

Abstract
Membrane proteins represent ≈30% of the proteome of both prokaryotes and eukaryotes. Unique to cell surface receptors is their biogenesis pathway, which involves vesicular trafficking from the endoplasmic reticulum through the Golgi apparatus and to the cell surface. Increasing evidence suggests specific regulation of biogenesis for different membrane receptors, hence affecting their surface expression. We report the development of a pulse–chase assay to monitor function recovery after chemobleaching (FRAC) to probe the transit time of the Kir2.1 K+ channel to reach the cell surface. Our results reveal that the channel activity is contributed by a small fraction of channel protein, providing evidence of activity-silent “sleeping” molecules on the cell surface. This method distinguishes molecular density from functional density, and the assay strategy is generally applicable to other membrane receptors. The ability of the reported method to access the biogenesis pathways in a high-throughput manner facilitates the identification and evaluation of molecules affecting receptor trafficking.
Keywords: covalent modification, fluorescence recovery after photobleaching, ion channels, signaling, trafficking
The rate of membrane receptor trafficking is critical for the steady-state level of surface expression and is tightly coupled to signaling events (1–3). Within a given molecule, such as K+ channels, various trafficking sequence motifs have been identified that contribute to the specificity of trafficking behavior (4). For a given receptor, recent evidence suggests that the rate of trafficking may change according to the physiological state, such as during cellular aging (5). To assess the transit time from the endoplasmic reticulum (ER) to the cell surface, a conventional approach is to measure the time of ER-to-Golgi transition, which is thought to be the rate-limiting step. The remaining steps of biogenesis usually take a very short time, ranging from seconds to minutes (6). The ER-to-Golgi transition time usually is determined by pulse–chase labeling combined with monitoring a shift of molecular weight as a result of glycosylation. This technique has been particularly useful for studies to determine specific organelle transitions, such as a rate change from ER to Golgi by forward transport signals (7). However, for proteins with limited or no glycosylation, this approach is not applicable. Furthermore, many receptors and ion channels undergo a stationary step in their trafficking cascade after exiting from trans-Golgi and before cell-surface expression. For example, only a fraction of synthesized nicotinic acetylcholine receptor matures and expresses on the cell surface (8). The combination of different transport rates, maturation pathways, and posttranslational modifications warrants a more in-depth consideration of methods to directly monitor the rate by which a given receptor complex populates and/or repopulates the cell surface after exiting from the Golgi apparatus.
In addition to the ER–Golgi trafficking, one critical spatial transition underlying diverse biological activity is the regulation of protein expression on the cell surface (for review, see ref. 9). Fluorescence recovery after photobleaching (FRAP) was developed for imaging the movement of biological molecules in cells (10, 11). This method has been improved greatly with extensive use of genetically coded fluorophores, such as green fluorescent protein (GFP), providing a powerful means to address questions regarding protein localization, activity, interactions, and dynamics with living cells (12). This approach has high spatial resolution at the single-cell level. However, the combination of optic detection and the photobleaching methodology limits its applicability in certain areas, especially evaluation of global surface expression and coupling the imaging signal to functionality of targeted molecules.
Here, we employ a chemical bleaching strategy to irreversibly nullify targeted cell surface receptors. By monitoring the replenished activity after inactivation, the technology allows for measurement of time transition of receptor recovery on cell surface. Hence, we termed the procedure “function recovery after chemobleaching” (FRAC). The methodology takes advantage of a chemical-modification strategy generic to almost all membrane proteins. Application of FRAC to monitor Kir2.1 K+ channels revealed that only a small fraction of ion channel protein on the cell surface contributes to the bulk of channel activity, providing evidence for the difference of protein density and functional density. This result underscores the need to monitor cell-surface protein by both activity and imaging techniques.
Methods
Materials. The methanethiosulfonate reagents, such as [2-(trimethylammonium)ethyl] methanethiosulfonate bromide (MTSET), were purchased from Toronto Research Chemicals (Downsview, ON, Canada). Brefeldin A (BFA), sodium butyrate, and RbCl were purchased from Sigma.
Transfection and Stable Cell Lines. Expression vectors were constructed by using pCDNA3.1(+) (Invitrogen), and the Kir2.1 cDNA was cloned into HindIII and NotI sites. For the two Kir2.1 cDNA clones used in the study, one has the hemagglutinin (HA) epitope inserted at the first extracellular loop between M1 and P-loop to facilitate surface detection (13), referred as Kir2.1. The other has both the HA epitope and a peptide, FRGRSWTY, fused at the C terminus to facilitate surface expression (S.S. and M.L., unpublished data), which is referred to as Kir2.1Y. The transfection into HEK 293 cells was carried out with FuGENE 6 (Roche Diagnostics). Stable clones were generated by using neomycin resistance. Stable clones with high expression level of channel proteins were selected by using immunoprecipitation and flow cytometry. These stable cell lines were maintained in 50/50 DMEM/F12 medium containing 10% FBS, penicillin/streptomycin, l-glutamine, and 500 μg/ml G418.
Flow Cytometry. HEK 293 cells stably expressing Kir2.1 or Kir2.1Y channel were seeded at ≈3 × 105 cells per well in a six-well plate and allowed to grow for 16–20 h in complete growth medium with 5 mM sodium butyrate at 37°C and 5% CO2. Cells were washed with 1× PBS and harvested by incubation with 0.5 mM EDTA in 1× PBS for 5–10 min at room temperature. Cells then were washed twice with Hanks' balanced salt solution (HBSS) plus 5 mM Hepes (pH 7.3) and 2% FBS and incubated with rat anti-HA monoclonal antibody (Roche Diagnostics) on ice for 1 h. Cells then were washed twice again with HBSS staining medium and incubated with FITC-labeled goat anti-rat IgG antibody (Jackson ImmunoResearch) for 15 min on ice. Finally, the cells were washed twice with HBSS staining medium, and the channel-surface expression was measured by FACSCalibur (Becton Dickinson) with cellquest software (Becton Dickinson).
Rb+ Flux Assay and Atomic Absorption Spectrometry. All assays shown were performed at room temperature in poly(l-lysine) (0.1 mg/ml)-coated 24-well microplates. For experiments with transiently transfected cells, ≈1 × 105 cells were seeded in each well the day before transfection (0.3 μg of DNA per well). Sodium butyrate at 5 mM was added 6–8 h after transfection. The assays were performed 24 h after transfection. HEK 293 cells stably expressing either Kir2.1 or Kir2.1Y channel were seeded with a cell density of 2 × 105 cells per well. To enhance expression, 5 mM sodium butyrate was added 4–6 h after seeding. Cells then were cultured continuously for 16–20 h before experiments.
Rb+ Influx. To perform the assay, cells first were incubated in 50/50 DMEM/F12 complete growth medium containing 5 mM RbCl for the indicated time periods before the medium was quickly aspirated. Cells then were washed twice quickly with non-Rb+ DMEM/F12 medium and lysed with 0.5 ml of 0.1% SDS per well. The Rb+ concentration in cell lysates was measured by atomic absorption spectrophotometry (ICR8000, Aurora Biomed, Vancouver), according to the manufacturer's user manual.
Rb+ Efflux. To load Rb+ into cells, we incubated cells in 0.5 ml per well 50/50 DMEM/F12 complete growth medium containing ≈5 mM RbCl at 37°C for 3–4 h. Cells then were washed twice quickly with 1 ml per well non-Rb+ medium to remove residual Rb+. To perform the assay, 0.5 ml of non-Rb+ complete growth medium per well was added to the cells, which were incubated at room temperature for 15 min unless otherwise indicated in the legends of Figs. 1, 2, 3, 4, 5. The supernatants from each well were transferred quickly to a new 24-well plate at the end of incubation. Cells then were lysed with 0.5 ml of 0.1% SDS per well. The supernatants and cell lysates were diluted further with distilled water, and the diluted samples were transferred to a 96-well microplate. The Rb+ concentration in each sample was analyzed by using the ICR 8000. Rb+ efflux (%) was determined as
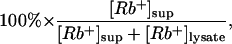 |
[1] |
where [Rb+]sup stands for the Rb+ concentration in the supernatant, and [Rb+]lysate stands for the Rb+ concentration in the cell lysate.
Fig. 1.

A schematic diagram outlining the FRAC assay.
Fig. 2.

Rb+ flux assays of HEK 293 cells transiently and stably expressing Kir2.1Y channel. (a and b) In Rb+ influx (a) and efflux (b), 293 cells were transiently transfected with Kir2.1Y expression vector (▪)or empty vector (•). Data obtained with 2.5 mM MTSET treatments are shown as □ or ○. (c) Rb+ efflux of 293 cells stably expressing Kir2.1Y channel with (□) or without (▪) MTSET treatment were measured. (d and e) Determination of MTSET treatment conditions. (d) Rb+ efflux (%) of 293 cells stably expressing Kir2.1Y channel (filled bars) and mock 293 cells (open bars) were quantified after being treated in triplicates with 5 mM MTSET for 0, 2.5, 5 and 10 min at room temperature. (e)Rb+ efflux (%) of 293 cells stably expressing Kir2.1Y channel (filled bars) and mock 293 cells (open bars) were quantified after being treated in duplicate with 0, 0.5, 1, 2.5 and 5 mM MTSET for 5 min at room temperature.
Fig. 3.

Detection of Kir2.1Y and Kir2.1 channel functional surface expression. (a) K+ channel activity measured by Rb+ efflux assayed at the indicated time periods. Results are for 293 cells stably expressing either Kir2.1 (Upper)or Kir2.1Y (Lower) channel after MTSET treatment (□), Rb+ efflux of 293 cells stably expressing Kir2.1 channel without MTSET treatment (▪) and mock 293 cells treated with (○) or without (•) MTSET. (b) Flow cytometry analyses of 293 cells stably expressing Kir2.1Y channel with MTSET treatment at different time points (0, 3, and 6 h) before staining with anti-HA antibody to quantify the surface expression of Kir2.1Y. FL, fluorescence intensity in a logarithmic scale. Events refers to the number of cells.
Fig. 4.

Temperature-dependent activity recovery of Kir2.1Y channel determined by Rb+ efflux. (a–c) Activity recovery of Kir2.1Y stable cell line after MTSET treatment (□), Rb+ efflux of Kir2.1Y stable cell line without MTSET treatment (▪), and Rb+ efflux of mock 293 cells after MTSET treatment (○)or without MTSET treatment (•). The recovery experiments were carried out at 15°C (a), 22°C (b), and 30°C (c). All measurements were performed in triplicate as described in Methods. (d) Normalized recovery rates from 0 to 5 h at 15°C (▪), 22°C (□), 30°C (▴), and 37°C (⋄) (from Fig. 3a). The Rb+ efflux of Kir2.1Y without MTSET treatment was set to 100%.
Fig. 5.

BFA effect on activity recovery of Kir2.1Y channel. (a) We incubated 293 cells stably expressing Kir2.1Y (hatched bars) and mock 293 cells (open bars) with 1 μM BFA at 37°C for 3 h after a 5-min MTSET treatment or without MTSET treatment. In control samples, neither MTSET nor BFA were added. Each column is the average of two samples. (b) Dose-dependent effect by BFA treatment. Results show 293 cells stably expressing Kir2.1Y that were incubated with BFA only (▪), cells stably expressing Kir2.1Y that were treated with MTSET before incubation with BFA (□), mock 293 cells incubated with BFA only (•), and mock 293 cells treated with MTSET before incubation with BFA (○). All experiments were performed in duplicate with 3-h incubation. The Rb+ efflux (%) is plotted against different concentrations of BFA at 37°C before Rb+ efflux assay.
MTSET Treatment. Fresh 100× MTSET stock solution (typically 250 mM) was prepared with cold sterile distilled water on the same day that the experiments were performed and stored at 4°C through the day. For a typical experiment, 5 μL of 100× stock solution was added to 0.5 ml of cell medium per well, and cells were incubated at room temperature for 5 min (unless otherwise indicated). The cell medium then was aspirated quickly at the end of incubation, and cells were washed twice with fresh medium to remove the residual MTSET.
Rb+ Efflux Recovery. After incubation in 50/50 DMEM/F12 complete growth medium containing 5 mM RbCl for 3 h, cells first were treated with 2.5 mM MTSET for 5 min; the residual MTSET was removed quickly by washing the cells twice with DMEM/F12 medium containing 5 mM RbCl. Then the cells were incubated at the indicated temperature in 5% CO2 for different time periods. At each indicated time point, cells were washed quickly again with Rb+-free medium, and the efflux assay was performed as described above.
Results and Discussion
To develop an assay that allows for monitoring the transit time of nacently surfaced receptor, our rationale is to first nullify (pulse) all functional molecules on the cell surface and then to monitor the recovery of activity at different time periods (chase). One thus may determine the rate required to populate the cell surface with newly inserted molecules (Fig. 1). Designing this assay requires consideration of at least three criteria. (i) A functional assay monitors activity only from molecules on the cell surface. Preferably, the assay will not affect protein half-lives on the cell surface. (ii) The assay time should be relatively short compared with the time required to repopulate the cell surface with functional molecules. (iii) The assay requires a rapid, quantitative, and irreversible inactivation of the functional proteins already on the cell surface, thereby allowing detection of the activity contributed by only the newly arrived (or newly activated) proteins on the cell surface.
Kir2.1 encodes an inward rectifier K+ channel consisting of four subunits that align a centrally positioned hydrophilic pore to conduct K+ ions (14). One of the attractive features of the Kir2.1 channel is that it has an open probability of nearly 100% at resting potential, thereby permitting an option of monitoring the activity without depolarization. Assays for detecting K+ channel activities include electrophysiological recording, fluorescence-based measurements, and Rb+ flux assay (for review, see ref. 15). All three types of assays can be achieved within minutes. The primary distinction concerns whether the assay reads signals from individual cells or a population of cells and whether the cells remain intact during the entire assay procedure. We chose the Rb+ flux assay because of its ability to assay a large population of cells for extended time periods without rupture of cell membrane. The flux activity linearly reflects the conducting ion passage. Rb+ is a nonphysiological ion but is permeable to almost all K+ channels, which affords general applicability. In addition, the Rb+ assays described here were carried out under conditions that do not tamper with the membrane potential, hence avoiding a stimulation of membrane fusion that could be caused by depolarization-induced Ca2+ influx.
To nullify the K+ channels already expressed on the cell surface, several approaches have been considered. These include antibody binding induced inhibition (16), pore blockers (such as tetraethylammonium), and covalent modifications by, for example, methanethiosulfonate (MTS) reagents (17). Of the three possibilities, covalent modification is superior because MTS reagents are irreversible under typical physiological redox conditions and would not cause clustering that often leads to other subsequent events, including endocytosis.
Earlier reports using electrophysiological recording have shown that membrane-nonpermeable MTS reagents such as MTSET reduce or abolish the current of Kir2.1 by covalently modifying the reduced form of cysteine residues already present in the channel protein or introduced by site-directed mutagenesis (18). It is not known whether or to what extent the modifications by MTS reagents will affect channel activity measured by a Rb+ flux assay. We tested several cysteine positions of Kir2.1 mutants by using MTSET, a positively charged cysteinyl sulfhydryl-specific reagent. Our results show that wild-type Kir2.1 may be quantitatively nullified by MTSET. Notably, a mutation of C149, the only cysteine on the extracellular side, leads to a loss of sensitivity to MTSET both by electrophysiological recording and Rb+ assay (data not shown). This data is consistent with results from electrophysiological recording (18), further confirming the fact that MTSET acts on the extracellular cysteine at the 149 position.
To monitor the specificity of the Rb+ assay and the effectiveness of Rb+ detection by atomic absorption spectrometry (19), we measured the Rb+ uptake signals of transiently transfected cells at different times of incubation. Comparison of transfected cells with nontransfected cells revealed an ≈2-fold signal-to-noise ratio (Fig. 2a) in a 10-min uptake. Treatment with MTSET before the assay abolished >80% of the specific signal. Prolonging uptake time increased the total signal but did not improve the signal-to-noise ratio. It should be noted that the influx measures the absolute level (mg/L) of Rb+ inside the cell. Thus, it is also a function of cell number in a given assay and dependent on Na+-K+ ATPase. We then compared the uptake (i.e., influx) with the efflux assay, which reflects the ratio of Rb+ released in the supernatant to total Rb+ in the loaded cells (see Methods and Fig. 2b). To further improve the signal-to-noise ratio, cell lines were generated that stably express either the Kir2.1 or Kir2.1Y channels (see Methods). The efflux assay using a stable Kir2.1Y line displayed a significant improvement of a nearly 4- to 6-fold signal-to-noise ratio (Fig. 2c). When cells were treated with MTSET, only background signals were observed. To determine the optimal conditions, we performed a titration by using different concentrations and time courses of the MTSET treatment. The results suggest an optimal treatment with 2.5 mM MTSET for 5 min (Fig. 2 d and e). These results demonstrate the functional expression of Kir2.1 channels, the sensitivity of the Rb+ efflux assay, and the feasibility of nullifying the channel activity on the cell surface by MTSET.
To evaluate FRAC, we generated two cell lines expressing either the Kir2.1 or Kir2.1Y where the protein expression level of Kir2.1Y is ≈2-fold higher than that of Kir2.1 (data not shown). Both cell lines were treated first with MTSET to nullify the channels on the cell surface. After its removal, the treated cells were assayed after incubations of the indicated periods of time for recovery. The half-maximal functional recovery time (FR1/2) at 37°C for both Kir2.1 and Kir2.1Y was 1 h (Fig. 3a). The functional expression reached 90% of the initial level 5 h after chemobleaching with MTSET. During the chase period, the Rb+ loaded cells without MTSET treatment gave rise to similar signals (Fig. 3a). The background Rb+ efflux of nontransfected cells remained consistent with or without MTSET treatment (Fig. 3a). The result shows that recovery was specific to the cells expressing the recombinant channels. To monitor the level of Kir2.1Y protein on the cell surface, flow cytometry analyses were carried out by using live cells stained with anti-HA antibody (see Methods). The total amount of protein signals remained essentially constant throughout the entire experimental time period of 5 h (Fig. 3b).
The source of functional recovery may originate from “reactivation” of the existing surface channel protein and/or from newly arrived vesicles. To test whether vesicular transport was responsible for delivering the recovered activity, the FRAC experiments were carried out in parallel but at different recovery temperatures (Figs. 3a and 4 a–c). It is known that lowering the temperature can stall the ER-to-Golgi or cis- to trans-Golgi transitions (20). In either case, only limited recovery should be observed, presumably caused by the vesicles in transit between trans-Golgi and the cell surface. Fig. 4 shows that the recovery of activity was progressively slowed when temperatures decreased from 37°C (FR1/2 = 1 h) to 30°C (FR1/2 = 2 h) to 22°C (FR1/2 = 3.5 h). At 15°C, which typically blocks the ER-to-Golgi transition (20), the activity was recovered partially, up to 40% of the initial level (Fig. 4a). Normalized recovery suggests a residual activity at 0 h (Fig. 4d). The activity may be contributed by a rapid recovery of the functional channels during the Rb+ assay, because all Rb+ efflux assays were performed at 22°C. These experiments support the notion that reduction of vesicular transport at progressively lower temperatures was causal to the different FR1/2 values.
Vesicular trafficking is sensitive to a variety of pharmacological agents. BFA is a hydrophobic membrane-permeable fungal toxin, which was reported to inhibit multiple steps of vesicular trafficking by causing disruption of Golgi apparatus and, to some extent, ER-to-Golgi transition (21). BFA has been used to block protein secretion (22), and it is thought to act on GTP exchange factors, which activate a family of small GTPases known as ADP-ribosylation factors (23–25). Hence, the application of BFA in the above FRAC assay could test the role of vesicular transport in the recovery of channel activity on the cell surface.
Experiments were carried out to compare the extent of activity recovery in 3 h under conditions of no treatment, MTSET only, and treatment with MTSET followed by BFA (Fig. 5a). The activity of nontreated cells remained consistent over a 3-h incubation. The MTSET-treated preparations displayed ≈70% recovery, consistent with data shown in Fig. 3a. When BFA was added immediately after the MTSET treatment, <20% recovery was observed. The BFA effect was specific to the recovery because without MTSET treatment, the 3-h incubation with BFA did not affect the activity for channels that were already on the cell surface (Fig. 5a).
Fig. 5b shows the dose-dependent effect of BFA treatment on the recovery of activity at 3 h. The results suggest that 0.1–0.2 μM BFA treatment gave intermediate effects. Prolonged incubation with 0.2 μM BFA for up to 8 h did not yield further improvements in recovery (data not shown). The BFA effect reaches a plateau at concentrations of >0.6 μM. In the absence of MTSET treatment, the surface channel activity displayed no sensitivity to BFA concentrations (Fig. 5b), demonstrating the specificity of BFA effects for recovery.
Incubation of BFA for the extended time of 3 h did not result in a detectable reduction of Rb+ efflux activity in the absence of MTSET treatment for Kir2.1Y (Fig. 5). In the case of Kir2.1, we observed reproducible ≈30% reduction over the same period of incubation (data not shown). The difference may stem from rate of endocytosis and total number of channel protein on cell surface. For Kir2.1Y, the combination of quantitative elimination of surface channel activity by MTSET, the >90% activity recovery, and little detectable reduction of Rb+ efflux in the presence of BFA then would predict a 2-fold increase of protein signal on the cell surface. To the contrary, Fig. 3b provides no support of a comparable increase at the protein level. Thus, the source of the recovered activity likely originated from newly surfaced channel molecules, which represent only a small fraction of total channel protein on the cell surface. It would be particularly interesting to investigate, especially in polarized cells, any potential spatial distribution preference for the freshly replenished channel protein coupled with FRAC.
The reported method allows for a direct measurement of the transition for repopulating functional channel protein on the cell surface. Of particular interest is the observation of consistent channel activity over a long time period in the absence of ER and Golgi vesicular transport (Fig. 5). When the surfaced channel molecules were irreversibly nullified, the cells were able to repopulate the surface with newly arrived functional channels within hours. In the 3-h recovery experiments, ≈70% of activity was recovered, but it was not accompanied by a significant increase of channel protein on the cell surface (Fig. 3). Together, these data provide evidence that the Rb+ efflux by Kir2.1Y channels on the cell surface is contributed by only a small fraction of the channel protein, suggesting the existence of a substantial fraction of “sleeping” channels. These sleeping channels are detectable at the protein level but functionally null to the Rb+ assay.
The FRAC experiments reported here allow for specific determination of the time required for a cell to populate its surface with functional channels and receptors. The electrophysiological measurement has allowed for the determination of overall conductance of the two cell lines, hence permitting an estimation of 5,000–7,000 conducting channels per cell for Kir2.1Y and 3,000–4,000 conducting channels for Kir2.1 (data not shown). It is known that different membrane proteins express with much variation in terms of molecular and functional density on the cell surface. The total recovery time likely reflects different incremental steps of biogenesis. The transit time measurement could provide a key parameter to differentiate their trafficking properties and potentially even discrete steps. For example, the fraction of BFA-insensitive recovery may represent vesicles that have exited ER and cis-Golgi compartments (Fig. 5a). Similarly, application of cycloheximide in FRAC experiments, which inhibits new protein synthesis, an earlier step than that inhibited by BFA, resulted in only 40% activity recovery (data not shown), compared with 20% activity recovery found with BFA (Fig. 5a). Hence, assays may provide resolution to isolate and evaluate compounds and cDNAs that affect these pathways, which could be useful for clinical intervention and mechanistic studies.
It is well known that cysteine substitution is a rather tolerable mutation. Site-directed mutagenesis allows for engineering a site into receptors of interest to confer sensitivity to the MTS reagent treatment (17). There are certainly other available methods in addition to MTS agents that may be used to “inactivate” receptors. For example, some receptors have antagonists with extremely high affinity and/or an unusually long off-rate. Dizocilpine (MK-801) is an open channel blocker for the N-methyl-d-aspartate (NMDA) receptor. It is conceivable that MK-801 combined with a calcium-based assay would allow for determination of the rate by which NMDA receptors repopulate the neuron surface. The spatial resolution of Ca2+-based imaging technologies may offer a comparison of rates for recovery in different subcellular domains of a single cell.
Of the >400 ion channel genes in the human genome, at least 167 are annotated to encode K+ channels. K+ channels are critical to a variety of biological processes ranging from neuronal excitability to oncogenesis. High-throughput assays to monitor channel activities and trafficking are of great value (15). Recently, a few cardiac K+ channels, particularly human ether-ago-go-related gene (hERG)-encoded K+ channels, have become subjects of recommended safety testing for all drug candidates, because many approved drugs have been found to inhibit hERG, which causes acquired long QT syndrome. These include therapeutic agents such as antiarrhythmics, antihistamines, antipsychotics, and antibiotics (26). The interactions of some of these compounds with the hERG K+ channels prolong cardiac repolarization, hence QT prolongation (long QT). In some cases, long QT induces torsade de pointes, which potentially could cause cardiac sudden death (27). The activity of hERG channels can be monitored by the Rb+ assay (28). There is an increasing demand of profiling compounds' effect on hERG channel activity in earlier stages of drug development. The reported assay should be useful to provide insights into possible roles of these drugs and candidate compounds in affecting the trafficking of the hERG channel protein.
Acknowledgments
We thank Dr. Yoshiro Kubo (National Institute for Physiological Sciences, Okazaki, Japan) for providing us with cysteine substitution mutants of Kir2.1 and members of the M.L. laboratory for valuable comments on the manuscript. Fig. 1 was illustrated by Bang Wong. The work is supported by grants from the National Institutes of Health (to M.L.) and an Established Investigator Award (M.L.) and a postdoctoral training award (H.S.) from the American Heart Association.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: BFA, brefeldin A; ER, endoplasmic reticulum; FRAC, function recovery after chemobleaching; FRAP, fluorescence recovery after photobleaching; HA, hemagglutinin; MTS, methanethiosulfonate; MTSET, [2-(trimethylammonium)ethyl] methanethiosulfonate bromide.
References
- 1.Pfeffer, S. (2003) Cell 112, 507-517. [DOI] [PubMed] [Google Scholar]
- 2.Ellgaard, L. & Helenius, A. (2003) Nat. Rev. Mol. Cell Biol. 4, 181-191. [DOI] [PubMed] [Google Scholar]
- 3.Mellman, I. & Warren, G. (2000) Cell 100, 99-112. [DOI] [PubMed] [Google Scholar]
- 4.Ma, D. & Jan, L. Y. (2002) Curr. Opin. Neurobiol. 12, 287-292. [DOI] [PubMed] [Google Scholar]
- 5.Blanpied, T. A., Scott, D. B. & Ehlers, M. D. (2003) Neurobiol. Aging 24, 1095-1104. [DOI] [PubMed] [Google Scholar]
- 6.Wieland, F. T., Gleason, M. L., Serafini, T. A. & Rothman, J. E. (1987) Cell 50, 289-300. [DOI] [PubMed] [Google Scholar]
- 7.Nishimura, N. & Balch, W. E. (1997) Science 277, 556-558. [DOI] [PubMed] [Google Scholar]
- 8.Merlie, J. P. & Lindstrom, J. (1983) Cell 34, 747-757. [DOI] [PubMed] [Google Scholar]
- 9.Bonifacino, J. S. & Glick, B. S. (2004) Cell 116, 153-166. [DOI] [PubMed] [Google Scholar]
- 10.Axelrod, D., Ravdin, P., Koppel, D. E., Schlessinger, J., Webb, W. W., Elson, E. L. & Podleski, T. R. (1976) Proc. Natl. Acad. Sci. USA 73, 4594-4598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Edidin, M., Zagyansky, Y. & Lardner, T. J. (1976) Science 191, 466-468. [DOI] [PubMed] [Google Scholar]
- 12.Reits, E. A. & Neefjes, J. J. (2001) Nat. Cell Biol. 3, E145-E147. [DOI] [PubMed] [Google Scholar]
- 13.Shikano, S. & Li, M. (2003) Proc. Natl. Acad. Sci. USA 100, 5783-5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang, J., Jan, Y. N. & Jan, L. Y. (1995) Neuron 15, 1441-1447. [DOI] [PubMed] [Google Scholar]
- 15.Xu, J., Chen, Y. & Li, M. (2004) Targets 3, 32-38. [Google Scholar]
- 16.Zhou, B. Y., Ma, W. & Huang, X. Y. (1998) J. Gen. Physiol. 111, 555-563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karlin, A. & Akabas, M. H. (1998) Methods Enzymol. 293, 123-145. [DOI] [PubMed] [Google Scholar]
- 18.Kubo, Y., Yoshimichi, M. & Heinemann, S. H. (1998) FEBS Lett. 435, 69-73. [DOI] [PubMed] [Google Scholar]
- 19.Terstappen, G. C. (1999) Anal. Biochem. 272, 149-155. [DOI] [PubMed] [Google Scholar]
- 20.Saraste, J. & Kuismanen, E. (1984) Cell 38, 535-549. [DOI] [PubMed] [Google Scholar]
- 21.Lippincott-Schwartz, J., Yuan, L. C., Bonifacino, J. S. & Klausner, R. D. (1989) Cell 56, 801-813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sciaky, N., Presley, J., Smith, C., Zaal, K. J. M., Cole, N., Moreira, J. E., Terasaki, M., Siggia, E. & Lippincott-Schwartz, J. (1997) J. Cell Biol. 139, 1137-1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chardin, P. & McCormick, F. (1999) Cell 97, 153-155. [DOI] [PubMed] [Google Scholar]
- 24.Jackson, C. L. & Casanova, J. E. (2000) Trends Cell Biol. 10, 60-67. [DOI] [PubMed] [Google Scholar]
- 25.Nebenfuhr, A., Ritzenthaler, C. & Robinson, D. G. (2002) Plant Physiol. 130, 1102-1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.De Ponti, F., Poluzzi, E. & Montanaro, N. (2000) Eur. J. Clin. Pharmacol. 56, 1-18. [DOI] [PubMed] [Google Scholar]
- 27.Antzelevitch, C. & Shimizu, W. (2002) Curr. Opin. Cardiol. 17, 43-51. [DOI] [PubMed] [Google Scholar]
- 28.Cheng, C. S., Alderman, D., Kwash, J., Dessaint, J., Patel, R., Lescoe, M. K., Kinrade, M. B. & Yu, W. (2002) Drug Dev. Ind. Pharm. 28, 177-191. [DOI] [PubMed] [Google Scholar]