

Abstract
The purpose of this study was to identify the biochemical and genetic defect in l-2-hydroxyglutaric aciduria, a neurometabolic disorder characterized by the presence of elevated concentrations of l-2-hydroxyglutaric acid in urine, plasma, and cerebrospinal fluid. Evidence is provided for the existence in rat tissues of a FAD-dependent enzyme catalyzing specifically the oxidation of l-2-hydroxyglutarate to α-ketoglutarate. This enzyme is mainly expressed in liver and kidney but also at lower levels in heart, brain, and other tissues. Subcellular fractionation indicates that the liver enzyme is present in mitochondria, where it is bound to membranes. Based on this information, a database search led to the identification of a gene encoding a human hypothetical protein homologous to bacterial FAD-dependent malate dehydrogenases and targeted to mitochondria. The gene encoding this protein, present on chromosome 14q22.1, was found to be in a region homozygous in patients with l-2-hydroxyglutaric aciduria from two consanguineous families. Three mutations that replaced a highly conserved residue (Lys-71-Glu and Glu-176-Asp) or removed exon 9 were identified in homozygous state in patients from three distinct families and were found to cosegregate with the disease. It is concluded that l-2-hydroxyglutarate is normally metabolized to α-ketoglutarate in mammalian tissues and that l-2-hydroxyglutaric aciduria is caused by mutations in the gene that most likely encodes l-2-hydroxyglutarate dehydrogenase. The pathological findings observed in this metabolic disorder must therefore be due to a toxic effect of l-2-hydroxyglutarate on the central nervous system.
Keywords: inborn error of metabolism, leukoencephalopathy, ataxia
As a neurometabolic disorder, l-2-hydroxyglutaric aciduria is characterized by the presence of elevated concentrations of l-2-hydroxyglutaric acid in plasma, cerebrospinal fluid, and urine (1–3). Clinically, the affection is characterized by progressive ataxia, mental deficiency with subcortical leukoencephalopathy, and cerebellar atrophy. As of 1996, >40 patients were known worldwide (3). The disease appears to be recessively transmitted, but the enzymatic defect leading to this condition is presently unknown, largely because of poor knowledge on the origin and fate of l-2-hydroxyglutarate. A NAD-dependent dehydrogenase acting on this compound has been reported to be present in liver (4). However, this enzyme displays a very high Km for l-2-hydroxyglutarate (≈10 mM), making it unsuitable to consume this substrate at physiologically relevant concentrations (probably of the order of 1 μM). Furthermore, it is not deficient in patients with l-2-hydroxyglutaric aciduria (5). l-2-hydroxyglutarate dehydrogenase activity transferring its electrons to pyocyanin was partially characterized ≈70 years ago by Weil-Malherbe (6), who found it to be mostly active in heart, kidney, and brain. However, no oxidation of l-2-hydroxyglutarate by rat liver mitochondria was later found by Lindahl and coworkers (7). Furthermore, rat tissues apparently do not contain any l-2-hydroxyglutarate oxidase activity (4).
In an attempt to identify enzymes acting on d- and l-2-hydroxyglutarate, we recently studied the detritiation of dl-2-hydroxy[2-3H]glutarate in rat liver extracts (8). Chromatography of such extracts allowed us to separate two main peaks of enzymes acting on dl-2-hydroxy[2-3H]glutarate. The activity of the enzyme present in the flow-through fractions was competed by much lower concentrations of l-than of d-2-hydroxyglutarate and the converse was true with the enzyme eluted with high salt concentrations. This finding suggested that these enzymes acted on l-2-hydroxyglutarate and d-2-hydroxyglutarate, respectively. Accordingly, the second enzyme was purified and shown to be a d-2-hydroxyglutarate dehydrogenase, transferring its electrons to artificial acceptors, and its sequence was identified by mass spectrometry analysis of a trypsin digest and confirmed by overexpression of the recombinant protein (8). The purpose of the present work was to characterize the enzyme able to detritiate l-2-hydroxyglutarate with the goal of identifying the defect in l-2-hydroxyglutaric aciduria.
Patients and Methods
The study was approved by the ethical committee of the medical faculty at the Université Catholique de Louvain. Written consent was obtained from all participants.
Patients. Family 1 is a Belgian family with consanguineous (first cousin) parents. The parents and four of the children had no neurological symptoms. The second and fourth children, both female, presented with mental retardation, seizures, and signs of ataxia, more severe in the second than in the fourth child. Brain MRI showed a (sub)cortical leukoencephalopathy and a diffuse atrophy in both cases. High levels of l-2-hydroxyglutaric acid were found in urine (291 and 401 mg/g creatinine) of both affected children but not in urine from the parents or the other siblings.
Family 2 is a Tunisian family with consanguineous (first cousin) healthy parents. One of the two children, a girl, had a slowly progressive neurodegenerative disorder with mild mental retardation, macrocephaly, ataxic gait with cerebellar signs, and seizures. MRI of the brain showed bilateral symmetrical abnormal signal in the subcortical white matter and dentate nuclei. Urinary organic acid analysis demonstrated increased excretion of l-2-hydroxyglutaric acid in the affected child but not in her healthy sibling.
Family 3 is a several-generation family of Lebanese origin. In a first-cousin marriage, both children were affected. At 2.5 years of age, the older child had an ataxic gait as the major symptom and had suffered a few episodes of seizures in the past. MRI revealed bilateral abnormal signal in the subcortical white matter. The younger child, investigated at 9 months of age, was asymptomatic and showed bilateral hyperintensity of the putamen and globus pallidum. In a second-cousin marriage, one of five children was affected. Five affected individuals were also identified in an 11-child kindred born to parents with no known consanguinity; two of these affected individuals had children who were healthy. Urinary organic acid analysis demonstrated increased excretion of l-2-hydroxyglutaric acid in all affected patients.
Enzyme Purification and Preparation of Tissular Extracts. l-2-hydroxyglutarate dehydrogenase was partially purified from frozen rat liver by chromatography on DEAE Sepharose (see Fig. 1A), as described for d-2-hydroxyglutarate dehydrogenase in ref. 8. In further purification attempts, frozen rat liver (1 g) was homogenized in two volumes of 20 mM Hepes, pH 7.1/25 mM KCl/1 μg/ml leupeptin/1 μg/ml antipain. We added 2% polyethyleneglycol 8000 to the homogenate and eliminated the precipitate by a 10-min centrifugation at 10,000 × g. We then added 6% polyethyleneglycol 8000 to the supernatant and collected the precipitate by centrifugation and resuspended it with a Down's homogenizer in 2 ml of 10 mM Tris·HCl, pH 8.5, containing 1% Triton X-100. After centrifugation to eliminate insoluble material, 1 ml of the supernatant was applied on a 1-ml Q-Sepharose column equilibrated with 20 mM Tris, pH 8.5/0.25% Triton X-100 (buffer A). The column was washed with buffer A, and the protein was eluted with a stepwise (50, 100, 150, 200, 300, and 500 mM) NaCl gradient in buffer A. l-2-hydroxyglutarate dehydrogenase activity was determined with the spectrophotometric assay. The peak fraction was used to test the effect of FAD and FMN by using the radiochemical assay.
Fig. 1.

Elution profile of l-2-hydroxyglutarate dehydrogenase from a DEAE-Sepharose column (A) and substrate saturation curve (B). (A) A rat liver extract prepared from 10 g of liver was chromatographed on DEAE Sepharose. The release of tritium from dl-2-hydroxy[2-3H]glutarate (○) (8), l-2-hydroxyglutarate dehydrogenase activity (•), and A280 (▵) were determined in the indicated fractions. (B) The activity was determined with the spectrophotometric assay on fraction 21 of the DEAE-Sepharose column in the presence of the indicated substrates. Protein was measured according to the Bradford method (15).
Tissue extracts were prepared by homogenizing frozen tissues with four volumes of 20 mM Hepes, pH 7.1, containing 1 μg/ml leupeptin and 1 μg/ml antipain in a Potter–Elvehjem apparatus. The extracts were used as such (without a centrifugation step) to determine the l-2-hydroxyglutarate dehydrogenase activity with the spectrophotometric and radiochemical assays. Subcellular fractionation of liver was carried out as described in refs. 8 and 9.
Enzyme Assays. The spectrophotometric assay was performed at 30°C by comparing the reduction of iodonitrotetrazolium (INT) in the absence and presence of the indicated concentrations (usually 100 μM) of l-2-hydroxyglutarate. The assay mixture contained 50 mM Hepes (pH 7.1), 0.01% cremophor, and 1.5 mM INT. The assay was initiated by the addition of 5–20 μl of tissue homogenate, cell fraction, or partially purified preparation of enzyme. A500 was measured for at least 40 min. Tissues contain endogenous compounds that react with INT; therefore, it was mandatory to run a blank in the absence of substrate. The presence of endogenous reducing agents also means that this assay could not be used to detect low activities of l-2-hydroxyglutarate dehydrogenase. A molar absorbance of 19,300 at 500 nm (10) for the INT–formazan complex was used to compute activities.
l-2-hydroxyglutarate dehydrogenase was also measured with a radiochemical assay by using dl-2-hydroxy[2-3H]glutarate (8). Because the latter is a racemic mixture, both d- and l-2-hydroxyglutarate dehydrogenases are detected in this way, and the specificity of the assay depends on prior separation of the two enzymes. Km determinations on the partially purified enzyme were performed in an assay mixture containing 20 mM Hepes, pH 7.1, 25 mM KCl, 1 mM MgCl2, 200,000 cpm dl-2-hydroxy[2-3H]glutarate, variable concentrations of l-2-hydroxyglutarate, and, when mentioned,1.5 mM INT. The assay was initiated by the addition of the enzymatic preparation and arrested by addition of perchloric acid. Tritiated water was separated from unreacted substrate as described in ref. 8. The radiochemical assay can also be used to determine specifically the l-2-hydroxyglutarate dehydrogenase activity in tissue extracts provided that d-2-hydroxyglutarate dehydrogenase is inhibited. Because the Km of the latter enzyme is low (≈3 μM) and barely affected by INT, its action on d-2-hydroxy[2-3H]glutarate can be inhibited by including 1 mM d-2-hydroxyglutarate in the assay. The assay mixture (250 μl), therefore, comprises 20 mM Hepes, pH 7.1, 25 mM KCl, 1 mM MgCl2, 100 μM l-2-hydroxyglutarate, 200,000 cpm dl-2-hydroxy[2-3H]glutarate, 1 mM d-2-hydroxyglutarate, 1.5 mM INT (included to increase the Vmax of the enzyme and facilitate comparison with the spectrophotometric assay), and 20 μl of tissue extracts.
Microsatellite Analysis and Mutation Detection. Genomic DNA was isolated from leukocytes with the QIAmp blood kit (Qiagen, Hilden, Germany), according to the manufacturer's instructions. Microsatellite markers close to the putative l-2-hydroxyglutarate dehydrogenase gene were selected from the University of California (Santa-Cruz) Genome Bioinformatics web site (http://genome.ucsc.edu). Genotyping by using 32P-labeled oligonucleotides was performed as described in ref. 11.
Single-strand conformation polymorphisms (12) and heteroduplex (13) analysis were used to screen all 10 exons and the flanking intronic sequences of the putative l-2-hydroxyglutarate dehydrogenase gene. PCR reactions were performed by using primers (Table 1, which is published as supporting information on the PNAS web site) synthesized by Eurogentec (Ougrée, Belgium) in 20-μl volumes on 10–20 ng of genomic DNA template, as described by ref. 14 by using 1 unit of DNA polymerase (BIOTOOLS B&M Labs, Madrid, Spain). PCR products to be sequenced were purified with the QIAquick PCR purification kit (Qiagen) and sequenced directly on both strands on a CEQ2000 sequencer (Beckman Coulter).
Results
Biochemical Characterization of l-2-Hydroxyglutarate Dehydrogenase. We previously reported that chromatography of a frozen rat liver extract on DEAE Sepharose separates two main enzymes catalyzing the release of tritiated water from dl-2-hydroxy[2-3H]glutarate (Fig. 1A). One enzyme, present in the flow-through fractions, appears to be specific for the l-isomer, because the detritiation it catalyses is inhibited by lower concentrations of unlabeled l-2-hydroxyglutarate than of d-2-hydroxyglutarate. The second main peak corresponds to d-2-hydroxyglutarate dehydrogenase (8). As shown in Fig. 1B, the enzyme present in the first peak catalyzed the reduction of INT in the presence of l-2-hydroxyglutarate, although not in the presence of d-2-hydroxyglutarate, l-malate, or l-lactate, indicating that it is a l-2-hydroxyglutarate dehydrogenase. With the radiochemical assay, the kinetic properties depended highly on the presence of the electron acceptor INT. Km values of 3.5 μM and 150 μM and Vmax values of 0.012 and 0.10 nmol·min–1·mg–1 protein were observed with 0 and 1.5 mM INT, respectively. The effect of INT to increase Km and Vmax is presumably due to the fact that it facilitates electron disposal. The ≈10-fold lower Vmax obtained with the radiochemical assay when performed in the presence of INT as compared with the spectrophotometric assay (≈1 nmol·min–1·mg–1 protein) is most likely due to a primary isotope effect. Assays of α-ketoglutarate with l-glutamate dehydrogenase, performed as described in ref. 8, indicated that l-2-hydroxyglutarate was oxidized to the corresponding 2-keto acid (data not shown).
In further attempts to purify the enzyme from rat liver, we noticed that it quantitatively precipitated with low concentrations (6–8%) of polyethyleneglycol in the absence but not the presence of Triton X-100, suggesting that it was membrane-bound. Further purification attempts were therefore performed with an 8% polyethyleneglycol pellet that was resuspended in a medium containing Triton X-100 and applied onto a DEAE-Sepharose column. Under these conditions, the enzyme was retained by the column and eluted with the salt gradient with a 35% recovery provided that Triton X-100 was present in the elution medium (results not shown). With such a preparation, it was found that the enzyme was stimulated by FAD by ≈30% but not by FMN (Fig. 2). Attempts to purify the enzyme further were unsuccessful, largely because of its instability.
Fig. 2.

Effect of FAD and FMN on the activity of l-2-hydroxyglutarate dehydrogenase. The rat liver enzyme was partially purified by chromatography on a Q-Sepharose column in the presence of detergents. It was assayed with the radiochemical assay in the presence of 100 μM unlabeled l-2-hydroxyglutarate, 1.5 mM INT, and the indicated concentrations of FAD or FMN.
Subcellular Localization and Tissue Distribution. Analysis of subcellular liver fractions obtained by differential centrifugation (9) with the spectrophotometric assay indicated l-2-hydroxyglutarate dehydrogenase activities of 0.51, 0.12, 0.0, and 0.03 nmol·min–1·mg–1 protein in the mitochondrial, light-mitochondrial, microsomal, and soluble fractions, indicating a mitochondrial localization. We also determined the tissue distribution of this enzyme both with the spectrophotometric assay and with the radiochemical assay (which was more sensitive, despite the primary isotopic effect). The highest activities were observed in liver and kidney, whereas in heart, brain, skeletal muscle, and spleen, the activities were ≈40%, 20%, 10%, and 5% of those observed in liver or kidneys (Fig. 3). It should be noted that the spectrophotometric assay did not allow us to measure accurately the activities in skeletal muscle and spleen.
Fig. 3.

Tissue distribution of l-2-hydroxyglutarate dehydrogenase. The activity was determined in homogenates from the indicated rat tissues with the spectrophotometric (spectr.) assay (Upper) or the radiochemical assay (Lower).
Identification of a Putative l-2-Hydroxyglutarate Dehydrogenase in Genome Databases. The experiments reported above indicated that l-2-hydroxyglutarate was metabolized in mammalian tissues by a FAD-dependent, membrane-bound mitochondrial dehydrogenase. No sequence of FAD-dependent l-2-hydroxyglutarate dehydrogenase is presently known. However, bacteria contain a FAD-dependent, membrane-bound enzyme that oxidizes l-malate, a close structural analog of l-2-hydroxyglutarate, to oxaloacetate (16). psi-blast searches (17) starting from the sequence of the Corynebacterium glutamicum enzyme (GenBank accession no. O69282) allowed us to identify after two iterations a human (C14orf160, GenBank accession NP_079160) and a closely related mouse protein (GenBank accession no. NP_663418.1) sharing ≈19% sequence identity with bacterial FAD-dependent l-malate dehydrogenase. Interestingly, the sequences encoding these enzymes comprise a mitochondrial targeting peptide (score of 0.908 for the human enzyme, as predicted with target p; see ref. 18), as well as an N-terminal (after removal of the mitochondrial targeting peptide) hydrophobic domain. These enzymes were therefore likely to encode l-2-hydroxyglutarate dehydrogenase. The human and mouse enzymes are aligned in Fig. 4 with putative proteins from Synechocystis sp. and Escherichia coli, sharing ≈40% sequence identity with the human protein. The human cDNA (GenBank accession no. NM_024884.1), which encodes the putative l-2-hydroxyglutarate dehydrogenase, has a length of 2040 bp, its initiator codon starts at nucleotide 80, and it encodes a 463-aa protein. The corresponding gene (GeneBank accession no. 79944) is located on chromosome 14q22.1, extends over ≈75 kbp and comprises 10 exons (Fig. 5A). The sequence for this gene can be obtained at www.ensembl.org under accession number ENSG00000087299.
Fig. 4.

Alignment of the putative human l-2-hydroxyglutarate dehydrogenase with homologous proteins from different species. The alignment shows sequences from the following species: Homo sapiens (Hs; GenBank accession no. NP-079160.1); Mus musculus (Mm; GenBank accession no. NP-663418.1); Synechocystis sp. (Sy; GenBank accession no. NP-442886.1), and E. coli (Ec; GenBank accession no. NP-289209.1). The coding region of the human cDNA was amplified from liver RNA by RT-PCR, and its sequence was confirmed (GenBank accession no. AY757363). The putative cleavage site (><) for the mitochondrial presequence in the human sequence as determined withtarget p (18), and the mutations found in the three different families (E, D, and x) and a conserved stretch of hydrophobic residues (h) are indicated above the alignment.
Fig. 5.

Structure of the putative l-2-hydroxyglutarate dehydrogenase gene (A) and cosegregation of mutations in this gene with l-2-hydroxyglutaric aciduria in three consanguineous families (B–D). (A) The polymorphic markers used to study the L2HGDH locus in families 1 and 2. (B–D) Alleles of these polymorphic markers and of the L2HGDH gene are indicated below each investigated individual in the families. In family 1 (B), the Δ allele corresponds to the removal of exon 9. In families 2 (C) and 3 (D), the one-letter code is used to designate either Lys (K) or Glu (E) at position 81 and Glu or Asp (D) at position 176. In the large, 11-child-kindred family, the first and last affected children had healthy children (data not shown).
Homozygosity Mapping of the Candidate Gene and Identification of Mutations. The availability of unrelated consanguineous families comprising two (family 1) and one (family 2) patient(s) with l-2-hydroxyglutaric aciduria enabled us to determine whether the gene (which we propose to designate L2HGDH) encoding the putative l-2-hydroxyglutarate dehydrogenase was linked to l-2-hydroxyglutaric aciduria by using homozygosity mapping. l-2-hydroxyglutaric aciduria being a rare recessive disorder, it is indeed likely that patients with consanguineous parents have inherited twice the same copy of the mutated gene and that they are also homozygous for neighboring polymorphic markers. Polymorphic markers were therefore selected on both sides of the candidate gene at distances of 0.4 and 1.3 Mbp, respectively. As shown in Fig. 5, the candidate gene was indeed found to be present in a region homozygous in patients of both families but not in the parents or unaffected siblings.
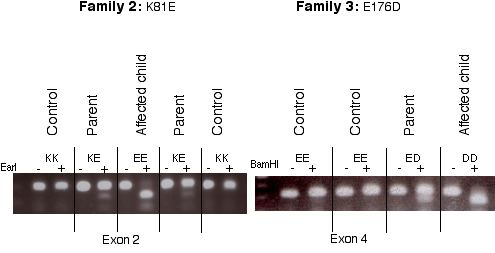
All 10 exons of the candidate gene were therefore amplified in the patients and the parents in the two families and in a third, large family from which we had obtained DNA samples in the meantime. The amplification products were analyzed by single-strand conformation polymorphisms and heteroduplex analysis, and the exons showing migration shifts were sequenced. This approach led us to find that the patient in family 2 was homozygous for a mutation in exon 2 (A320G) that replaces the highly conserved Lys-81 with a Glu and that all eight patients from family 3 were homozygous for a mutation in exon 4 (G607T) that replaces the highly conserved Glu-176 with an Asp (Figs. 4 and 5). Each of these mutations introduces an additional restriction site (EarI and BamHI, respectively), which allows their easy identification on PCR products (Fig. 6, which is published as supporting information on the PNAS web site). In family 1, no significant mutation was found in exons 1–8 and 10, but exon 9 could not be amplified, suggesting a deletion. Primers situated 2.4 kb upstream and 1.9 kb downstream of exon 9 allowed the amplification of a product of 4.5 kb in controls and 3 kb in the patients and their parents, indicating a 1.5-kb deletion (data not shown). Sequencing demonstrated that the deletion took place between two perfect 22-bp repeats (TGGGATTA-CAGGTGTGAGCCAC) that started 175 bp upstream and 1,227 bp downstream of exon 9, respectively. For all three families, the parents of the patients were heterozygous for the mutations identified in the children, and no healthy sibling was homozygous for these mutations. None of these three mutations could be found in 50 controls.
In addition to these mutations, several polymorphisms were identified. Some of them (C238T and A706G in the Ile-53 and Ala-209 codons, respectively) did not modify the encoded amino acid. Another substitution (T132G) led to the nonconservative replacement of Leu-18 with an Arg. However, Leu-18 is not a conserved amino acid, being a Cys in the mouse sequence. Furthermore, Leu-18 is present in a region of the protein that is presumably removed by processing (Fig. 4). Finally, some of the (healthy) parents were homozygous either for a Leu or an Arg at this position. These findings indicate that the Leu-18-Arg substitution is a polymorphism without pathological significance. We verified also that the heteroduplex/single-strand conformation polymorphisms analysis approach did not lead us to miss mutations by sequencing all exons that did not show migration shifts in at least one patient and one obligate heterozygote per family.
Discussion
Properties of the Enzyme. Here we describe the properties of a mitochondrial and membrane-bound enzyme that catalyses the conversion of l-2-hydroxyglutarate to α-ketoglutarate. This enzyme is slightly but significantly stimulated by FAD, suggesting that this compound serves as a cofactor. The lack of a higher degree of stimulation is presumably due to the fact that FAD is tightly bound, as is often the case with FAD-linked enzymes. The association of the dehydrogenase with the membrane suggests that it transfers its reducing equivalents to the respiratory chain, most probably at the level of ubiquinone, which, considering the oxidoreduction potential of this acceptor compared with that of 2-hydroxyglutarate (19), should favor the oxidation of l-2-hydroxyglutarate. The enzyme appears to be specific for l-2-hydroxyglutarate because it does not act on l-lactate, l-malate, or d-2-hydroxyglutarate, and it can be measured either through the release of tritiated water from dl-2-hydroxy[2-3H]glutarate or by the reduction of an artificial electron acceptor. Comparisons of the two assays indicate that the enzyme acts ≈10 times less rapidly on the radiolabeled substrate than on unlabeled l-2-hydroxyglutarate, indicating a primary isotope effect of ≈10. This rather high value indicates that the breaking of the C2–H bond is a rate-limiting step in the reaction, which also appears to be the case for two other flavoproteins acting on α-hydroxy acids: d-2-hydroxyglutarate dehydrogenase, for which a tritium isotope effect of 4.5 has been observed (8), and d–lactate oxidase, which shows a deuterium isotopic effect of 8 (20). In contrast to another report (7), our work confirms the existence of a l-2-hydroxyglutarate dehydrogenase in mammalian tissues (6). Our work also provides additional biochemical characterization that has led to the identification of the gene mutated in l-2-hydroxyglutaric aciduria (see below).
Identification of the Gene Mutated in l-2-Hydroxyglutaric Aciduria. It is likely that l-2-hydroxyglutarate dehydrogenase deficiency is responsible for l-2-hydroxyglutaric aciduria. However, the activity of this enzyme is below the detection of our assay methods in fibroblasts, lymphoblasts, or leukocytes, which prevents us from checking this hypothesis directly. Knowing some of the properties of l-2-hydroxyglutarate dehydrogenase, we identified a human gene encoding a protein related to bacterial FAD-dependent malate dehydrogenase and predicted to be targeted to the mitochondria. This gene was found to be mutated in all patients with l-2-hydroxyglutaric aciduria that we investigated. Two of the mutations affect highly conserved residues (see Fig. 4) and a third one removes a complete exon, being therefore most likely responsible for a loss of enzymatic activity. These findings suggest that this gene encodes l-2-hydroxyglutarate dehydrogenase or a subunit of this enzyme. Preliminary attempts to overexpress this enzyme in human embryonic kidney cells did not produce an active enzyme in a reproducible manner (R.R. and M.V.-d.-C., unpublished results), preventing us to prove definitely the identification of l-2-hydroxyglutarate dehydrogenase. This lack of success may be due to the fact that l-2-hydroxyglutarate dehydrogenase is a membrane protein and that such proteins are notoriously difficult to overexpress. Another interesting possibility is that l-2-hydroxyglutarate dehydrogenase may be composed of two or more different subunits, meaning that coexpression of the different subunits would be needed to get a functional enzyme. If the latter is true, some cases of l-2-hydroxyglutaric aciduria may be due to mutations in a different gene than the one that we have identified. Our finding that the gene encoding the putative l-2-hydroxyglutarate dehydrogenase is mutated in all of the three families that we have investigated suggests, however, that it is most often, if not always, implicated in l-2-hydroxyglutaric aciduria.
Pathophysiological Implications. The finding that l-2-hydroxyglutaric aciduria is due to a defect in a putative l-2-hydroxyglutarate dehydrogenase indicates that the pathological symptoms observed in this disease are not due to the lack of formation of a (hypothetical) compound for which l-2-hydroxyglutarate would be a precursor. α-Ketoglutarate can indeed be amply provided by other sources. The deleterious consequences of l-2-hydroxyglutarate dehydrogenase deficiency are therefore most likely the consequence of the accumulation of l-2-hydroxyglutarate. This compound has been shown to induce oxidative stress (21) and to inhibit mitochondrial creatine kinase in the cerebellum (22). The structural analogy of l-2-hydroxyglutarate with l-glutamate suggests that it could also interfere with glutamatergic transmission. Of interest is the recent observation that l-2-hydroxyglutarate significantly increased l-[3H]glutamate uptake into synaptosomes and synaptic vesicles, without altering synaptosomal glutamate release and glutamate binding to synaptic plasma membranes (23).
Whatever the mechanism of this toxicity, it would be most interesting to identify the source of l-2-hydroxyglutarate because this could help identify strategies to decrease its formation and thereby impair the progression of the disease in patients with l-2-hydroxyglutaric aciduria. An endogenous source for l-2-hydroxyglutarate is likely: It is known that at least one isozyme of l-lactate dehydrogenase, called L-LDH X and found in rat testis but not detected in liver, muscle, or heart (24), converts α-ketoglutarate to l-2-hydroxyglutarate. This reaction is thermodynamically favorable (19), being comparable with the NAD-linked conversions of pyruvate and oxaloacetate to the corresponding l-2-hydroxyacids, lactate and malate. Because l-2-hydroxyglutarate has apparently no physiological role in mammals, the function of the FAD-linked l-2-hydroxyglutarate dehydrogenase might be one of “metabolic repair” (conceptually analogous to DNA and protein repair), in this case the elimination of a toxic compound that forms because of the imperfectness of some enzymes.
The finding (25) that l-2-hydroxyglutarate accumulates to a much larger extent in fibroblasts derived from patients with l-2-hydroxyglutaric aciduria than from controls (230 nmol versus 30 nmol per 96 h × mg protein) supports an endogenous source for this compound. Our hypothesis is that the formation of l-2-hydroxyglutarate, most likely through reduction of α-ketoglutarate by NAD- or NADP-linked enzymes, is compensated by its removal by the FAD-linked l-2-hydroxyglutarate dehydrogenase in fibroblasts from controls but not from patients. Calculations indicate that enzymatic activities well below the detection level of our assays in fibroblasts would be enough to account for these observations.
In conclusion, we have identified a gene encoding a mitochondrial l-2-hydroxyglutarate dehydrogenase linked to FAD and found that this gene is mutated in l-2-hydroxyglutaric aciduria. The symptoms of this affection are therefore most likely due to the toxic effects of l-2-hydroxyglutarate, the source of which still needs to be identified.
Supplementary Material
Acknowledgments
This work was supported by the Concerted Research Action program of the Communauté Française de Belgique, the Interuniversity Attraction Poles Program of the Belgian Science Policy, and the Fonds de la Recherche Scientifique Médicale. M.V.-d.-C. is tenured investigator of the Fonds National de la Recherche Scientifique. R.R. is Fellow of the Research Formation in Industry and Agriculture, Belgium.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviation: INT, iodonitrotetrazolium.
Data deposition: The sequence reported in this paper has been deposited in the GenBank database (accession no. AY757363).
Note Added in Proof. Using homozygosity mapping, another group of investigators (26) recently identified the gene mutated in l-2-hydroxyglutaric aciduria and proposed to name it Duranin. This gene is the same as that reported in the present paper.
References
- 1.Duran, M., Kamerling, J. P., Bakker, H. D., van Gennip, A. H. & Wadman, S. K. (1980) J. Inherited Metab. Dis. 3, 109–112. [DOI] [PubMed] [Google Scholar]
- 2.Barth, P. G., Hoffmann, G. F., Jaeken, J., Wanders, R. J., Duran, M., Jansen, G. A., Jakobs, C., Lehnert, W., Hanefeld, F., Valk, J., et al. (1993) J. Inherited Metab. Dis. 16, 753–761. [DOI] [PubMed] [Google Scholar]
- 3.Chen, E., Nyhan, W. L., Jakobs, C., Greco, C. M., Barkovich, A. J., Cox, V. A. & Packman, S. (1996) J. Inherited Metab. Dis. 19, 335–343. [DOI] [PubMed] [Google Scholar]
- 4.Jansen, G. A. & Wanders, R. J. (1993) Biochim. Biophys. Acta 1225, 53–56. [DOI] [PubMed] [Google Scholar]
- 5.Wanders, R. J., Vilarinho, L., Hartung, H. P., Hoffmann, G. F., Mooijer, P. A., Jansen, G. A., Huijmans, J. G., de Klerk, J. B., ten Brink, H. J., Jakobs C., et al. (1997) J. Inherited Metab. Dis. 20, 725–726. [DOI] [PubMed] [Google Scholar]
- 6.Weil-Malherbe, H. (1937) Biochem. J. 31, 2080–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lindahl, G., Lindstedt, G. & Lindstedt, S. (1967) Arch. Biochem. Biophys. 119, 347–352. [DOI] [PubMed] [Google Scholar]
- 8.Achouri, Y., Noël, G., Vertommen, D., Rider, M. H., Veiga-da-Cunha, M. & Van Schaftingen, E. (2004) Biochem. J. 381, 35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Duve, C., Pressman, B. C., Gianetto, R., Wattiaux, R. & Appelmans, F. (1955) Biochem. J. 60, 614–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Munujos, P., Coll-Canti, J., Gonzalez-Sastre, F. & Gella, F. J. (1993) Anal. Biochem. 212, 506–509. [DOI] [PubMed] [Google Scholar]
- 11.Boon, L. M., Mulliken, J. B., Vikkula, M., Watkins, H., Seidman, J., Olsen, B. R. & Warman, M. L. (1994) Hum. Mol. Genet. 3, 1583–1587. [DOI] [PubMed] [Google Scholar]
- 12.Orita, M., Iwahana, H., Kanazawa, H., Hayashi, K. & Sekiya, T. (1989) Proc. Natl. Acad. Sci. USA 86, 2766–2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nagamine, C. M., Chan, K. & Lau, Y. F. (1989) Am. J. Hum. Genet. 45, 337–339. [PMC free article] [PubMed] [Google Scholar]
- 14.Veiga-da-Cunha, M., Gerin, I., Chen, Y. T., de Barsy, T., de Lonlay, P., Dionisi-Vici, C., Fenske, C. D., Lee, P. J., Leonard, J. V., Maire, I., et al. (1998) Am. J. Hum. Genet. 63, 976–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bradford, M. M. (1976) Anal. Biochem. 72, 248–254. [DOI] [PubMed] [Google Scholar]
- 16.Molenaar, D., van der Rest, M. E. & Petrovic, S. (1998) Eur. J. Biochem. 254, 395–403. [DOI] [PubMed] [Google Scholar]
- 17.Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W. & Lipman, D. J. (1997) Nucleic Acids Res. 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Emanuelsson, O., Nielsen, H., Brunak, S. & von Heijne, G. (2000) J. Mol. Biol. 300, 1005–1016. [DOI] [PubMed] [Google Scholar]
- 19.Buckel, W. & Miller, S. L. (1987) Eur. J. Biochem. 164, 565–569. [DOI] [PubMed] [Google Scholar]
- 20.Cromartie, T. H. & Walsh, C. (1975) Biochemistry 14, 3482–3489. [DOI] [PubMed] [Google Scholar]
- 21.Latini, A., Scussiato, K., Rosa, R. B., Leipnitz, G., Llesuy, S., Bello-Klein, A., Dutra-Filho, C. S. & Wajner, M. (2003) J. Neurosci. Res. 74, 103–110. [DOI] [PubMed] [Google Scholar]
- 22.Da Silva, C. G., Bueno, A. R., Schuck, P. F., Leipnitz, G., Ribeiro, C. A., Wannmacher, C. M., Wyse, A. T. & Wajner, M. (2003) Int. J. Dev. Neurosci. 21, 217–224. [DOI] [PubMed] [Google Scholar]
- 23.Junqueira, D., Brusque, A. M., Porciuncula, L. O., Rotta, L. N., Ribeiro, C. A., Frizzo, M. E., Dutra-Filho, C. S., Wannmacher, C. M., Wyse, A. T., Souza, D. O., et al. (2003) Metab. Brain Dis. 18, 233–243. [DOI] [PubMed] [Google Scholar]
- 24.Schatz, L. & Segal, H. L. (1969) J. Biol. Chem. 244, 4393–4397. [PubMed] [Google Scholar]
- 25.Struys, E. A., Verhoeven, N. M., Roos, B., Jakobs, C. (2003) Clin. Chem. 49, 1133–1138. [DOI] [PubMed] [Google Scholar]
- 26.Topcu, M., Jobard, F., Halliez, S., Coskun, T., Yalcinkaya, C., Gerceker, F. O., Wanders, R. J., Prud'homme, J. F., Lathrop, M., Ozguc, M., et al. (2004) Hum. Mol. Genet., in press. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}