 | 0.0023±0.0006 | 0.0012 ± 0.0007 | 0.0004 ± 0.0003 |
| Ni | 0.09 ± 0.03 | 0.346 ± 0.007 | 0.08 ± 0.02 | 0.0025 ± 0.0005 |
| Zn | 0.65 ± 0.3 | 0.27 ± 0.20 | 0.17 ± 0.1 | 0.027 ± 0.0007 |
Values are molar metal/protein ratio. Values are an average of at least two replicates
It was previously shown that the total soluble D512A lacked the initial hydroxyl group on Trp581 25. The posttranslational modifications to form the active D512A LodA were likely not detected because, as we now show, the active protein comprises only about 5% of that fraction. The metal analysis suggests the possibility that the D512A mutation eliminates the specificity for binding copper versus other metals prior to CTQ formation, and that only when copper is bound does the initial hydroxylation occur allowing subsequent LodB-dependent CTQ formation in the cell, which results in a more stable and active D512A LodA. Consistent with this notion is the observation that the moles of copper per protein in the total soluble D512 LodA in Table 1 is about 8% of the moles of copper per protein in the further purified fraction of active D512A LodA. Thus, the total amount of copper present in the total soluble D512A LodA can be largely attributed to the 5% which is present in the portion which is the active D512A LodA. Copper was also present in WT LodA with 1.2 ± 0.5 Cu per protein, with no other metals at statistically relevant occupancies. These results implicate copper as the bound metal responsible for the spectral feature centered at 315 nm in WT LodA as well (Figure 2D). After treatment of WT LodA with EDTA, copper was absent, consistent with it being weakly bound to the enzyme.
Effect of bound copper on CTQ fluorescence
Important questions that are raised by the results described above are whether the copper is bound to a specific site in these proteins, rather than merely non-specific binding, and whether copper can rebind to the site after it has been removed. This is of particular interest since the crystal structures of LodA 31 did not show the presence of copper. It was possible in the current study to use fluorescence spectroscopy to probe the environment of the CTQ cofactor in order to address these questions. The absorbance spectrum of CTQ in LodA has two overlapping absorbance maxima, at 375 nm and 420 nm. Each of these was examined for fluorescence. In WT LodA and active D512A LodA excitation at either 420 nm or 375 nm did not yield any significant fluorescence emission. Each protein was then also tested after EDTA treatment to remove the bound-metal. Again, excitation at 420 nm did not yield any fluorescence emission. However, excitation at 375 nm did yield fluorescence emission with a maximum at 430 nm in both WT LodA and active D512A LodA (Figure 3).
Figure 3.

Fluorescence emission spectra of different forms of WT LodA and D512A LodA that results from excitation of CTQ at 375 nm. A. Fluorescence of active D512A LodA before (solid line) and after EDTA treatment (dotted line). B. Fluorescence of WT LodA before (solid line) and after EDTA treatment (dotted line).
Quinones are typically not fluorescent due to effective intersystem crossing; however modified quinones have been known to fluoresce from an π-π* transition with an intense absorption due to a charge transfer into the carbonyl of the C=C–C=O bond by electron donating substituents attached to the conjugated carbon 32, 33. In contrast, the dominant visible absorbance of most tryptophan-derived quinones likely corresponds to an n-π* transition of the same C=C–C=O bond, which is known to have no fluorescence 32. Since excitation at 420 nm band did not yield fluorescence emission, this absorbance feature is most likely due to the n-π* transition. Excitation of the 375 nm band did yield fluorescence emission with a maximum at 430 nm. As CTQ contains a strongly electron donating sulfur substituent, the 375 nm absorbance feature is likely due to a π-π* transition associated with charge transfer from the p orbital of the cysteine sulfur.
That fluorescence is only observed after EDTA treatment suggests that the bound copper is quenching that fluorescence in WT LodA and active D512A LodA prior to its removal. This quenching of the λex375 fluorescent feature indicates that some or all of the copper present within these proteins is close enough to the quinone carbonyl or crosslinked-cysteine sulfur to quench the CTQ fluorescence. To obtain support for this idea, copper was added back to the copper-free WT LodA and D512A LodA. The addition of copper did quench the fluorescence. In order to determine the Kd for copper binding to the specific site which results in fluorescence quenching, the percent fluorescence quenching was determined over a range of concentrations of added copper (Figure 4). Analysis of these data to Eq. 1 yielded Kd values of 101 ± 29 μM and 62 ± 22 μM, respectively, for WT LodA and active D512A LodA. The advantage of monitoring copper binding by the fluorescence quenching is that it only detects the binding of copper to the specific site in proximity to CTQ and not other non-specific binding. This is important because in this case, the specific binding site has relatively weak affinity. As such, is not possible to distinguish copper binding to this site from other non-specific binding by metal analysis, since EDTA treatment removes all of the copper. Metal analysis (Table 1) indicated a greater molar metal/protein ratio for the active D512A LodA that for WT LodA. This may be a consequence of a more spacious and accessible active site in D512A LodA which would result from the conversion of Asp to Ala, thus allowing additional copper to remain loosely bound.
Figure 4.

Equilibrium binding of copper to copper-depleted WT LodA and D512A LodA. The Copper-depleted WT LodA (A) and D512A LodA (B) were prepared and then incubated with varied concentrations of CuSO4 as described under Experimental Procedures. Fluorescence quenching was determined from the relative fluorescence at 440 nm with excitation at 375 nm. The curves are the fits of the data by Eq 1.
Effect of bound copper on LodA activity
The results described above raise the question of whether bound copper is required for the enzymatic activity of LodA. The question is relevant because in enzymes with the tyrosine-derived cofactors TPQ and LTQ, copper is required both for cofactor biogenesis and for activity in the enzyme after cofactor maturation. The activities of WT LodA and active D512A LodA were compared before and after EDTA treatment (Table 2). In neither case was there any loss of activity. In fact, a small increase in activity was observed for WT LodA.
Table 2.
Effect of EDTA-treatment on the activity of WT LodA and D512A LodA
| WT LodA (kcat, s−1) | Active D512A LodA (kcat,s−1) | |
|---|---|---|
| As Isolated | 0.29 ± 0.01 | 0.20 ± 0.03 |
| EDTA-treated | 0.35 ± 0.02 | 0.20 ± 0.01 |
Values are an average of at least two replicates
DISCUSSION
The hydroxylation of aromatic rings is a difficult synthetic reaction requiring very strong oxidants, and is often complicated by lack of specificity, excessive oxidations, and production of undesired reactive by-products. Oxidation of aromatic amino acid residues in proteins occurs as a consequence of oxidative stress, and reaction with reactive oxygen species or free radicals. There are few examples in nature of mechanisms for site-specific hydroxylation of specific aromatic amino acid residues in proteins. One class of enzymes which accomplishes this task is the iron and tetrahydropterin-dependent amino acid hydroxylases 34, 35. These enzymes utilize a protein-bound FeIV=O oxidant to perform the oxidative chemistry on unmodified phenylalanine and tryptophan residues. In the other known protein class, the protein-derived TPQ and LTQ cofactors of the copper-dependent tyrosine-derived amine oxidases are formed by an auto-catalytic mechanism in which a bound copper is used to hydroxylate a “pre-activated” substrate, the phenol moiety of tyrosine residue 2.
The first step in the biosynthesis of the tryptophan-derived quinone cofactors, TTQ and CTQ, is the auto-catalytic hydroxylation of a specific Trp residue at the C-7 position 36 on the indole side-chain. The mechanism by which this occurs has remained elusive. The current results strongly suggest that the initially translated LodA protein which lacks CTQ and any posttranslational modifications requires copper for this auto-catalytic hydroxylation. It follows that structural features of the precursor protein, notably residue 512, are required to selectively bind the copper prior to the maturation of the enzyme cofactor, and direct the activity of the bound copper. This is in contrast to the mechanism of biosynthesis of the tyrosine-derived quinone cofactors where copper is catalyzing the insertion of a second oxygen into an aromatic ring of a tyrosine. Thus, the mechanism by which copper is used to hydroxylate the Trp581 in LodA must be different because it involves insertion of the first oxygen into an un-activated aromatic ring system. This reaction requires a higher oxidation state of copper, comparable to the high-valent iron which is utilized by the tetrahydropterin-dependent amino acid hydroxylases. This suggests a highly unusual reaction mechanism for the hydroxylation reaction in LodA.
While metal analysis confirms the presence of at least one copper in WT LodA, the three reported crystal structures of LodA (PDB entries 3WEU, 3WEV and 2YMW) show no copper to be present. However, one of the structures (PDB entry 2YMW) identifies two putative sodium binding sites, one of which is in close proximity to the sulfur of Cys516. If copper were to bind here, this would be consistent with the property of quenching the quinone fluorescence in WT LodA and the active D512A LodA. In any case, the three crystal structures of LodA exhibit the presence of several structural waters, and crystallization additives such as ethanol and ethylene glycol. The presence of both water and additives in the structures is significant as it suggests the copper in the as isolated protein could be removed or leached out during crystallization, or even during storage after purification. For example, glycerol which is used as a cryoprotectant in crystallography and storage of frozen protein stocks could potentially chelate and remove bound copper. This could explain why copper has not been previously reported to be present in LodA.
In the TPQ-dependent copper amine oxidases copper is ligated by three His residues, which is a common type II copper site, and the copper also interacts with the phenyl oxygen of the Tyr side-chain that it modifies 37. In LodA, multiple His residues are not present in the proximity of CTQ, and the unmodified Trp581 seems an unlikely candidate for a copper ligand. This suggests a unique type of copper site in the unmodified LodA precursor. Possible ligands in LodA that have been implicated by this study and others include Asp512 and Cys516 25, the residue that crosslinks to the hydroxylated Trp581 to form CTQ. Cysteine sulfurs exhibit reactivity towards copper and have been reported to bind tightly to Cu(I) and to reduce Cu(II) to its Cu(I) state 38. In this respect, it is noteworthy that a C516A LodA variant was shown to lack any posttranslational modifications and so the initial hydroxylation did not occur in the absence of this Cys 25.
While copper is utilized less often than iron for difficult biological oxidation reactions, it is capable of Fenton-like chemistry when reacting with oxygen and peroxide. As with traditional iron Fenton chemistry, the copper Fenton-like chemistry generates reactive hydroxyl radicals and the Cu(I)/Cu(II) reaction is known to generate nonspecific aromatic hydroxylation 39, 40. The LodA precursor contains one or more coppers in either a Cu(I) state, or Cu(II) state that could be reduced via Cys516, prior to CTQ formation. A direct reaction to peroxide could generate a reactive hydroxyl radical or a Cu(III)-OH species, either of which would be suitable for oxidizing the Trp581 ring. This peroxide reaction could be performed with or without the need of external reductants, while a reaction with oxygen would need at least two electron reducing equivalents to generate an ideal reactive oxidant (•OH or Cu(III)-OH). With this in mind, three possible mechanisms for Trp hydroxylation which utilize copper may be considered: a radical mediated mechanism, a single electron transfer (SET) from substrate mechanism, and an organometallic intermediate mechanism (Figure 5).
Figure 5.

Possible mechanisms for tryptophan hydroxylation involving cysteine and copper. Cys515 is indicated to be the ligand for copper in the proposed mechanisms because previous studies showed that mutation of this Cys resulted in expression of LodA lacking any modifications25.
Each of the three mechanisms begins with Cu(II) binding to Cys516 which would be able to stabilize a Cu (I) metal – S0 ligand bond via a charge-transfer-stabilized complex. While theoretically possible, the radical and SET mechanisms seem unlikely. The radical mediated mechanism is feasible within a protein active site; however, it will not be as selective in its choice of substrate due to the free hydroxyl radical. Instead the radical mediated mechanism may indiscriminately attack other amino acids within the protein. Lack of selectivity is also a disadvantage with the SET mechanism. While the SET mechanism may be selective for tryptophan, a tryptophan radical cation is susceptible to Cu(III)-OH oxidation on any of the atoms of the aromatic side chain, not just the C7 carbon. Thus, the most likely mechanism for the required hydroxylation at a specific site on the Trp581 is to proceed through an organometallic intermediate. In the proposed organometallic mechanism, the Cu(III)-OH is a high potential species which is capable of oxidizing Trp581. It is proposed that its coordination with Cys516 positions it so that it selectively reacts with the OH moiety attaching to the C7 position of the Trp ring. While the proposed organometallic intermediate mechanism is uncommon, it has been reported that Cu(III) can stabilize intermediates with an aromatic ring via such an organometallic complex 41. Once the initial hydroxylation is achieved, the remaining steps in CTQ biosynthesis may be catalyzed by LodB in the cell resulting in the isolation of the mature active LodA.
While the organometallic mechanism describes the chemical reactions that could lead to the initial hydroxylation of Trp581, other structural features of the protein must be responsible for the site specificity of the oxygen insertion. Asp512 likely is important as the D512A mutation results in only ~5% of the isolated protein being posttranslationally modified, with 95% of the protein lacking the initial hydroxylation. Also, it is shown that the total soluble D512A LodA exhibits no significant specificity for binding copper over other metals. It was previously shown that mutagenesis of other residues, such as Cys448, in the active-site and substrate channel of LodA effect the levels of mature CTQ present in the enzyme as well as kinetic parameters for the catalytic reaction 42. Thus, perturbations of the positions of other residues in the active site which are caused by the D512A mutation or binding of a metal other than copper could also abolish the initial hydroxylation.
The results of this study reveal an unusual role for a highly structurally conserved amino acid residue, Asp512. A structurally conserved Asp residue is present in an equivalent position in each of the previously reported structures of tryptophylquinone enzymes: the CTQ-bearing glycine oxidase (GoxA)31 and quinohemoprotein amine dehydrogenase11, and the TTQ-bearing methylamine and aromatic amine dehydrogenases43, 44. Prior to this study, it had not been possible to assess the role of this residue in catalysis because mutation of the residue compromised the cofactor formation21, 25. In this study, it was possible to isolate at least a small amount of active D512A LodA. The activity of this variant was comparable to that of WT LodA ruling out a critical role for this residue in catalysis. Another novel observation in this study is that copper binds, albeit weakly to the mature LodA in proximity to CTQ. Copper binds to the active D512A LodA with a similar affinity. The Kd for copper binding to the mature enzymes seems too high to be physiologically relevant, and indeed its removal has little effect on activity. It is likely that copper binding to the unmodified precursor of LodA prior to CTQ formation is much stronger and that its affinity decreases after CTQ formation. In conclusion, it appears that the reason that this conserved Asp residue is critical for LodA, and possibly all tryptophylquinone enzymes, is not because it is required for catalysis but because it is necessary for CTQ biosynthesis, more specifically to facilitate the initial copper-dependent hydroxylation of a specific Trp residue.
Acknowledgments
The authos thank Yu Tang for providing technical assistance and Melissa Gilbert for analytical work. This research was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number R37GM41574 (VLD).
ABBREVIATIONS
- CTQ
cysteine tryptophylquinone
- MADH
methylamine dehydrogenase
- SET
single electron transfer
- TTQ
tryptophan tryptophylquinone
- WT
wild-type
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have approved the final version of the manuscript.
References
- 1.Davidson VL. Generation of protein-derived redox cofactors by posttranslational modification. Mol Biosyst. 2011;7:29–37. doi: 10.1039/c005311b. [DOI] [PubMed] [Google Scholar]
- 2.Klinman JP, Bonnot F. Intrigues and intricacies of the biosynthetic pathways for the enzymatic quinocofactors: PQQ, TTQ, CTQ, TPQ, and LTQ. Chem Rev. 2014;114:4343–4365. doi: 10.1021/cr400475g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davidson VL. Protein-derived cofactors. Expanding the scope of post-translational modifications. Biochemistry. 2007;46:5283–5292. doi: 10.1021/bi700468t. [DOI] [PubMed] [Google Scholar]
- 4.Janes SM, Mu D, Wemmer D, Smith AJ, Kaur S, Maltby D, Burlingame AL, Klinman JP. A new redox cofactor in eukaryotic enzymes: 6-hydroxydopa at the active site of bovine serum amine oxidase. Science. 1990;248:981–987. doi: 10.1126/science.2111581. [DOI] [PubMed] [Google Scholar]
- 5.Wang SX, Mure M, Medzihradszky KF, Burlingame AL, Brown DE, Dooley DM, Smith AJ, Kagan HM, Klinman JP. A crosslinked cofactor in lysyl oxidase: redox function for amino acid side chains. Science. 1996;273:1078–1084. doi: 10.1126/science.273.5278.1078. [DOI] [PubMed] [Google Scholar]
- 6.Matsuzaki R, Fukui T, Sato H, Ozaki Y, Tanizawa K. Generation of the topa quinone cofactor in bacterial monoamine oxidase by cupric ion-dependent autooxidation of a specific tyrosyl residue. FEBS Lett. 1994;351:360–364. doi: 10.1016/0014-5793(94)00884-1. [DOI] [PubMed] [Google Scholar]
- 7.Moore RH, Spies MA, Culpepper MB, Murakawa T, Hirota S, Okajima T, Tanizawa K, Mure M. Trapping of a dopaquinone intermediate in the TPQ cofactor biogenesis in a copper-containing amine oxidase from Arthrobacter globiformis. J Am Chem Soc. 2007;129:11524–11534. doi: 10.1021/ja0731165. [DOI] [PubMed] [Google Scholar]
- 8.Bollinger JA, Brown DE, Dooley DM. The formation of lysine tyrosylquinone (LTQ) is a self-processing reaction. Expression and characterization of a Drosophila lysyl oxidase. Biochemistry. 2005;44:11708–11714. doi: 10.1021/bi0504310. [DOI] [PubMed] [Google Scholar]
- 9.Cai D, Klinman JP. Copper amine oxidase: heterologous expression, purification, and characterization of an active enzyme in Saccharomyces cerevisiae. Biochemistry. 1994;33:7647–7653. doi: 10.1021/bi00190a019. [DOI] [PubMed] [Google Scholar]
- 10.McIntire WS, Wemmer DE, Chistoserdov A, Lidstrom ME. A new cofactor in a prokaryotic enzyme: tryptophan tryptophylquinone as the redox prosthetic group in methylamine dehydrogenase. Science. 1991;252:817–824. doi: 10.1126/science.2028257. [DOI] [PubMed] [Google Scholar]
- 11.Datta S, Mori Y, Takagi K, Kawaguchi K, Chen ZW, Okajima T, Kuroda S, Ikeda T, Kano K, Tanizawa K, Mathews FS. Structure of a quinohemoprotein amine dehydrogenase with an uncommon redox cofactor and highly unusual crosslinking. Proc Natl Acad Sci U S A. 2001;98:14268–14273. doi: 10.1073/pnas.241429098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Y, Graichen ME, Liu A, Pearson AR, Wilmot CM, Davidson VL. MauG, a novel diheme protein required for tryptophan tryptophylquinone biogenesis. Biochemistry. 2003;42:7318–7325. doi: 10.1021/bi034243q. [DOI] [PubMed] [Google Scholar]
- 13.Gomez D, Lucas-Elio P, Sanchez-Amat A, Solano F. A novel type of lysine oxidase: L-lysine-epsilon-oxidase. Biochim Biophys Acta. 2006;1764:1577–1585. doi: 10.1016/j.bbapap.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 14.Campillo-Brocal JC, Lucas-Elio P, Sanchez-Amat A. Identification in Marinomonas mediterranea of a novel quinoprotein with glycine oxidase activity. Microbiologyopen. 2013;2:684–694. doi: 10.1002/mbo3.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davidson VL, Wilmot CM. Posttranslational biosynthesis of the protein-derived cofactor tryptophan tryptophylquinone. Annu Rev Biochem. 2013;82:531–550. doi: 10.1146/annurev-biochem-051110-133601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sehanobish E, Campillo-Brocal JC, Williamson HR, Sanchez-Amat A, Davidson VL. Interaction of GoxA with Its Modifying Enzyme and Its Subunit Assembly Are Dependent on the Extent of Cysteine Tryptophylquinone Biosynthesis. Biochemistry. 2016;55:2305–2308. doi: 10.1021/acs.biochem.6b00274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pearson AR, de la Mora-Rey T, Graichen ME, Wang Y, Jones LH, Marimanikkupam S, Aggar SA, Grimsrud PA, Davidson VL, Wilmot CW. Further insights into quinone cofactor biogenesis: Probing the role of MauG in methylamine dehydrogenase TTQ formation. Biochemistry. 2004;43:5494–5502. doi: 10.1021/bi049863l. [DOI] [PubMed] [Google Scholar]
- 18.Jensen LM, Sanishvili R, Davidson VL, Wilmot CM. In crystallo posttranslational modification within a MauG/pre-methylamine dehydrogenase complex. Science. 2010;327:1392–1394. doi: 10.1126/science.1182492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li X, Jones LH, Pearson AR, Wilmot CM, Davidson VL. Mechanistic possibilities in MauG-dependent tryptophan tryptophylquinone biosynthesis. Biochemistry. 2006;45:13276–13283. doi: 10.1021/bi061497d. [DOI] [PubMed] [Google Scholar]
- 20.Yukl ET, Liu F, Krzystek J, Shin S, Jensen LM, Davidson VL, Wilmot CM, Liu A. Diradical intermediate within the context of tryptophan tryptophylquinone biosynthesis. Proc Natl Acad Sci U S A. 2013;110:4569–4573. doi: 10.1073/pnas.1215011110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jones LH, Pearson AR, Tang Y, Wilmot CM, Davidson VL. Active site aspartate residues are critical for tryptophan tryptophylquinone biogenesis in methylamine dehydrogenase. J Biol Chem. 2005;280:17392–17396. doi: 10.1074/jbc.M500943200. [DOI] [PubMed] [Google Scholar]
- 22.Campillo-Brocal JC, Chacon-Verdu MD, Lucas-Elio P, Sanchez-Amat A. Distribution in microbial genomes of genes similar to lodA and goxA which encode a novel family of quinoproteins with amino acid oxidase activity. BMC Genomics. 2015;16 doi: 10.1186/s12864-015-1455-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chacon-Verdu MD, Gomez D, Solano F, Lucas-Elio P, Sanchez-Amat A. LodB is required for the recombinant synthesis of the quinoprotein L -lysine-epsilon-oxidase from Marinomonas mediterranea. Appl Microbiol Biot. 2014;98:2981–2989. doi: 10.1007/s00253-013-5168-3. [DOI] [PubMed] [Google Scholar]
- 24.Gomez D, Lucas-Elio P, Solano F, Sanchez-Amat A. Both genes in the Marinomonas mediterranea lodAB operon are required for the expression of the antimicrobial protein lysine oxidase. Mol Microbiol. 2010;75:462–473. doi: 10.1111/j.1365-2958.2009.07000.x. [DOI] [PubMed] [Google Scholar]
- 25.Chacon-Verdu MD, Campillo-Brocal JC, Lucas-Elio P, Davidson VL, Sanchez-Amat A. Characterization of recombinant biosynthetic precursors of the cysteine tryptophylquinone cofactors of l-lysine-epsilon-oxidase and glycine oxidase from Marinomonas mediterranea. Biochim Biophys Acta. 2015;1854:1123–1131. doi: 10.1016/j.bbapap.2014.12.018. [DOI] [PubMed] [Google Scholar]
- 26.Sehanobish E, Shin S, Sanchez-Amat A, Davidson VL. Steady-state kinetic mechanism of LodA, a novel cysteine tryptophylquinone-dependent oxidase. FEBS Lett. 2014;588:752–756. doi: 10.1016/j.febslet.2014.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leontievsky AA, Vares T, Lankinen P, Shergill JK, Pozdnyakova NN, Myasoedova NM, Kalkkinen N, Golovleva LA, Cammack R, Thurston CF, Hatakka A. Blue and yellow laccases of ligninolytic fungi. FEMS Microbiol Lett. 1997;156:9–14. doi: 10.1111/j.1574-6968.1997.tb12698.x. [DOI] [PubMed] [Google Scholar]
- 28.Andrew CR, Yeom H, Valentine JS, Karlsson BG, Bonander N, Vanpouderoyen G, Canters GW, Loehr TM, Sandersloehr J. Raman-spectroscopy as an indicator of Cu-S bond-length in Type-1 and Type-2 Copper cysteinate proteins. J Am Chem Soc. 1994;116:11489–11498. [Google Scholar]
- 29.Feig AL, Lippard SJ. Reactions of nonheme Iron(II) centers with dioxygen in biology and chemistry. Chem Rev. 1994;94:759–805. [Google Scholar]
- 30.Padiglia A, Medda R, Pedersen JZ, Agro AF, Lorrai A, Murgia B, Floris G. Effect of metal substitution in copper amine oxidase from lentil seedlings. J Biol Inorg Chem. 1999;4:608–613. doi: 10.1007/s007750050384. [DOI] [PubMed] [Google Scholar]
- 31.Okazaki S, Nakano S, Matsui D, Akaji S, Inagaki K, Asano Y. X-Ray crystallographic evidence for the presence of the cysteine tryptophylquinone cofactor in L-lysine epsilon-oxidase from Marinomonas mediterranea. J Biochem. 2013;154:233–236. doi: 10.1093/jb/mvt070. [DOI] [PubMed] [Google Scholar]
- 32.Diaz AN. Absorption and Emission-Spectroscopy and Photochemistry of 1,10-Anthraquinone Derivatives - a Review. J Photoch Photobio A. 1990;53:141–167. [Google Scholar]
- 33.Cory RM, McKnight DM. Fluorescence spectroscopy reveals ubiquitous presence of oxidized and reduced quinones in dissolved organic matter. Environ Sci Technol. 2005;39:8142–8149. doi: 10.1021/es0506962. [DOI] [PubMed] [Google Scholar]
- 34.Fitzpatrick PF. Tetrahydropterin-dependent amino acid hydroxylases. Annu Rev Biochem. 1999;68:355–381. doi: 10.1146/annurev.biochem.68.1.355. [DOI] [PubMed] [Google Scholar]
- 35.Roberts KM, Fitzpatrick PF. Mechanisms of tryptophan and tyrosine hydroxylase. IUBMB Life. 2013;65:350–357. doi: 10.1002/iub.1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pearson AR, Marimanikkuppam S, Li X, Davidson VL, Wilmot CM. Isotope labeling studies reveal the order of oxygen incorporation into the tryptophan tryptophylquinone cofactor of methylamine dehydrogenase. J Am Chem Soc. 2006;128:12416–12417. doi: 10.1021/ja064466e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dove JE, Schwartz B, Williams NK, Klinman JP. Investigation of spectroscopic intermediates during copper-binding and TPQ formation in wild-type and active-site mutants of a copper-containing amine oxidase from yeast. Biochemistry. 2000;39:3690–3698. doi: 10.1021/bi992225w. [DOI] [PubMed] [Google Scholar]
- 38.Rigo A, Corazza A, di Paolo ML, Rossetto M, Ugolini R, Scarpa M. Interaction of copper with cysteine: stability of cuprous complexes and catalytic role of cupric ions in anaerobic thiol oxidation. J Inorg Biochem. 2004;98:1495–1501. doi: 10.1016/j.jinorgbio.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 39.Hage JP, Llobet A, Sawyer DT. Aromatic hydroxylation by Fenton reagents (reactive intermediate [Lx+FeIIOOH(BH+)], not free hydroxyl radical (HO)) Bioorg Med Chem. 1995;3:1383–1388. doi: 10.1016/0968-0896(95)00123-x. [DOI] [PubMed] [Google Scholar]
- 40.Maskos Z, Rush JD, Koppenol WH. The hydroxylation of tryptophan. Arch Biochem Biophys. 1992;296:514–520. doi: 10.1016/0003-9861(92)90605-v. [DOI] [PubMed] [Google Scholar]
- 41.McCann SD, Stahl SS. Copper-catalyzed aerobic oxidations of organic molecules: Pathways for two-electron oxidation with a four-eectron oxidant and a one-eectron redox-active catalyst. Accounts Chem Res. 2015;48:1756–1766. doi: 10.1021/acs.accounts.5b00060. [DOI] [PubMed] [Google Scholar]
- 42.Sehanobish E, Chacon-Verdu MD, Sanchez-Amat A, Davidson VL. Roles of active site residues in LodA, a cysteine tryptophylquinone dependent epsilon-lysine oxidase. Arch Biochem Biophys. 2015;579:26–32. doi: 10.1016/j.abb.2015.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen LY, Doi N, Durley RCE, Chistoserdov AY, Lidstrom ME, Davidson VL, Mathews FS. Refined crystal structure of methylamine dehydrogenase from paracoccus denitrificans at 1.75 angstrom resolution. J Mol Biol. 1998;276:131–149. doi: 10.1006/jmbi.1997.1511. [DOI] [PubMed] [Google Scholar]
- 44.Roujeinikova A, Scrutton NS, Leys D. Atomic level insight into the oxidative half-reaction of aromatic amine dehydrogenase. J Biol Chem. 2006;281:40264–40272. doi: 10.1074/jbc.M605559200. [DOI] [PubMed] [Google Scholar]